ALECENSA 150 mg 224 sert kapsül Kısa Ürün Bilgisi
{ Alektinib Hidroklorür }
1. BEŞERİ TIBBİ ÜRÜNÜN ADI
ALECENSA 150 mg sert kapsül
2. KALİTATİF VE KANTİTATİF BİLEŞİM
Etkin madde
Alektinib (161,33 mg Alektinib hidroklorüre eşdeğer) 150 mg
Yardımcı maddeler
Laktoz (monohidrat) | 33,67 mg (inek sütünden elde edilmektedir) |
Sodyum (laurilsülfat) | 6 mg |
Dehidre etil alkol | Baskı mürekkebinde % 21 oranında (0,49 mikrogram) bulunmaktadır |
Yardımcı maddeler için 6.1'e bakınız.
3. FARMASÖTİK FORMU
Sert kapsül.
Kapağının üzerinde siyah mürekkeple "ALE" yazılı ve gövdesinin üzerinde siyah mürekkeple “150 mg†yazılı 19,2 mm uzunluğunda beyaz sert kapsül.
4. KLİNİK ÖZELLİKLER
4.1. Terapötik endikasyonlar
ALECENSA'nın, yetişkin anaplastik lenfoma kinaz (ALK) pozitifliği (standardize FISH veya RT-PCR veya yeni nesil dizileme yöntemleri ile tespit edilen rearanjman/füzyon varlığı; veya immünhistokimya ALK pozitifliği) saptanan, ileri evre veya metastatik küçük hücreli dışı akciğer kanserli hastaların birinci basamak tedavisinde, monoterapi olarak progresyona kadar kullanımı endikedir.
ALECENSA, daha önce krizotinib ile tedavi edilmiş, yetişkin, anaplastik lenfoma kinaz (ALK)-pozitif lokal ileri veya metastatik küçük hücreli dışı akciğer kanseri (KHDAK) hastalarında monoterapi olarak progresyona kadar kullanılır.
4.2. Pozoloji ve uygulama şekli
ALECENSA tedavisi, kanser hastalarının tedavisinde deneyimli bir hekim tarafından
başlatılmalı ve denetlenmelidir.
ALK-pozitif KHDAK hastaların seçilmesi için valide bir ALK testi gereklidir. ALECENSA tedavisine başlanmadan önce ALK-pozitif KHDAK durumu tespit edilmelidir.
Pozoloji:
ALECENSA için önerilen doz, yemeklerle birlikte günde iki kez alınacak 600 mg'dır (dört adet 150 mg kapsül) (toplam günlük 1200 mg doz).
Altta yatan şiddetli karaciğer yetmezliği olan hastalar (Child-Pugh C), günde iki kez yemeklerle birlikte 450 mg'lık başlangıç dozu almalıdır (günde toplam 900 mg).
Tedavi süresi
Hastalık ilerlemediği veya kabul edilemez bir toksisite meydana gelmediği sürece ALECENSA tedavisine devam edilmelidir.
Unutulan veya geciken dozlar:
ALECENSA'nın alınması gereken dozlarından biri unutulursa, sonraki doza en az 6 saatten fazla bir süre varsa, unutulan doz telafi edilebilir. Hastalar, unutulan dozu telafi etmek için aynı anda iki doz almamalıdır. Hasta, ALECENSA aldıktan sonra kusarsa, doz tekrarı yapmamalı ve planlanan doz şemasına göre sonraki dozu almalıdır.
Doz ayarlamaları:
Genel:
Advers olayların kontrol altına alınması için dozun azaltılması, geçici bir süre boyunca ara verilmesi veya ALECENSA tedavisinin tamamen kesilmesi gerekebilir. ALECENSA dozu tolerabiliteye göre aşamalı bir şekilde günde iki kez 150 mg azaltılabilir. Eğer hasta günde iki kez 300 mg'lık dozu tolere edemezse ALECENSA tedavisi tamamen kesilmelidir.
ALECENSA dozunun değiştirilmesine ilişkin tavsiyeler Tablo 1 ve Tablo 2'de sunulmaktadır.
