ADMIRAZ 0.25 mg film kapl� tablet (84 tablet) K�sa �r�n Bilgisi
{ Siponimod Fumarik Asit }
1. BE�ER� TIBB� �R�N�N ADI
ADM�RAZ 0,25 mg film kapl� tablet
2. KAL�TAT�F VE KANT�TAT�F B�LE��M
Etkin madde
Siponimod fumarik asit 0,278 mg
(0,25 mg siponimoda kar��l�k gelmektedir)
Yard�mc� maddeler
Laktoz monohidrat (s���r kaynakl�) 62,197 mg
Soya lesitin 0,092 mg
Yard�mc� maddelerin tam listesi i�in b�l�m 6.1'e bak�n�z.
3. FARMAS�T�K FORMU
Film kapl� tablet
Bir y�z�nde logo ve di�er y�z�nde “T” bask�s� bulunan, yakla��k 6,1 mm �ap�nda, soluk k�rm�z�, yuvarlak, bikonveks, kenarlar� e�imli film kapl� tablet.
4. KL�N�K �ZELL�KLER
4.1. Terap�tik endikasyonlar
ADM�RAZ, eri�kinlerde, radyolojik veya klinik olarak aktif sekonder progresif multipl skleroz hastalar�n�n tedavisi i�in endikedir.
4.2. Pozoloji ve uygulama �ekli
Siponimod tedavisi, multipl skleroz tedavisinde deneyimli bir doktor taraf�ndan ba�lat�lmal� ve denetlenmelidir.
Tedaviye ba�lamadan �nce hastalar, CYP2C9 metaboliz�r durumlar�n� belirlemek i�in CYP2C9 i�in genotiplenmelidir (bkz. B�l�m 4.4, 4.5 ve 5.2).
CYP2C9*3*3 genotipi olan hastalarda siponimod kullan�lmamal�d�r (bkz. B�l�m 4.3, 4.4 ve 5.2).
CYP2C9*2*3 veya *1*3 genotipi olan hastalarda, �nerilen idame dozu g�nde bir kez al�nan 1 mg'd�r (0,25 mg'l�k d�rt tablet) (bkz. B�l�m 4.4 ve 5.2).
Di�er t�m CYP2C9 genotip hastalar�nda �nerilen siponimod idame dozu 2 mg'd�r. ADM�RAZ, g�nde bir defa al�n�r.
Pozoloji:
Tedaviye ba�lama
Tedaviye 5 g�n s�ren titrasyon paketi ile ba�lanmal�d�r. Tedavi, 1. ve 2. g�nlerde g�nde bir kez 0,25 mg ile ba�lar, ard�ndan 3. g�nde bir kez 0,5 mg, 4. g�nde 0,75 mg ve 5. g�nde 1,25 mg dozlar�yla devam edilir, ard�ndan 6. g�nde ba�layarak hastan�n re�ete edilen siponimod idame dozuna ula��l�r (bak�n�z Tablo 1).
Tedaviye ba�land�ktan sonraki ilk 6 g�n� boyunca, �nerilen g�nl�k doz g�nde bir kez sabah, a� veya tok karn�na al�nmal�d�r.
Tablo 1 �dame dozuna ula�mak i�in doz titrasyon rejimi
Titrasyon | Titrasyon dozu | Titrasyon rejimi | Doz |
G�n 1 | 0,25 mg | 1 x 0,25 mg |
Titrasyon |
G�n 2 | 0,25 mg | 1 x 0,25 mg | |
G�n 3 | 0,5 mg | 2 x 0,25 mg | |
G�n 4 | 0,75 mg | 3 x 0,25 mg | |
G�n 5 | 1,25 mg | 5 x 0,25 mg | |
G�n 6 | 2 mg | 1 x 2 mg | �dame |
Tedaviye ba�lama s�ras�nda ka��r�lan doz/dozlar
Tedavinin ilk 6 g�n� boyunca, bir titrasyon dozunun bir g�n i�in ka��r�lmas� durumunda, tedavinin yeni bir titrasyon paketi ile yeniden ba�lat�lmas� gerekir.
6. g�nden sonra ka��r�lan doz
Bir doz ka��r�l�rsa, re�ete edilen doz bir sonraki planlanan zamanda al�nmal�d�r; bir sonraki doz iki kat�na ��kar�lmamal�d�r.
Tedaviye ara verildikten sonra idame tedavisinin yeniden ba�lat�lmas�
�dame tedavisine 4 veya daha fazla g�nl�k doz s�resiyle ara verilirse, siponimodun yeni bir titrasyon paketi ile yeniden ba�lat�lmas� gerekir.
Uygulama �ekli:
A��z yolu ile al�n�r.
Siponimod a� veya tok kar�na al�n�r. Film kapl� tabletler suyla birlikte b�t�n halde yutulmal�d�r.
�zel pop�lasyonlara ili�kin ek bilgiler:
B�brek yetmezli�i:
Klinik farmakoloji �al��malar�na dayanarak, b�brek yetmezli�i olan hastalarda doz ayarlamas�na gerek yoktur (bkz. B�l�m 5.2).
Karaci�er yetmezli�i:
Siponimod, ciddi karaci�er yetmezli�i olan hastalarda (Child-Pugh s�n�f C) kullan�lmamal�d�r (bkz. B�l�m 4.3). Hafif veya orta �iddette karaci�er yetmezli�i olan hastalarda doz ayarlamas�na gerek olmamakla birlikte, bu hastalarda tedaviye ba�larken dikkatli olunmal�d�r (bkz. B�l�m 4.4 ve 5.2).
Pediyatrik pop�lasyon:
0 ila 18 ya� aras� �ocuklarda ve ad�lesanlarda siponimodun g�venlili�i ve etkilili�i hen�z belirlenmemi�tir. Veri bulunmamaktad�r.
Geriyatrik pop�lasyon:
Siponimod 65 ya� ve �st� hastalarda �al���lmam��t�r. Klinik �al��malar 61 ya��na kadar olan hastalar� i�ermektedir. Siponimod, g�venlilik ve etkililik hakk�nda yeterli veri bulunmad���ndan ya�l�larda dikkatle kullan�lmal�d�r (bkz. B�l�m 5.2).
4.3. Kontrendikasyonlar
Etkin madde
�mm�n yetmezlik sendromu
Progresif multifokal l�koensefalopati veya kriptokokal menenjit �yk�s�
Aktif maligniteler
�iddetli karaci�er yetmezli�i (Child-Pugh s�n�f C)
�nceki 6 ayda miyokard enfarkt�s� (MI), unstabil angina pektoris, inme/ge�ici iskemik atak (TIA), dekompanse kalp yetmezli�i (yatarak tedavi gerektiren) veya New York Kalp Derne�i (NYHA) s�n�f III/IV kalp yetmezli�i (bkz. b�l�m 4.4) olan hastalar
Kalp pili kullanm�yorlarsa ikinci derece Mobitz tip II atriyoventrik�ler (AV) blok, ���nc� derece AV blok, sino-atriyal kalp blo�u veya hasta sin�s sendromu �yk�s� olan hastalar (bkz. B�l�m 4.4)
CYP2C9*3 (CYP2C9*3*3) genotipi (zay�f metabolize edici) i�in homozigot olan hastalar
Gebelik s�ras�nda ve etkili do�um kontrol� kullanmayan �ocuk do�urma potansiyeli olan kad�nlar (bkz. B�l�m 4.4 ve 4.6)
4.4. �zel kullan�m uyar�lar� ve �nlemleri
Enfeksiyonlar
Enfeksiyon riski
Siponimodun temel farmakodinamik etkisi, periferik lenfosit say�s�n� doza ba�l� olarak ba�lang�� de�erinin %20-30'una azaltmakt�r. Bu, lenfoid dokulardaki lenfositlerin geri d�n��l� sekestrasyonundan kaynaklanmaktad�r (bkz. B�l�m 5.1).
Siponimodun ba����kl�k sistemi �zerindeki etkileri enfeksiyon riskini art�rabilir (bkz. B�l�m 4.8).
Tedaviye ba�lamadan �nce, yeni bir tam kan say�m� (CBC) (yani son 6 ay i�inde veya �nceki tedavinin kesilmesinden sonra) mevcut olmal�d�r. CBC de�erlendirmeleri tedavi s�ras�nda periyodik olarak da �nerilir. Klinik �al��malarda mutlak lenfosit say�s� <0,2 x 10/l olan hastalarda siponimod dozu azalt�ld���ndan, mutlak lenfosit say�lar�n�n <0,2 x 10/l olarak �l��lmesi halinde, siponimod dozu 1 mg'a d���r�lmelidir. Halihaz�rda siponimod 1 mg alan bir hastada do�rulanm�� mutlak lenfosit say�s� <0,2 x 10/l ise 0,6 x 10/l'ye ula��ncaya kadar siponimod tedavisi kesilmelidir. Lenfosit say�s� bu de�ere ula�t���nda tedaviye tekrar ba�lanmas� d���n�lebilir.
�iddetli aktif enfeksiyonu olan hastalarda tedavinin ba�lat�lmas�, bu durum d�zelene kadar ertelenmelidir. Periferik lenfosit say�s� �zerindeki azalt�c� etkiler gibi rezid�el farmakodinamik etkiler, ila� kesildikten sonra 3 ila 4 haftaya kadar s�rebilece�inden, bu s�re zarf�nda enfeksiyon i�in dikkatli olmaya devam edilmelidir (bkz. A�a��da “Siponimod tedavisinin durdurulmas�” b�l�m�).