Tablo 1 Doz azaltma şeması
Doz azaltma şeması | Doz düzeyi |
Doz | 600 mg günde iki kez |
İlk doz azaltımı | 450 mg günde iki kez |
İkinci doz azaltımı | 300 mg günde iki kez |
Tablo 2 Belirli advers ilaç reaksiyonları için doz değişikliği önerileri (bkz. Bölüm 4.4 ve 4.8)
CTCAE Derecesi | ALECENSA tedavisi |
İnterstisyel akciğer hastalığı (ILD)/pnömonit: herhangi bir şiddet derecesinde | ILD/pnömonit için potansiyel başka nedenler belirlenemezse tedavi hemen durdurulmalı ve kalıcı olarak kesilmelidir. |
Başlangıç veya ï‚£ Derece 1'e (normal üst limitin ≤3 katı artış) dönene dek tedavi geçici olarakkesilmeli, daha sonra düşük dozda başlatılmalıdır(BakınızTablo1). |
CTCAE Derecesi | ALECENSA tedavisi |
Kolestaz veya hemolizin eşlik etmediği ALT veya AST yükselmesi Derece ≥ 2 (normal üst limitin >3 katı artış ile total bilirubinde normal üst limitin >2 katı artış), | ALECENSA tedavisi kalıcı olarak kesilmelidir. |
Bradikardi Derece 2 veya Derece 3 (semptomatik, şiddetli veya tıbben önemli, tıbbi müdahele gerekli) | (Asemptomatik) bradikardi ï‚£ Derece 1 veya kalp hızı ≥ 60 bpm'e (atım/dakika) dönene dek tedavi geçici olarak kesilmelidir. Anti- hipertansif ilaçlarla bradikardiye neden olduğu bilenen eşzamanlı tıbbi ürünler değerlendirilmelidir.
Katkıda bulunan eşzamanlı ilaç belirlenmiş ve kesilmişse veya dozu ayarlanmışsa, ï‚£ Derece 1 (asemptomatik) bradikardi veya kalp hızı ≥ 60 bpm'e (atım/dakika) dönünce tedavi önceki dozda başlatılmalıdır.
Katkıda bulunan hiçbir ilaç tanımlanmamış veya katkıda bulunan eşzamanlı ilaç kesilmemişse veya doz ayarlanmamışsa, ≤ Derece 1 (asemptomatik) bradikardi veya kalp hızı ≥ 60 bpm'e (atım/dakika) dönünce tedavi düşük dozda başlatılmalıdır (Bakınız Tablo 1). |
Bradikardi Derece 4 (hayatı tehdit edici sonuçlar, acil müdahale gerekli) | Katkıda bulunan hiçbir eşzamanlı ilaç tanımlanmamışsa tedavi tamamen kesilmelidir. Katkıda bulunan eşzamanlı ilaç tanımlanmış ve kesilmişse veya doz ayarlanmışsa, ï‚£ Derece 1 (asemptomatik) bradikardi veya ≥ 60 bpm kalp hızına dönünce, klinik endikasyona göre sık takip edilerek, tedavi düşük dozda başlatılmalıdır (Bakınız Tablo 1). Tekrar ettiği durumda tedavi kalıcı olarak kesilmelidir. |
Kreatin fosfokinaz yükselmesi > Normal üst limitin 5 katı artış | Başlangıç veya normal üst limitin ï‚£2,5 katına dönene dek tedavi geçici olarak kesilmeli, daha sonra aynı dozda devam edilmelidir. |
Kreatin fosfokinaz yükselmesi > Normal üst limitin 10 katı artış veya ikinci kez normal üst limitin > 5 katı artış | Başlangıç veya normal üst limitin ï‚£2,5 katına dönene dek tedavi geçici olarak kesilmeli, daha sonra düşük dozda başlatılmalıdır. (Bakınız Tablo 1) |
<10 g/dL'lik hemoglobin düşüklüğü ile görülen Derece≥ 2 hemolitik anemi | İyileşme sağlanana kadar tedavi geçici olarak kesilmeli, daha sonra tedavi düşük dozda başlatılmalıdır (Bakınız Tablo 1) |
ALT: Alanin transaminaz, AST: Aspartat transaminaz, CTCAE = Advers Olaylar için NCI Ortak Terminoloji Kriteri
Uygulama şekli:
ALECENSA oral kullanım içindir. Sert kapsüller bütün olarak yutulmalı, açılmamalı veya çözdürülmemelidir.