Hastalara enfeksiyon belirtilerini derhal doktorlar�na bildirmeleri s�ylenmelidir. Tedavi s�ras�nda enfeksiyon semptomlar� olan hastalarda etkili tan� ve tedavi stratejileri uygulanmal�d�r. Bir hastada ciddi bir enfeksiyon geli�irse, siponimod ile tedavinin ask�ya al�nmas� d���n�lmelidir.
Siponimod i�in kriptokokal menenjit (CM) vakalar� bildirilmi�tir. CM ile uyumlu semptom ve bulgular� olan hastalara derhal tan�sal de�erlendirme yap�lmal�d�r. CM ekarte edilene kadar siponimod tedavisi ask�ya al�nmal�d�r. CM te�hisi konulursa, uygun tedavi ba�lat�lmal�d�r.
Geli�tirme program�nda siponimod i�in progresif multifokal l�koensefalopati (PML) vakas� bildirilmemi�tir; bununla birlikte, ba�ka bir S1P resept�r mod�lat�r� i�in PML rapor edilmi�tir. Doktorlar, PML'yi d���nd�recek klinik semptomlar veya manyetik rezonans g�r�nt�leme (MRG) bulgular� konusunda dikkatli olmal�d�r. PML'den ��pheleniliyorsa, PML ekarte edilinceye kadar siponimod tedavisi ask�ya al�nmal�d�r.
Herpes viral enfeksiyonu
Tedavi s�ras�nda herhangi bir zamanda siponimod ile herpes viral enfeksiyonu vakalar� (varicella zoster vir�slerinin [VZV] neden oldu�u menenjit veya meningoensefalit vakalar� dahil) meydana gelmi�tir. Herpes menenjit veya meningoensefalit meydana gelirse, siponimod kesilmeli ve ilgili enfeksiyon i�in uygun tedavi uygulanmal�d�r. Hekim taraf�ndan onaylanm�� su�i�e�i �yk�s� olmayan veya VZV'ye kar�� tam bir a��lama s�reci belgelenmemi� hastalar siponimod ba�lamadan �nce VZV antikorlar� a��s�ndan test edilmelidir (a�a��da “A��lama” b�l�m�ne bak�n�z).
A��lama
Siponimod ile tedaviye ba�lamadan �nce antikor negatif hastalar i�in tam k�r su�i�e�i a��lamas� �nerilir; bunun ard�ndan a��laman�n tam etkisinin ortaya ��kmas�n� beklemek �zere tedavinin ba�lat�lmas� 1 ay ertelenmelidir (bkz. B�l�m 4.8).
Hastalar siponimod al�rken ve tedaviyi b�rakt�ktan sonra 4 hafta boyunca canl� zay�flat�lm�� a��lar�n kullan�m�ndan ka��n�lmal�d�r (bkz. B�l�m 4.5).
Siponimod tedavisi s�ras�nda a��lar daha az etkili olabilir. Planl� a��lamadan 1 hafta �nce ba�layarak 4 hafta sonras�na kadar tedavinin kesilmesi �nerilir. Siponimod tedavisini a��lama
i�in durdururken, hastal�k aktivitesinin olas� geri d�n��� g�z �n�nde bulundurulmal�d�r
(a�a��da “Siponimod tedavisinin durdurulmas�” b�l�m�ne bak�n�z).
Anti-neoplastik, imm�nomod�lat�r veya imm�nos�presif tedavilerle birlikte tedavi
Bu t�r bir tedavi s�ras�nda edinsel ba����kl�k sistemi etkileri riski nedeniyle anti-neoplastik, imm�nomod�lat�r veya imm�nos�presif tedaviler (kortikosteroidler dahil) bir arada dikkatle uygulanmal�d�r (bkz. B�l�m 4.5).
Makula �demi
Faz III klinik �al��mada g�rme semptomlar�n�n e�lik etti�i veya etmedi�i makula �demi, siponimod tedavisinde (%1,8) plaseboya (%0,2) k�yasla daha s�k bildirilmi�tir (bkz. B�l�m 4.8). Olgular�n �o�u tedavinin ilk 3-4 ay�nda meydana gelmi�tir. Bu nedenle, tedavinin ba�lamas�ndan 3-4 ay sonra oftalmolojik bir de�erlendirme �nerilir. Makula �demi vakalar� daha uzun s�reli tedavide de meydana geldi�inden, hastalar siponimod tedavisi s�ras�nda herhangi bir zamanda ortaya ��kabilecek g�rme bozukluklar�n� bildirmelidir ve makula da dahil olmak �zere fundusun de�erlendirilmesi �nerilir.
Makula �demi olan hastalarda, bu durum d�zelinceye kadar siponimod tedavisi ba�lat�lmamal�d�r.
Siponimod, makula �demi riskinde potansiyel bir art�� nedeniyle diyabet, �veit veya altta yatan/birlikte var olan retina hastal��� �yk�s� olan hastalarda dikkatli kullan�lmal�d�r (bkz. B�l�m 4.8). Makula �demini tespit etmek i�in bu hastalar�n tedaviye ba�lamadan �nce ve siponimod tedavisi s�ras�nda d�zenli olarak oftalmolojik bir de�erlendirmeye tabi tutulmas� �nerilir.
Makula �demi olan hastalarda siponimod tedavisinin devam� de�erlendirilmemi�tir. Bir hastada makula �demi geli�irse siponimodun kesilmesi �nerilir. Durumun d�zelmesinin ard�ndan siponimodun yeniden ba�lat�lmas� gerekip gerekmedi�ine dair kararda, her hastadaki potansiyel faydalar ve riskler dikkate al�nmal�d�r.
Bradiaritmi
Kalp at�m h�z�nda azalma
Siponimod tedavisinin ba�lat�lmas� kalp at�m h�z�nda ge�ici bir azalmaya neden olur (bkz. B�l�m 4.8 ve 5.1) ve bu nedenle tedavinin ba�lang�c�nda, 6. g�nde idame dozuna ula�mak �zere, bir titrasyon �emas� uygulan�r (bak�n�z b�l�m 4.2).
�lk titrasyon dozundan sonra, kalp at�m h�z�n�n azalmas� bir saat i�inde ba�lar ve 1. g�ndeki d���� yakla��k 3 ila 4 saatte maksimum seviyeye ula�m��t�r. Art�rmal� titrasyon devam ettik�e, sonraki g�nlerde kalp at�m h�z�ndaki d����lerin daha fazla oldu�u g�r�l�r ve 1. g�ndeki (ba�lang��) de�erden maksimum d����e 5 ila 6. g�nde ula��l�r. Mutlak saatlik ortalama kalp at�m h�z�nda en y�ksek g�nl�k doz sonras� azalma 1. g�nde g�r�l�r, nab�z dakikada ortalama 5- 6 vuru� (bpm) azal�r. Sonraki g�nlerde doz sonras� d����ler daha az belirgindir. Dozlara devam edildik�e kalp at�m h�z� 6. g�nden sonra artmaya ba�lar ve tedavinin ba�lamas�ndan sonraki 10 g�n i�inde plasebo d�zeylerine ula��r.
Dakikada 40 bpm'nin alt�ndaki kalp at�m h�zlar� nadiren g�zlenmi�tir. Bradikardi ya�ayan hastalar genellikle asemptomatik olmu�tur. Birka� hastada ba� d�nmesi ve kardiyak olmayan g���s a�r�s� dahil olmak �zere hafif ve orta �iddette semptomlar g�r�lm��t�r; bunlar m�dahale olmadan 24 saat i�inde d�zelmi�tir (bkz. B�l�m 4.8). Gerekirse, siponimodun neden oldu�u kalp at�m h�z�ndaki azalma,parenteraldozlardaatropinveya izoprenalin ile tersine �evrilebilir.
Atriyoventrik�ler iletim
Siponimod tedavisinin ba�lat�lmas�, doz titrasyonu s�ras�nda kalp at�m h�z�nda g�zlenen azalmaya benzer ge�ici bir patern izleyen ge�ici atriyoventrik�ler iletim gecikmeleri ile ili�kilendirilmi�tir. Atriyoventrik�ler iletim gecikmeleri �o�u durumda birinci derece atriyoventrik�ler (AV) bloklar (elektrokardiyogramda uzam�� PR aral���) olarak kendini g�stermi�tir. Klinik �al��malarda, tedavinin ba�lamas� s�ras�nda hastalar�n %1,7'sinden az�nda, genellikle Mobitz tip I (Wenckebach) olmak �zere ikinci derece AV bloklar� g�zlenmi�tir. �letim anormallikleri tipik olarak ge�ici ve asemptomatik olmu�, 24 saat i�inde d�zelmi� ve tedavinin kesilmesini gerektirmemi�tir.
�nceden belirli kardiyak rahats�zl�klar� olan hastalarda tedaviye ba�lama �nerisi
�nlem olarak, a�a��daki kardiyak rahats�zl�klar� olan hastalar, ilk siponimod dozundan sonra 6 saat boyunca bradikardi belirtileri ve semptomlar� a��s�ndan izlenmelidir (ayr�ca bkz. B�l�m 4.3):
sin�s bradikardisi (kalp at�m h�z� <55 bpm/dakika),
4.5. Di�er t�bbi �r�nler ile etkile�imler ve di�er etkile�im �ekilleri
Antineoplastik, ba����kl�k mod�le edici veya imm�nos�presif tedaviler
Siponimod; antineoplastik, imm�nomod�lat�r veya imm�nos�presif tedavilerle kombinasyon halinde �al���lmam��t�r. Bu t�r bir tedavi s�ras�nda ve bu t�bbi �r�nlerin herhangi birinin durdurulmas�ndan sonraki haftalarda ek ba����kl�k etkileri riski nedeniyle, birlikte uygulama s�ras�nda dikkatli olunmal�d�r (bkz. B�l�m 4.4).