Yiyeceklerle beraber alınmalıdır (bkz. Bölüm 5.2).
Özel popülasyonlara ilişkin ek bilgiler:
Karaciğer yetmezliği:
Altta yatan hafif (Child-Pugh A) veya orta derecede (Child-Pugh B) karaciğer bozukluğu olan hastalarda başlangıç dozu ayarlaması gerekmemektedir. Altta yatan şiddetli karaciğer yetmezliği olan hastalar (Child-Pugh C), günde iki kez 450 mg'lık başlangıç dozu almalıdır (günde toplam 900 mg) (bkz. Bölüm 4.2). Karaciğer yetmezliği olan tüm hastalar için uygun izlem (karaciğer fonksiyon belirteçleri gibi) önerilmektedir (bkz. Bölüm 4.4).
Böbrek yetmezliği:
Hafif veya orta derecede böbrek yetmezliği olan hastalarda doz ayarlaması gerekmemektedir. ALECENSA, şiddetli böbrek yetmezliğiu olan hastalarda incelenmemiştir. Bununla birlikte, alektinibin böbrekle atılımı ihmal edilebilir boyutta olduğundan, şiddetli böbrek yetmezliğiu olan hastalarda doz ayarlaması gerekmemektedir (bkz. Bölüm 5.2).
Pediyatrik popülasyon:
Çocuklarda ve adolesanlarda (<18 yaş) ALECENSA'nın güvenliliği ve etkililiğine dair veri bulunmamaktadır.
Geriyatrik popülasyon (≥ 65 yaş):
65 yaş ve üzeri hastalarda ALECENSA'nın güvenliliği ve etkililiğine ilişkin kısıtlı veriler, yaşlı hastalarda doz ayarlamasının gerekli olmadığını düşündürmektedir (bkz. Bölüm 5.2). 80 yaş üzeri hastalar için veri yoktur.
Aşırı vücut ağırlığı (>130 kg):
ALECENSA için farmakokinetik simülasyonları aşırı vücut ağırlığına (yani >130 kg) sahip hastalarda düşük maruziyete işaret etmemekle birlikte, alektinib yaygın dağılıma sahiptir ve alektinibe ilişkin klinik çalışmalara vücut ağırlıkları 36,9-123 kg aralığında yer alan hastalar kaydedilmiştir. Vücut ağırlığı 130 kg'ın üzerinde olan hastalar için veri yoktur.
4.3. Kontrendikasyonlar
Etkin madde
4.4. Özel kullanım uyarıları ve önlemleri
İnterstisyel akciğer hastalığı (ILD) / Pnömonit:
ALECENSA ile yapılan klinik çalışmalarda ILD/pnömonit vakaları bildirilmiştir (bkz. Bölüm 4.8). Hastalar, pnömonit göstergesi pulmoner belirtiler için yakından takip edilmelidir. ILD/pnömonit tanısı konulan hastalarda ALECENSA tedavisine hemen ara verilmelidir ve eğer başka ILD/pnömonit potansiyel nedenleri tanımlanmamışsa tedavi kalıcı olarak kesilmelidir (bkz. Bölüm 4.2).
Hepatotoksisite
ALECENSA ile yürütülen pivotal klinik çalışmalarda yer alan hastalarda, alanin aminotransferaz (ALT) ve aspartat aminotransferazda (AST) normalin üst limitinin 5 katından fazla artışın yanı sıra bilirubinde normalin üst limitinin 3 katından fazla artış meydana gelmiştir (bkz. Bölüm 4.8). Bu olayların büyük çoğunluğu tedavinin ilk 3 ayı içinde meydana gelmiştir. Pivotal ALECENSA klinik çalışmalarında, Derece 3-4 AST/ALT artışı görülen üç hastada ilaca bağlı karaciğer hasarı görülmüştür. ALECENSA klinik çalışmalarında tedavi edilen bir hastada normal alkalin fosfataz eşliğinde ALT veya AST'de normalin üst limitinin 3 katı veya daha yüksek ve total bilirubinde normalin üst limitinin 2 katı ya da daha yüksek olan eşzamanlı artış meydana gelmiştir.