�r�n bilgilerinde a��klanan alemtuzumab ba����kl�k bask�lay�c� etkilerinin �zellikleri ve s�resi nedeniyle, tedavinin yararlar� her bir hasta i�in risklerden a��k�a daha a��r basmad�k�a, alemtuzumab sonras� siponimod ile tedaviye ba�lanmas� �nerilmez (bkz. B�l�m 4.4).
Anti-aritmik t�bbi �r�nler, QT uzatan t�bbi �r�nler, kalp at�m h�z�n� d���rebilecek t�bbi �r�nler Tedavi ba�lang�c�nda s�n�f Ia (�rn., kinidin, prokainamid) veya s�n�f III (�rn. amiodaron, sotalol) anti-aritmik t�bbi �r�nler, bilinen aritmojenik �zelliklere sahip QT uzatan t�bbi �r�nler, kalp at�m h�z�n� d���ren kalsiyum kanal blokerleri (verapamil veya diltiazem gibi) veya kalp at�m h�z�n� azaltabilecek di�er maddeler (�rn. ivabradin veya digoksin) kullanmakta olan hastalarda, kalp at�m h�z� �zerindeki potansiyel ilave etkiler nedeniyle, siponimod e�zamanl� olarak kullan�lmamal�d�r (bkz. b�l�m 4.4). Bu t�bbi �r�nlerin siponimod ile birlikte kullan�m� ile ilgili veri mevcut de�ildir. Tedaviye ba�lama s�ras�nda bu maddelerin birlikte kullan�lmas� ciddi bradikardi ve kalp blo�u ile ili�kili olabilir. Kalp at�m h�z� �zerindeki potansiyel katk� etkisi nedeniyle, siponimod tedavisi genellikle bu maddelerle e�zamanl� tedavi edilen hastalarda ba�lat�lmamal�d�r (bkz. B�l�m 4.4). Siponimod ile tedavi d���n�l�rse, kalp at�m h�z�n� d���rmeyen t�bbi �r�nlere ge�i� veya tedavinin ba�lat�lmas� i�in uygun izlem ile ilgili olarak bir kardiyologdan tavsiye al�nmal�d�r.
Beta blokerler
Beta bloker alan hastalarda kalp at�m h�z�n� d���rme �zerine ilave etkiler nedeniyle siponimod ba�lat�ld���nda dikkatli olunmal�d�r (bkz. B�l�m 4.4). Stabil dozlarda siponimod alan hastalarda beta-bloker tedavisi ba�lat�labilir.
Siponimod ve propranolol�n birlikte uygulanmas�n�n olumsuz kronotropik etkisi, �zel bir farmakodinamik/g�venlilik �al��mas�nda de�erlendirilmi�tir. Siponimod farmakokinetik/ farmakodinamik kararl� durum �zerine propranolol eklenmesi, propranolol farmakokinetik/farmakodinamik kararl� durumun �st�ne siponimod eklenmesine k�yasla daha az belirgin negatif kronotropik etkilere (aditiften daha az) sahip olmu�tur (aditif HR etkisi).
A��lama
Canl� zay�flat�lm�� a��lar�n kullan�m� enfeksiyon riski ta��yabilir ve bu nedenle siponimod tedavisi s�ras�nda ve tedaviden sonra 4 haftaya kadar bunlar kullan�lmamal�d�r (bkz. B�l�m 4.4).
Siponimod ile tedavi s�ras�nda ve tedaviden sonra 4 haftaya kadar a��lama daha az etkili olabilir. Siponimod tedavisi a��lamadan 1 hafta �ncesinden 4 hafta sonras�na kadar duraklat�l�rsa, a��laman�n etkilili�inin tehlikeye girmedi�i kabul edilmektedir (bkz. B�l�m 4.4). �zel bir faz I sa�l�kl� g�n�ll� �al��mas�nda, grip a��lar�yla birlikte siponimod tedavisi veya daha k�sa tedavi duraklamas� (a��lamadan 10 g�n ile 14 g�n �ncesi aras�) plaseboya k�yasla daha d���k yan�t oranlar�na (yakla��k %15 ila %30 daha d���k) neden olmu�, di�er yandan bir PPV 23 a��s�n�n etkilili�i, e�zamanl� siponimod tedavisinden olumsuz etkilenmemi�tir (bkz. b�l�m 4.4).
![]()
Siponimod esas olarak sitokrom P450 2C9 (CYP2C9) (%79,3) ve daha az �l��de sitokrom P450
3A4 (CYP3A4) (%18,5) taraf�ndan metabolize edilir. CYP2C9 bir polimorfik enzimdir ve CYP3A veya CYP2C9 inhibit�rlerinin veya ind�kleyicilerinin varl���nda ila�-ila� etkile�imi (DDI) etkisinin CYP2C9 genotipine ba�l� oldu�u tahmin edilmektedir.
CYP2C9 ve CYP3A4 inhibit�rleri
Siponimod ile orta derecede CYP2C9 ve orta veya g��l� CYP3A4 inhibisyonuna neden olan t�bbi �r�nlerin birlikte kullan�lmas� siponimod maruziyetinde �nemli bir art�� nedeniyle �nerilmez. Bu e�zamanl� ila� rejimi, ayr� bir orta veya g��l� CYP3A4 inhibit�r� ile kombinasyon halinde orta derecede bir CYP2C9/CYP3A4 �ift inhibit�r�n� (�rn. flukonazol) veya orta bir CYP2C9 inhibit�r�n� i�erebilir.
CYP2C9*1*1 genotipi olan sa�l�kl� g�n�ll�lerde kararl� durumda 200 mg/g�n flukonazol (orta CYP2C9/g��l� CYP3A4 inhibit�r�) ile tek doz siponimod 4 mg'�n birlikte uygulanmas�, siponimodun e�ri alt�ndaki alan�nda (EAA) 2 kat art��a neden olmu�tur. Fizyolojik temelli farmakokinetik (PBPK) modelleme kullan�larak yap�lan bir ila� etkile�im potansiyeli de�erlendirilmesine g�re, CYP2C9*2*2 genotipi olan hastalar d���nda t�m genotipler genelinde herhangi bir CYP3A4 ve CYP2C9 inhibit�r� tipiyle siponimodun EAA's�nda maksimum 2 kat art�� �ng�r�lmektedir. CYP2C9*2*2 hastalar�nda, orta CYP2C9/CYP3A4 inhibit�rlerinin varl���nda siponimodun EAA's�nda 2,7 kat art�� beklenmektedir.
CYP2C9 ve CYP3A4 ind�kleyicileri
Siponimod, �o�u CYP2C9 ve CYP3A4 ind�kleyici tipi ile kombine edilebilir. Bununla birlikte, siponimod maruziyetinde beklenen bir azalma nedeniyle, siponimod kombine edildi�inde tedavinin uygunlu�u ve olas� yarar� g�z �n�nde bulundurulmal�d�r:
genotipten ba��ms�z olarak t�m hastalarda g��l� CYP3A4/orta CYP2C9 ikili ind�kleyicileri (�rn. karbamazepin) veya ayr� bir g��l� CYP3A4 ind�kleyicisi ile kombinasyon halinde orta derecede bir CYP2C9 ind�kleyicisi ile.
4.6. Gebelik ve laktasyon
Gebelik kategorisi: D
�ocuk do�urma potansiyeli bulunan kad�nlar/Do�um kontrol� (Kontrasepsiyon) Siponimod, etkili do�um kontrol y�ntemi kullanmayan �ocuk do�urma potansiyeli olan kad�nlarda kontrendikedir (bkz. B�l�m 4.3). Bu nedenle, �ocuk do�urma potansiyeli olan kad�nlarda tedaviye ba�lamadan �nce gebelik testi sonucu negatif ��kmal� ve fet�s i�in ciddi risk konusunda dan��manl�k sa�lanmal�d�r. �ocuk do�urma potansiyeli olan kad�nlar, tedavi s�ras�nda ve son siponimod dozunu takip eden en az on g�n boyunca etkili bir do�um kontrol y�ntemi kullanmal�d�r (bkz. B�l�m 4.4).
Ayr�ca Doktor E�itim Paketinde �zel �nlemler de yer almaktad�r. Siponimod kad�n hastalara re�ete edilmeden �nce ve tedavi s�ras�nda bu �nlemler uygulanmal�d�r.
Gebeli�i planlamak i�in siponimod tedavisini durdururken, hastal�k aktivitesinde olas� bir geri d�n�� g�z �n�nde bulundurulmal�d�r (bkz. B�l�m 4.4).
Gebelik d�nemi
Gebe kad�nlarda siponimod kullan�m�yla ilgili veri bulunmamaktad�r ya da s�n�rl� veri mevcuttur. Hayvan �al��malar�, g�nl�k 2 mg dozda insan maruziyeti ile kar��la�t�r�labilir maruziyet d�zeylerinde embriyo-fetal �l�mler ve iskelet veya visseral malformasyonlar dahil olmak �zere s��anlarda ve tav�anlarda siponimod kaynakl� embriyotoksisite ve fetotoksisite ve s��anlarda teratojenisite g�stermi�tir (bkz. B�l�m 5.3). Ek olarak, ba�ka bir sfingozin-1-fosfat resept�r mod�lat�r� ile sahip olunan klinik deneyim, gebelik s�ras�nda uyguland���nda genel pop�lasyonda g�zlenen orana k�yasla 2 kat daha fazla b�y�k konjenital malformasyon riski g�stermi�tir.
Sonu� olarak, siponimod gebelik s�ras�nda kontrendikedir (bkz. B�l�m 4.3). Siponimod, gebelik planlanmadan en az 10 g�n �nce durdurulmal�d�r (bkz. B�l�m 4.4). Bir kad�n tedavi s�ras�nda gebe kal�rsa, siponimod kesilmelidir. Tedaviyle ili�kili fet�se zararl� etki riski konusunda t�bbi tavsiye verilmeli ve ultrasonografi muayeneleri yap�lmal�d�r.
Laktasyon d�nemi
Siponimod veya ana metabolitlerinin anne s�t�ne ge�ip ge�medi�i bilinmemektedir. Siponimod ve metabolitleri s��anlar�n s�t�ne ge�er. Siponimod emzirme d�neminde kullan�lmamal�d�r.
�reme yetene�i/Fertilite
Siponimodun insan fertilitesi �zerindeki etkisi de�erlendirilmemi�tir. Siponimodun s��anlarda ve maymunlarda erkek �reme organlar� veya s��anlarda fertilite parametreleri �zerinde hi�bir etkisi olmam��t�r.
4.7. Ara� ve makine kullan�m� �zerindeki etkiler
Siponimodun ara� ve makine kullan�m� �zerinde hi�bir etkisi yoktur veya etkileri g�z ard� edilebilir d�zeydedir. Bununla birlikte, siponimod ile tedaviye ba�larken bazen ba� d�nmesi meydana gelebilir. Bu nedenle, hastalar, siponimod tedavisine ba�lanan ilk g�n boyunca ara� veya makine kullanmamal�d�r (bkz. B�l�m 4.4).
4.8. �stenmeyen etkiler
G�venlilik profilinin �zeti
En yayg�n advers ila� reaksiyonlar� ba� a�r�s� (%15) ve hipertansiyondur (%12,6).
Advers ila� reaksiyonlar�n�n tablo halinde �zeti
Her sistem organ s�n�f� i�inde, advers ila� reaksiyonlar�, en s�k reaksiyonlar �nce olacak �ekilde s�kl��a g�re s�ralanmaktad�r. Ek olarak, her advers ila� reaksiyonu i�in kar��l�k gelen s�kl�k kategorisi a�a��daki sisteme dayanmaktad�r: �ok yayg�n (≥1/10); yayg�n (≥1/100 ila ≤1/10); yayg�n olmayan (≥1/1.000 ila ≤1/100); seyrek (≥1/10.000 ila ≤1/1.000); �ok seyrek (<1/10.000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor).
Enfeksiyonlar ve enfestasyonlar | |
Yayg�n | Herpes zoster |
Bilinmiyor | Kriptokokal menenjit |
�yi huylu, habis ve belirtilmemi� neoplazmalar (kistler ve polipler dahil) | |
Yayg�n | Melanositik nevus Bazal h�creli karsinom |
Yayg�n olmayan | Skuam�z h�creli karsinom |
Kan ve lenf sistemi hastal�klar� | |
Yayg�n | Lenfopeni |
Sinir sistemi hastal�klar� | |
�ok yayg�n | Ba� a�r�s� |
Yayg�n | Ba� D�nmesi |
N�bet | |
Tremor | |
G�z hastal�klar� | |
Yayg�n | Makula �demi |
Kalp hastal�klar� | |
Yayg�n | Bradikardi |
Atriyoventrik�ler blok (birinci ve ikinci derece) | |
Vask�ler hastal�klar� | |
�ok yayg�n | Hipertansiyon |
Gastrointestinal hastal�klar | |
Yayg�n | Bulant� |
�shal | |
Kas-iskelet bozukluklar�, ba� doku ve kemik hastal�klar� | |
Yayg�n | Ekstremitede a�r� |
Genel bozukluklar ve uygulama b�lgesine ili�kin hastal�klar | |
Yayg�n | Periferik �dem |
Asteni | |
Ara�t�rmalar | |
Karaci�erfonksiyontestindeart�� 3Q3NRZ1AxZmxXRG83ZW56 | |
Yayg�n | Solunum fonksiyon testinde azalma |
Tablo 2 Advers reaksiyonlar�n tablo halinde listesi
Se�ilmi� advers reaksiyonlar�n tan�m�
Enfeksiyonlar
SPMS hastalar�ndaki Faz III klinik �al��mada, toplam enfeksiyon oran�, siponimod hastalar� ile plasebo hastalar� aras�nda kar��la�t�r�labilir olmu�tur (s�ras�yla %49,0'a kar��l�k %49,1). Bununla birlikte, plasebo (%0,7) ile kar��la�t�r�ld���nda siponimod (%2,5) tedavisinde herpes zoster enfeksiyonu oran�nda bir art�� bildirilmi�tir.
Tedavi s�ras�nda herhangi bir zamanda siponimod ile varicella zoster vir�slerinin neden oldu�u menenjit veya meningoensefalit vakalar� ortaya ��km��t�r. Siponimod i�in kriptokokal menenjit (CM) vakalar� da bildirilmi�tir (bkz. b�l�m 4.4).
Makula �demi
Makula �demi siponimod alan hastalarda (%1,8) plasebo verilenlerden (%0,2) daha s�k bildirilmi�tir. Olgular�n �o�unlu�u siponimodun ba�lamas�ndan sonraki 3 ila 4 ay i�inde meydana gelmi� olmakla birlikte, siponimod ile tedavi edilen hastalarda 6 aydan daha uzun bir s�reyle vakalar bildirilmi�tir (bkz. B�l�m 4.4). Baz� hastalar bulan�k g�rme veya g�rme keskinli�inde azalma ile ba�vururken, baz�lar� asemptomatik olmu� ve rutin oftalmolojik muayenesinde tan� konmu�tur. Tedavinin kesilmesinden sonra genellikle makula �deminde d�zelme olmu� veya kendili�inden kaybolmu�tur. Yeniden tedavi ile birlikte tekrarlama riski de�erlendirilmemi�tir.
Bradiaritmi
Siponimod tedavisinin ba�lat�lmas�, kalp at�m h�z�nda ge�ici bir d����e neden olur ve ayr�ca atriyoventrik�ler iletim gecikmeleriyle de ili�kili olabilir (bkz. B�l�m 4.4). Siponimod ile tedavi edilen hastalar�n %6,2'sinde bradikardi bildirilirken bu oran plasebo ile %3,1 olmu�tur; AV blok ise siponimod ile tedavi edilen hastalar�n %1,7'sinde ve plasebo uygulanan hastalar�n
%0,7'sinde bildirilmi�tir (bkz. B�l�m 4.4).
Kalp at�m h�z�ndaki maksimum d����, dozdan sonraki ilk 6 saatte g�r�l�r.
�lk dozlama a�amas�nda ge�ici, doza ba�l� bir d���� g�zlenmi�tir ve ≥ 5 mg'l�k dozlarda bu etki plato yapm��t�r. Bradiaritmik olaylar (AV bloklar� ve sin�s duraklamalar�) siponimod tedavisi alt�nda plaseboya k�yasla daha y�ksek bir insidansla tespit edilmi�tir.
�o�u AV blo�u ve sin�s duraklamas�, 2 mg'l�k terap�tik dozun �zerinde meydana gelmi�tir ve doz titrasyonu yap�lmam�� ko�ullar alt�nda doz titrasyon ko�ullar�na k�yasla �nemli �l��de daha y�ksek insidans g�stermi�tir.
Siponimodun neden oldu�u kalp at�m h�z�ndaki azalma, atropin veya izoprenalin ile tersine �evrilebilmektedir.
Karaci�er fonksiyon testleri
Siponimod ile tedavi edilen MS hastalar�nda hepatik enzimlerde art�� (�o�unlukla ALT y�ksekli�i) bildirilmi�tir. SPMS hastalar�nda yap�lan faz III �al��mada, �zellikle karaci�er transaminaz (ALT/AST) ve GGT y�kselmelerine ba�l� olarak, plasebo (%3,1) hastalar� ile kar��la�t�r�ld���nda siponimod hastalar�nda (%11,3) daha s�k karaci�er fonksiyon testi art��lar� g�zlenmi�tir. Y�ksekliklerin �o�u tedaviye ba�lad�ktan sonraki 6 ay i�inde meydana gelmi�tir. Siponimod kesildikten sonra yakla��k 1 ay i�inde ALT d�zeyleri normale d�nm��t�r (bkz. B�l�m 4.4).
Kan bas�nc�
SPMS hastalar�ndaki faz III klinik �al��mada hipertansiyon, plasebo uygulanan hastalar (%9,0) ile kar��la�t�r�ld���nda siponimod kullanan hastalarda (%12,6) daha s�k bildirilmi�tir. Siponimod ile tedavi, tedavinin ba�lamas�ndan k�sa bir sonra ba�layarak sistolik ve diyastolik kan bas�nc�nda bir art��la sonu�lanm��, bu etki yakla��k 6 ayl�k tedaviden sonra maksimum d�zeye ula�m�� (sistolik 3 mmHg, diyastolik 1,2 mmHg) ve sonras�nda stabil kalm��t�r. Devam eden tedavi ile bu etki de devam etmi�tir.
N�betler
SPMS hastalar�ndaki faz III klinik �al��mada plasebodaki %0,4'l�k oran ile kar��la�t�r�ld���nda, siponimod ile tedavi edilen hastalar�n %1,7'sinde n�betler bildirilmi�tir.
Solunum etkileri
Siponimod tedavisi ile 1 saniyede zorlu ekspiratuvar hacimde (FEV) ve akci�erin karbon monoksit (DLCO) de�erleri i�in dif�zyon kapasitesinde k���k d����ler g�zlenmi�tir. SPMS hastalar�ndaki faz III klinik �al��mada tedavinin 3. ve 6. aylar�nda, siponimod grubunda FEV'de ba�lang��tan ortalama de�i�iklikler, her bir zaman noktas�nda -0,1 L iken plasebo grubunda de�i�iklik olmam��t�r. Bu g�zlemler, siponimod ile tedavi edilen ve kronik obstruktif akci�er hastal��� (KOAH) veya ast�m gibi solunum bozukluklar� olan hastalarda biraz daha y�ksek olmu�tur (FEV'de ba�lang��tan yakla��k 0,15 L ortalama de�i�iklik). Kronik tedavide bu azalma klinik olarak anlaml� advers olaylara d�n��memi�tir ve �ks�r�k veya dispne raporlar�ndaki art��la ili�kili de�ildir (bkz. B�l�m 5.1).
��pheli advers reaksiyonlar�n raporlanmas�
Ruhsatland�rma sonras� ��pheli ila� advers reaksiyonlar�n�n raporlanmas� b�y�k �nem ta��maktad�r. Raporlama yap�lmas�, ilac�n yarar / risk dengesinin s�rekli olarak izlenmesine olanak sa�lar. Sa�l�k mesle�i mensuplar�n�n herhangi bir ��pheli advers reaksiyonu T�rkiye Farmakovijilans Merkezi (T�FAM)'ne bildirmeleri gerekmektedir. (www.titck.gov.tr; e- posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks: 0 312 218 35 99)
4.9. Doz a��m� ve tedavisi
Sa�l�kl� olgularda 75 mg'l�k tek dozlardan sonra semptomatik bradikardi olu�umuna ba�l� olarak maksimum tolere edilen tek dozun 25 mg oldu�u belirlenmi�tir. Birka� olgu, 3 ila 4 g�n boyunca kas�ts�z olarak g�nde 200 mg'a kadar dozlar alm��t�r ve karaci�er fonksiyon testlerinde asemptomatik hafif ila orta derecede ge�ici art��lar ya�am��lard�r.
84 mg siponimod alan bir hastada (depresyon �yk�s� olan) karaci�er transaminazlar�nda hafif bir y�kselme g�r�lm��t�r.
Doz a��m� e�er siponimoda ilk maruziyet ise veya siponimodun doz titrasyon faz� s�ras�nda meydana gelirse, gece boyunca g�zlemi de i�erebilecek �ekilde bradikardinin belirti ve semptomlar�n� g�zlemlemek �nemlidir. Kalp at�m h�z� ve kan bas�nc�n�n d�zenli olarak �l��lmesi gerekir ve elektrokardiyogramlar �ekilmelidir (bkz. B�l�m 4.2 ve 4.4).
Siponimod i�in spesifik bir antidot yoktur. Diyaliz ya da plazma de�i�imi siponimodun v�cuttan anlaml� bir �ekilde uzakla�t�r�lmas�na neden olmaz.
5. FARMAKOLOJ�K �ZELL�KLER
5.1. Farmakodinamik �zellikler
Farmakoterap�tik grup: �mm�nosupresanlar, se�ici imm�nosupresanlar ATC kodu: L04AA42
Etki mekanizmas�
Siponimod, bir sfingozin-1-fosfat (S1P) resept�r mod�lat�r�d�r. Siponimod, S1P i�in be� G- protein ba�l� resept�rden (GPCR) ikisine (yani S1P1 ve S1P5) se�ici olarak ba�lan�r. Siponimod, lenfositler �zerindeki S1P1 resept�rleri �zerinde fonksiyonel bir antagonist olarak hareket ederek lenf d���mlerinden ��k��� �nler. Bu, T h�crelerinin santral sinir sistemine (SSS) yeniden dola��m�n� azaltarak merkezi enflamasyonu s�n�rland�r�r.
Farmakodinamik etkiler
Periferik kan lenfositlerinde azalma
Siponimod, lenfoid dokularda geri d�n��l� lenfosit sekestrasyonu nedeniyle, ilk dozdan sonraki 6 saat i�inde periferik kan lenfosit say�s�nda doza ba�l� bir azalmaya neden olur.
Devam eden g�nl�k dozlama ile birlikte lenfosit say�s� azalmaya devam ederek tipik bir CYP2C9*1*1 SPMS hastas�nda veya *1*2 Japon olmayan SPMS hastas�nda yakla��k %0,560 (0,271-1,08) h�cre/nL lenfosit say�s� �eklindeki dip medyan (%90 GA) de�erine ula��r (ba�lang�� de�erinin %20-30'una kar��l�k gelir). G�nl�k dozlama ile d���k lenfosit say�lar� korunur.
SPMS hastalar�n�n b�y�k �o�unlu�unda (%90), tedaviyi b�rakt�ktan sonraki 10 g�n i�inde lenfosit say�lar� normal aral��a d�ner. Siponimod tedavisini durdurduktan sonra periferik lenfosit say�s� �zerindeki rezid�el d���r�c� etkiler son dozdan sonra 3-4 haftaya kadar devam edebilir.
Kalp at�m h�z� ve ritmi
Siponimod, tedavi ba�lang�c�nda, kalp at�m h�z� ve atriyoventrik�ler iletimde ge�ici bir azalmaya neden olur (bkz. B�l�m 4.4 ve 4.8); bu etki, mekanik olarak, h�cresel hiperpolarizasyona ve azalt�lm�� uyar�labilirli�e yol a�an, G-protein kenetli i�e rektifiye potasyum (GIRK) kanallar�n�n aktivasyonu ile ili�kilidir. S1P1 resept�rlerindeki fonksiyonel antagonizmi nedeniyle, siponimodun ilk titrasyonu, idame dozu elde edilene kadar GIRK kanallar�n� ard���k olarak duyars�zla�t�r�r.
QT aral���n� uzatma potansiyeli
Siponimodun terap�tik (2 mg) ve supraterap�tik (10 mg) dozlar�n�n kardiyak repolarizasyon �zerindeki etkileri ayr�nt�l� bir QT �al��mas�nda ara�t�r�lm��t�r. Sonu�lar siponimod ile QT uzamas� ile ili�kili bir aritmojenik potansiyel ortaya koymam��t�r. Siponimod, doz sonras� 3 saat sonra plaseboya g�re d�zeltilmi� ba�lang�ca ayarl� ortalama QTcF'yi (ΔΔQTcF) 5 ms'den fazla art�rm��t�r ve maksimum ortalama etki s�ras�yla 7,8 ms (2 mg) ve 7,2 ms (10 mg) olmu�tur. B�t�n zaman noktalar�nda ΔΔQTcF i�in tek tarafl� %95 g�ven aral���n�n �st s�n�r� 10 ms'nin alt�nda kalm��t�r. Kategorik analiz, tedaviden kaynaklanan 480 ms'nin �zerinde QTc de�erleri olmad���n�, ba�lang�ca k�yasla 60 ms'den daha fazla QTc art��� olmad���n� ve d�zeltilmi� veya d�zeltilmemi� QT/QTc de�erinin 500 ms'yi a�mad���n� ortaya koymu�tur.
Pulmoner fonksiyon
28 g�n boyunca tek veya �oklu dozlarla siponimod tedavisi, zorlu vital kapasitenin %25 ila 75'inin (FEF %25-75) ekspirasyonu s�ras�nda 1 saniyede zorlu ekspiratuvar hacim (FEV) ve zorlu ekspiratuvar ak�� (FEF) ile �l��ld��� �zere, havayolu direncinde klinik olarak anlaml� art��larla ili�kili de�ildir. Terap�tik olmayan tekli dozlarda (> 10 mg) hafif bir azalt�lm�� FEV e�ilimi tespit edilmi�tir. Birden fazla siponimod dozu, %FEV ve FEF'te, doza ve g�nd�z saatlerine ba��ml� olmayan ve artm�� hava yolu direncine dair herhangi bir klinik bulgu ile ili�kili olmayan hafif ila orta de�i�iklikler ile ili�kilendirilmi�tir.
Klinik etkililik ve g�venlilik
Siponimodun etkilili�i, SPMS hastalar�nda g�nde bir kez 2 mg'l�k dozlar�n de�erlendirildi�i bir faz III �al��mas�nda ara�t�r�lm��t�r.
SPMS'de A2304 (EXPAND) �al��mas�
�al��ma A2304, relapslar�n yoklu�unda veya relapslardan ba��ms�z olarak �nceki 2 y�l i�inde belgelenmi� progresyon kan�t� olan, �al��maya kay�t �ncesindeki 3 ay i�inde relaps kan�t� bulunmayan ve �al��maya girdi�i tarihte ortanca Geni�letilmi� �z�rl�l�k Durum �l�e�i (EDSS) skoru 3,0 ila 6,5 olan SPMS hastalar�yla y�r�t�len randomize, �ift k�r, plasebo kontroll�, olay ve takip s�resi g�d�ml�, bir faz III bir �al��mas�d�r. Ba�lang��ta ortalama EDSS 6,0'd�r. 61 ya� �st� hastalar dahil edilmemi�tir. Hastal�k aktivitesi ile ilgili olarak, SPMS'de enflamatuvar aktivitenin karakteristik �zellikleri relaps veya g�r�nt�lemeyle ili�kili olabilir (yani Gd tutan T1 lezyonlar� veya aktif [yeni veya geni�leyen] T2 lezyonlar�).
Hastalar, g�nde bir kez siponimod 2 mg veya plasebo almak �zere 2:1 oran�nda randomize edilmi�tir. Taramada ve her 3 ayda bir ve relaps zaman�nda klinik de�erlendirmeler ger�ekle�tirilmi�tir. MRG de�erlendirmeleri taramada ve 12 ayda bir y�r�t�lm��t�r.
�al��man�n birincil sonlan�m noktas�, EDSS'de 3 ay boyunca devam eden ba�lang��tan en az 1 puanl�k art�� (ba�lang�� EDSS'si ≥ 5,5 olan hastalar i�in 0,5 puanl�k art��) olarak tan�mlanan 3 ayl�k do�rulanm�� �z�rl�l�k progresyonunun (CDP) zaman� olmu�tur. Ba�l�ca ikincil sonlan�m noktalar�, zamanl� 25-ad�m y�r�me testinde (T25W) 3 ayda do�rulanm�� ba�lang�ca k�yasla en az %20 k�t�le�me ve T2 lezyon hacminde ba�lang�ca k�yasla de�i�im olmu�tur. Ek ikincil sonlan�m noktalar� aras�nda, 6 ayl�k CDP'ye kadar ge�en s�re, beyin hacminde y�zde de�i�im ve enflamatuvar hastal�k aktivitesi (y�ll�k n�ks oran�, MRG lezyonlar�) �l��mleri yer alm��t�r. Sembol Say� Modalite Testi skorundaki bili�sel i�lem h�z�ndaki de�i�im ke�ifsel bir sonlan�m nokta olarak olmu�tur.
�al��ma s�resi her hasta i�in de�i�ken olmu�tur (ortanca �al��ma s�resi 21 ay, da��l�m: 1 g�n ila 37 ay).
�al��ma, 1.651 hastan�n siponimod 2 mg (N = 1.105) veya plaseboya (N = 546) randomize edilmesini i�ermi�tir; siponimod ile tedavi edilen hastalar�n %82'si ve plasebo ile tedavi edilen hastalar�n %78'i �al��may� tamamlam��t�r. Ba�lang��ta ortanca ya� 49, ortanca hastal�k s�resi 16 y�l ve ortanca EDSS skoru 6,0 olmu�tur. Hastalar�n %64'�nde �al��ma giri�inden �nceki 2 y�l i�inde relaps g�r�lmemi�tir ve %76's�nda ba�lang�� MRG taramas�nda gadolinyum (Gd) tutan lezyon yoktur. Hastalar�n %78'i daha �nce MS'leri i�in bir tedavi g�rm��t�r.
3 ayl�k ve 6 ayl�k CDP'nin ba�lama zaman�, siponimod i�in anlaml� olarak daha ge� olmu�, 3 ayl�k CDP riskinde plasebo ile kar��la�t�r�ld���nda %21 azalma (tehlike oran� [HR] 0,79, p = 0,0134) ve 6 ayl�k CDP riskinde plasebo ile kar��la�t�r�ld���nda %26'l�k azalma (HR 0,74, p = 0,0058) izlenmi�tir.
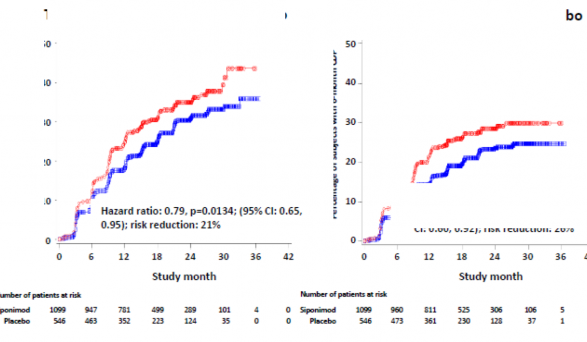
Plasebo kar��s�nda 3 ayl�k CDP'ye kadar ge�en s�re
Plasebo kar��s�nda 6 ayl�k CDP'ye kadar ge�en s�re
Riskli hasta say�s�
Riskli hasta say�s�
�al��ma ay�
�al��ma ay�
�ekil 1 EDSS-Kaplan-Meier e�rilerine dayal� 3 ayl�k ve 6 ayl�k CDP'li hastalar (tam analiz seti, �al��ma A2304)
Tablo 3 A2304 �al��mas�n�n klinik ve MRG sonu�lar�
Sonlan�m noktalar� | A2304 (EXPAND) | |
Siponimod 2 mg (n=1.099) | Plasebo (n=546) | |
Klinik sonlan�m noktalar� | ||
Birincil etkililik sonlan�m noktas�: 3 ayl�k do�rulanm�� �z�rl�l�k progresyonu olan hastalar�n oran� (birincil sonlan�m noktas�) | %26,3 | %31,7 |
Risk azalmas� | %21 (p=0,0134) | |
Zamanl� 25 ad�m y�r�me testinde 3 ayl�k do�rulanm�� %20 art��� olan hastalar�n oran� | %39,7 | %41,4 |
Risk azalmas� | %6 (p=0,4398) | |
6 ayl�k do�rulanm�� �z�rl�l�k progresyonu olan hastalar�n oran� | %19,9 | %25,5 |
Risk azalmas� | %26 [(p=0,0058)]6 | |
Y�ll�k relaps oran� (ARR) | 0,071 | 0,152 |
Oran azalmas� | %55 [(p<0,0001)]6 | |
MRG sonlan�m noktalar� | ||
T2 lezyon hacminde ba�lang�ca g�re de�i�im (mm) | +184 mm | +879 mm |
T2 lezyon hacmi de�i�imindeki fark | -695 mm (p<0,0001) | |
Ba�lang�ca g�re beyin hacmi de�i�im y�zdesi (%95 GA) | -%0,497 | -%0,649 |
Beyin hacmi de�i�imindeki y�zde fark | 0.152%[(p=0,0002)]6 | |
Gd tutan T1 a��rl�kl� lezyonlar�n ortalama k�m�latif say�s� (%95 GA) | 0,081 | 0,596 |
Oran azalmas� | 86%[(p<0,0001)]6 | |
Sembol Say� Modalite Testinde 4 puan k�t�le�me olan hastalar�n oran� | %16,0 | %20,9 |
Risk azalmas� | %25 [(p=0,0163)]6 | |
�al��madan elde edilen sonu�lar, 3 ayl�k ve 6 ayl�k CDP'ye kadar ge�en s�rede, cinsiyet, ya�, �al��ma �ncesi relaps aktivitesi, ba�lang�� MRG hastal�k aktivitesi, hastal�k s�resi ve ba�lang��ta �z�rl�l�k d�zeylerine dayal� tan�ml� alt gruplarda siponimod ile de�i�ken fakat istikrarl� bir risk azalmas� g�stermi�tir.
Aktif hastal��� olan hasta alt grubunda (n = 779) (�al��madan �nceki 2 y�l i�inde relaps� olan hastalar ve/veya ba�lang��ta Gd tutan T1 lezyonlar�n�n varl��� olarak tan�mlan�r) tedavi ba�lang�c�ndaki �zellikler genel pop�lasyona benzer olmu�tur. Ortanca ya� 47, ortanca hastal�k s�resi 15 y�l ve ba�lang��taki ortanca EDSS skoru 6,0'd�r.
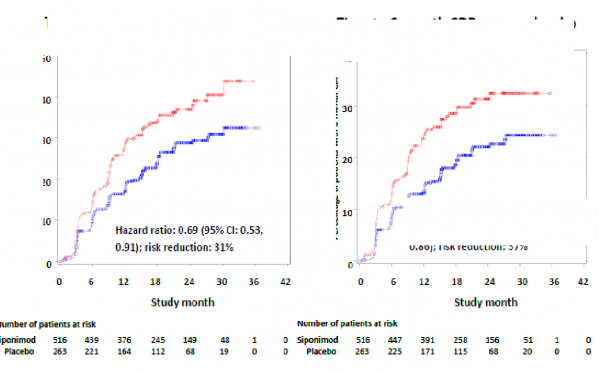
Siponimod ile tedavi edilen aktif hastal�kl� hastalarda 3 ayl�k ve 6 ayl�k CDP'nin ba�lama zaman�, plaseboya k�yasla s�ras�yla %31 (tehlike oran� [HR] 0,69; %95 GA: 0,53, 0,91) ve %37
(HR 0,63; %95 Cl: 0,47, 0,86) oranlar�nda daha ge� olmu�tur. ARR (do�rulanm�� relapslar)
plasebo ile kar��la�t�r�ld���nda %46 azalm��t�r (ARR oran� 0,54; %95 GA: 0,39, 0,77). 24 ay boyunca Gd tutan T1 a��rl�kl� lezyonlar�n k�m�latif say�s�nda ba��l oran azalmas�, plaseboya k�yasla %85 olmu�tur (vuku oran� 0,155; %95 GA: 0,104, 0,231). T2 lezyon hacmi de�i�imi ve beyin hacmi de�i�im y�zdesindeki (12 ve 24 ayl�k ortalamalar) plaseboya k�yasla farklar s�ras�yla -1163 mm (%95 GA: -1484, -843 mm) ve %0,141 (%95 GA: 0,020, %0,261)
bulunmu�tur.
�ekil 2 EDSS-Kaplan-Meier e�rilerine dayal� 3 ayl�k ve 6 ayl�k CDP'li hastalar - Aktif
Riskli hasta say�s�
Riskli hasta say�s�
�al��ma ay�
�al��ma ay�
SPMS'li alt grup (tam analiz seti, �al��ma A2304)
Plasebo kar��s�nda 3 ayl�k CDP'ye kadar
ge�en s�re (birincil sonlan�m noktas�)
Plasebo kar��s�nda 6 ayl�k CDP'ye kadar ge�en s�re
Hastal�k aktivitesi belirtileri ve semptomlar� olmayan (�al��madan �nceki 2 y�l i�inde relaps� olmayan hastalar ve/veya ba�lang��ta Gd tutan T1 lezyonlar� olmayan hastalar olarak tan�mlan�r) hasta alt grubunda (n = 827), 3 ayl�k ve 6 ayl�k CDP �zerindeki etkiler k���k olmu�tur (risk azalmalar� s�ras�yla %7 ve %13).
5.2. Farmakokinetik �zellikler
Genel �zelliklerEmilim:
Siponimodun �oklu oral uygulamas�ndan sonra maksimum plazma konsantrasyonlar�na
(Cmaks) ula�ma s�resi yakla��k 4 saattir (aral�k: 2 ila 12 saat). Siponimod emilimi geni�tir (≥
%70, idrarla at�lan radyoaktivite miktar�na ve d��k�daki sonsuzlu�a ekstrapole edilen metabolit
miktar�na ba�l� olarak). Siponimodun mutlak oral biyoyararlan�m� yakla��k %84't�r. 10 g�n
boyunca g�nde bir kez verilen 2 mg siponimod i�in, 10. g�nde ortalama 30,4 ng/ml C ve ortalama 558 h*ng/ml EAA de�erleri g�zlenmi�tir. Siponimodun g�nde bir kez �oklu uygulamalar�ndan sonra kararl� duruma yakla��k 6 g�n sonra ula��lm��t�r.
T'ta tek bir dozdan sonra 8 saatlik bir gecikmeye ra�men, g�da al�m�n�n siponimodun sistemik maruziyeti (C ve EAA) �zerinde hi�bir etkisi olmam��t�r, bu nedenle siponimod yemeklerden ba��ms�z olarak al�nabilir (bkz. B�l�m 4.2).
Da��l�m:
Siponimod v�cut dokular�na ortalama 124 litre da��l�m hacmi ile da��t�l�r. Plazmada bulunan siponimod fraksiyonu insanlarda %68'dir. Siponimod kan-beyin bariyerini kolayl�kla ge�er. Siponimodun proteinlere ba�lanmas� sa�l�kl� ki�ilerde ve karaci�er veya b�brek yetmezli�i olan hastalarda >%99,9'dur.
Biyotransformasyon:
Siponimod, esas olarak sitokrom P450 2C9 (CYP2C9) (%79,3) ve daha az bir �l��de sitokrom P450 3A4 (CYP3A4) (%18,5) taraf�ndan geni� �l��de metabolize edilir.
Ana metabolitler M3 ve M17'nin farmakolojik aktivitesinin, insanlarda siponimodun klinik etkisine ve g�venlili�ine katk�da bulunmas� beklenmemektedir.
In vitro ara�t�rmalar, siponimod ve onun ana sistemik metabolitleri M3 ve M17'nin, ara�t�r�lan t�m CYP enzimleri ve ta��y�c�lar� i�in g�nde bir kez 2 mg'l�k terap�tik dozda klinik olarak anlaml� herhangi bir ila�-ila� etkile�im potansiyeli g�stermedi�ini ve klinik ara�t�rma gerektirmedi�ini g�stermi�tir.
CYP2C9 polimorfiktir ve genotip, iki oksidatif metabolizma yolunun genel eliminasyona fraksiyonel katk�lar�n� etkiler. PBPK modellemesi, CYP2C9 genotipine ba��ml� diferansiyel bir inhibisyon ve CYP3A4 yolaklar�n�n ind�ksiyonunu g�stermektedir. �lgili genotiplerde CYP2C9 metabolik aktivitesi daha az oldu�undan, CYP3A4 ile etkile�en maddelerin siponimod maruziyeti �zerinde daha b�y�k bir etkisinin olmas� beklenmektedir (bkz. B�l�m 4.5).
Eliminasyon:
MS hastalar�nda 3,11 l/saatlik g�r�n�r bir sistemik klirens (CL/F) hesaplanm��t�r. Siponimodun g�r�nen eliminasyon yar�lanma �mr� yakla��k 30 saattir.
Siponimod, esas olarak metabolizma ve daha sonra safra/fe�al at�l�m yoluyla sistemik dola��mdan elimine edilir. �drarda de�i�memi� siponimod saptanmam��t�r.
Do�rusall�k / do�rusal olmayan durum:
Siponimod konsantrasyonu, 0,3 mg ila 20 mg/g�n aras�ndaki �oklu siponimod dozlar�ndan sonra dozla orant�l� bir �ekilde artar.
6 g�n s�reyle g�nde bir kez doz uygulamas�ndan sonra kararl� durum plazma konsantrasyonlar�na ula��l�r ve kararl� durum d�zeyleri ba�lang�� dozundan yakla��k 2 ila 3 kat daha y�ksektir. 6 g�n sonra 2 mg siponimod klinik terap�tik dozuna ula�mak i�in bir titrasyon rejimi kullan�l�r ve kararl� durum plazma konsantrasyonlar�na ula�mak i�in 4 g�n daha dozlama gerekir.
Belirli gruplardaki veya �zel pop�lasyonlardaki hastalardaki karakteristik �zellikler
CYP2C9 genotipi
CYP2C9 genotipi siponimod CL/F'yi etkiler. �ki pop�lasyon farmakokinetik analizi CYP2C9*1*1 ve*1*2 olgular�n�n b�y�k �l��de metabolize edici,*2*2 ve*1*3 olgular�n�n orta d�zeyde metabolize edici ve *2*3 ve*3*3 olgular�n�n zay�f metabolize edici olarak davrand���n� g�stermi�tir. CYP2C9*1*1 olgular�yla kar��la�t�r�ld���nda, CYP2C9*2*2,*1*3,*2*3 ve*3*3 genotipleri olan bireylerde CL/F de�erleri s�ras�yla %20, %35-38, %45-48 ve %74 daha d���kt�r. Dolay�s�yla siponimod maruziyeti, CYP2C9*2*2,*1*3,*2*3 ve*3*3 olgularda s�ras�yla %25, %61, %91 ve %284 daha y�ksektir. (bkz. Tablo 4) (bkz. b�l�m 4.2 ve 4.4).
CYP2C9 i�in daha az s�kl�kta meydana gelen ba�ka polimorfizmler de vard�r. Siponimodun farmakokineti�i bu t�r olgularda de�erlendirilmemi�tir. *5, *6, *8 ve *11 gibi baz� polimorfizmler, enzim fonksiyonunun azalmas� veya kayb� ile ili�kilidir. CYP2C9 *5, *6, *8 ve *11 allellerinin Afrika k�kenli pop�lasyonlarda yakla��k %10, Latinler/Hispaniklerde %2 ve Beyazlar ve Asyal�larda <%0.4 birle�ik s�kl��a sahip oldu�u tahmin edilmektedir.
Tablo 4 Siponimod CL/F ve sistemik maruziyet �zerinde CYP2C9 genotipetkisi
CYP2C9 genotipi | Beyaz olgularda s�kl�k |
Tahmini CL/F (L/h) |
CYP2C9*1*1 CL/F %'si | CYP2C9*1*1 kar��s�nda maruziyet art��� %'si |
B�y�k �l��de metabolize edici | ||||
CYP2C9*1*1 | 62-65 | 3,1-3,3 | 100 | - |
CYP2C9*1*2 | 20-24 | 3,1-3,3 | 99-100 | - |
Orta d�zeyde metabolize edici | ||||
CYP2C9*2*2 | 1-2 | 2,5-2,6 | 80 | 25 |
CYP2C9*1*3 | 9-12 | 1,9-2,1 | 62-65 | 61 |
Zay�f metabolize edici | ||||
CYP2C9*2*3 | 1,4-1,7 | 1,6-1,8 | 52-55 | 91 |
CYP2C9*3*3 | 0,3-0,4 | 0,9 | 26 | 284 |
Ya�l� hastalar
Pop�lasyon farmakokineti�inin sonu�lar�, ya�l� hastalarda (65 ya� ve �st�) doz ayarlamas�n�n gerekli olmad���n� g�stermektedir. Klinik �al��malara 61 ya��n �zerindeki hasta dahil edilmemi�tir. Siponimod ya�l�larda dikkatli kullan�lmal�d�r (bkz. B�l�m 4.2).
Cinsiyet
Pop�lasyon farmakokineti�inin sonu�lar�, cinsiyete dayal� doz ayarlamas�n�n gerekli olmad���n� g�stermektedir.
Irk/etnik k�ken
Tek doz farmakokinetik parametreler, sa�l�kl� Japon ve Beyaz bireyler aras�nda farkl�l�k g�stermemi�tir ki bu dasiponimodunfarmakokineti�i�zerinde etnik duyarl�l���n olmad���n�
B�brek yetmezli�i
Hafif, orta veya �iddetli b�brek yetmezli�i olan hastalarda siponimod doz ayarlamas�na gerek yoktur. Ortalama siponimod yar�lanma �mr� ve C (toplam ve plazma proteinlerine ba�lanmam��), ciddi b�brek yetmezli�i olan hastalar ile sa�l�kl� ki�iler aras�nda benzer olmu�tur. Toplam ve ba�lanmam�� EAA'lar sa�l�kl� olgulara k�yasla sadece biraz artm��t�r (%23-33). Son d�nem b�brek yetmezli�i veya hemodiyalizin siponimodun farmakokineti�i �zerindeki etkileri ara�t�r�lmam��t�r. Siponimodun y�ksek plazma protein ba�lanmas�na (>%99,9) ba�l� olarak, hemodiyalizin toplam ve ba�lanmam�� siponimod konsantrasyonunu de�i�tirmesi beklenmez ve bu hususlara dayanarak herhangi bir doz ayarlamas� �ng�r�lmez.
Karaci�er yetmezli�i
Siponimod, ciddi karaci�er yetmezli�i olan hastalarda kullan�lmamal�d�r (bkz. B�l�m 4.3). Hafif veya orta �iddette karaci�er yetmezli�i olan hastalarda siponimod i�in doz ayarlamas�na gerek yoktur. Ba�lanmam�� siponimod farmakokineti�i (EAA), incelenen 0,25 mg tek doz i�in sa�l�kl� olgulara k�yasla, orta ve �iddetli karaci�er yetmezli�i olanlarda s�ras�yla %15 ve %50 daha y�ksektir. Siponimodun ortalama yar� �mr� karaci�er yetmezli�inde de�i�memi�tir.
5.3. Klinik �ncesi g�venlilik verileri
Farelerde, s��anlarda ve maymunlarda tekrarl� doz toksisite �al��malar�nda, siponimod, lenfoid sistemi (lenfopeni, lenfoid atrofi ve azalt�lm�� antikor tepkisi) belirgin �ekilde etkilemi� olup bu, S1P1 resept�rlerindeki birincil farmakolojik aktivitesi ile tutarl�d�r (bkz. B�l�m 5.1).
Hayvan t�rlerindeki doz s�n�rlay�c� toksisiteler, farelerde nefrotoksisite, s��anlarda v�cut a��rl��� geli�imi ve maymunlarda advers SSS ve gastrointestinal etkiler olmu�tur. Kemirgenlerdeki toksisitenin ana hedef organlar� akci�er, karaci�er, tiroid, b�brek ve rahim/vajinad�r. Maymunlarda ayr�ca kas ve cilt �zerindeki etkiler g�zlenmi�tir. Bu toksisiteler, 2 mg/g�n idame dozunda EAA baz�nda insan maruziyetinden 30 kat daha y�ksek sistemik siponimod d�zeylerinde geli�mi�tir.
Siponimod herhangi bir fototoksik veya ba��ml�l�k potansiyeli g�stermemi�tir ve in vitro ve in vivo ko�ullarda genotoksik etki sergilememi�tir.
Karsinojenite
Karsinojenite ara�t�rmalar�nda, siponimod, farelerde lenfoma, hemanjiyom ve hemanjiyosarkomu ind�klerken erkek s��anlarda folik�ler adenom ve tiroid bezinin karsinomu saptanm��t�r. Bu t�m�r bulgular� ya fareye �zg� olarak kabul edilmi�tir ya da �zellikle hassas olan s��an t�rlerinde metabolik karaci�er adaptasyonlar�na atfedilebilirdir ve insanlar a��s�ndan ilgisi ��phelidir.
Fertilite ve �reme toksisitesi
Siponimod, g�nl�k 2 mg dozda insan sistemik maruziyetine (EAA) dayal� olarak yakla��k 19 kat g�venlilik marj�na kar��l�k gelen, test edilen en y�ksek doza kadar s��anlarda erkek ve di�i do�urganl���n� etkilememi�tir.
Siponimoddan etkilenen resept�r�n (sfenosin-1-fosfat resept�r�) embriyogenez s�ras�nda vask�ler olu�umda rol oynad��� bilinmektedir.
artm��t�r. S��anlarda d��, iskelet ve i� organ malformasyonlar� olan (�rn. yar�k damak ve �ekilsiz
klavik�ller, kardiyomegali ve �dem) daha fazla say�da fet�s kaydedilirken, tav�an fet�slerinde a��rl�kl� olarak iskelet ve i� organ varyasyonlar� g�zlenmi�tir.
S��anlarda yap�lan prenatal ve postnatal geli�im �al��mas�nda, daha y�ksek �l� yavru (�l� do�an veya do�um sonras� 4. g�nden �nce �l� bulunan) ve malforme yavru (�rogenital malformasyonlar� ve/veya artm�� anogenital mesafeli olan erkek yavrular; her iki cinsiyetten �demli, �i�mi� yumu�ak kafatasl� veya b�k�lm�� arka ayakl� yavrular) say�s� g�zlenmi�tir.
Embriyofetal (s��anlar ve tav�anlar) ve pre/postnatal (s��anlar) geli�im i�in ilgili NOAEL'lerde maruziyet d�zeyleri (EAA), g�nl�k 2 mg'l�k bir dozda insan sistemik maruziyetinin (EAA) alt�nda olmu�tur ve dolay�s�yla herhangi bir g�venlik pay� bulunmamaktad�r.
6. FARMAS�T�K �ZELL�KLER
6.1. Yard�mc� maddelerin listesi
Laktoz monohidrat (s���r kaynakl�) Mikrokristalin sel�loz Krospovidon (Tip A)
Gliserol dibehenat Kolloidal susuz silika
Polivinil alkol (k�smen hidrolize) Titanyum dioksit (E171)
K�rm�z� demir oksit (E172) Siyah demir oksit (E172) Talk
Soya lesitini (E322) Ksantan sak�z�
6.2. Ge�imsizlikler
Ge�erli de�ildir.
6.3. Raf �mr�
�r�n a��lmam�� olarak buzdolab�nda (2-8°C'de) saklanmak ko�ulu ile 24 ay raf �mr�ne sahiptir. �r�n a��ld�ktan sonra 25°C'nin alt�ndaki oda s�cakl���nda 3 ay saklanabilir.
6.4. Saklamaya y�nelik �zel tedbirler
2 ºC – 8 ºC aras�nda buzdolab�nda muhafaza ediniz.
�r�n a��ld�ktan sonra 25°C'nin alt�ndaki oda s�cakl���nda 3 ay saklanabilir.
6.5. Ambalaj�n niteli�i ve i�eri�i
C�zdan i�erisinde 12 film kapl� tablet i�eren PA/alu/PVC/alu blister ambalajlarda titrasyon paketleri.
84 ve 120 film kapl� tablet i�eren PA/alu/PVC/alu blister ambalajlarda paketler.
6.6. Be�eri t�bbi �r�nden arta kalan maddelerin imhas� ve di�er �zel �nlemler
Kullan�lmam�� olan �r�nler ya da at�k materyaller “T�bbi At�klar�n Kontrol� Y�netmeli�i” ve “Ambalaj At�klar�n�n Kontrol� Y�netmeli�i”ne uygun olarak imha edilmelidir.
 Do�um Sonras� Depresyonu
Do�um sonras� depresyonu, do�umdan sonra her on kad�ndan biri taraf�ndan
tecr�be edilen stresli bir durumdur.
Do�um Sonras� Depresyonu
Do�um sonras� depresyonu, do�umdan sonra her on kad�ndan biri taraf�ndan
tecr�be edilen stresli bir durumdur. |
 Pankreas Kanseri
Pankreas karn�n alt k�sm�nda yatay �ekilde bulunan bir organd�r. Sindirime yard�mc� olan enzimleri ve kan �ekerini y�netmeye yard�mc� olan hormonlar� v�cuda da��tmakla g�revlidir.
Pankreas Kanseri
Pankreas karn�n alt k�sm�nda yatay �ekilde bulunan bir organd�r. Sindirime yard�mc� olan enzimleri ve kan �ekerini y�netmeye yard�mc� olan hormonlar� v�cuda da��tmakla g�revlidir. |
 |
�nme �nme, beynin hasar g�rmesinin sonucudur. Bu hasar, beynin bir k�sm�ndaki ya bir kanama ya da akut kan eksikli�i nedeniyle o k�sm�n ge�ici ya da kal�c� olarak i�levini yapamamas�na yol a�ar. |
 |
Artrit Artrit, olduk�a yayg�n bir hastal�kt�r ancak iyi anla��lamam��t�r. Asl�nda �artrit� tek bir hastal���n ad� de�ildir; eklem a�r�s� veya eklem hastal�klar�n� adland�rman�n gayri resmi yoludur. |
 |
Omurilik zedelenmeleri Omurilik zedelenmesini takip eden birka� g�n i�inde, hi�kimse hasarin ne kadar olacagini tahmin edemez. Buradaki sorun, omuriligin herhangi bir zedelenmesinden hemen sonra, bir omurilik sokunun olusmasidir. |
�LA� GENEL B�LG�LER�
Farmanova Sa�l�k Hizmetleri Ltd. �ti
| Sat�� Fiyat� | 38608.6 TL [ 1 Dec 2025 ] |
| �nceki Sat�� Fiyat� | 38608.6 TL [ 24 Nov 2025 ] |
| Original / Jenerik | Original �la� |
| Re�ete Durumu | K�rm�z� Re�eteli bir ila�d�r. |
| Barkodu | 8699074091018 |
| Etkin Madde | Siponimod Fumarik Asit |
| �thal ( ref. �lke : Almanya ) ve Be�eri bir ila�d�r. |