ALT, AST dahil olmak üzere karaciğer fonksiyonu ve total bilirubin, başlangıçta ve daha sonra tedavinin ilk 3 ayı süresince her 2 haftada bir izlenmelidir. Advers reaksiyonlar 3 aydan daha geç bir zamanda da meydana gelebileceğinden, izleme periyodik olarak devam edilmeli, özellikle aminotransferaz ve bilirubin artışları görülen hastalar daha sık test ile izlenmelidir. Advers ilaç reaksiyonunun şiddetine bağlı olarak ALECENSA tedavisi, Tablo 2'de tarif edildiği şekilde geçici olarak kesilmeli ve azaltılmış bir dozda yeniden başlatılmalı ya da kalıcı olarak kesilmelidir (bkz. Bölüm 4.2).
Şiddetli kas ağrısı ve kreatin fosfokinaz (CPK) artışı
ALECENSA ile yürütülen pivotal çalışmalarda, Derece 3 olaylar da dahil olmak üzere kas ağrısı veya muskuloskeletal ağrı bildirilmiştir (bkz. Bölüm 4.8).
ALECENSA ile yürütülen pivotal çalışmalarda, Derece 3 olaylar da dahil olmak üzere CPK artışları bildirilmiştir (bkz. Bölüm 4.8). Klinik çalışmalarda (NP28761, NP28673, BO28984), Derece 3 CPK artışına kadar geçen medyan süre 14 gün olmuştur.
Hastalara açıklanmayan kas ağrısı, hassasiyeti veya güçsüzlüğünü bildirmeleri tavsiye edilmelidir. CPK düzeyleri tedavinin ilk ayında iki haftada bir ve semptom bildiren hastalarda klinik olarak endike olduğu şekilde değerlendirilmelidir. CPK artışının şiddetine bağlı olarak ALECENSA kesilmeli, ardından yeniden başlatılmalı veya dozu azaltılmalıdır (bkz. Bölüm 4.2).
Bradikardi
5 -
![]()
bradikardi veya yaşamı tehdit eden olaylar yaşayan hastalarda, bradikardiye yol açtığı bilinen
eşzamanlı ilaçların ve antihipertansif ilaçların incelenmesi ve ALECENSA tedavisinin Tablo 2'de belirtilen şekilde düzenlenmesi önerilir (bkz. Bölüm 4.2 ve 4.5, ‘P-gp ve BCRP substratları').
Hemolitik anemi
ALECENSA ile hemolitik anemi bildirilmiştir (bkz. Bölüm 4.8). Hemoglobin konsantrasyonu
10 g/dL'nin altındaysa ve hemolitik anemiden şüpheleniliyorsa, ALECENSA tedavisi kesilmeli ve uygun laboratuvar testleri başlatılmalıdır. Hemolitik anemi doğrulanırsa, Tablo 2'de açıklandığı üzere, ALECENSA iyileşme sağlandıktan sonra düşük bir dozda başlatılmalıdır (bkz. Bölüm 4.2).
Gastrointestinal perforasyon
ALECENSA ile tedavi edilen yüksek riskli hastalarda (Örn. divertikülit öyküsü, gastrointestinal sistemde metastazı, gastrointestinal perforasyon riski bilinen ilaçlarla birlikte tedavi) gastrointestinal perforasyon vakaları bildirilmiştir. Gastrointestinal perforasyon gelişen hastalarda ALECENSA tedavisinin kesilmesi düşünülmelidir. Hastalar gastrointestinal perforasyon semptomları ve işaretleri hakkında bilgilendirilmeli ve bu durumun gelişmesi durumunda acilen bir hekime danışmaları tavsiye edilmelidir.
Işığa duyarlılık
ALECENSA uygulaması sırasında güneş ışığına duyarlılık bildirilmiştir (bkz. Bölüm 4.8). ALECENSA kullanan hastalara, tedavi sırasında ve tedavi kesiminden sonra en az 7 gün boyunca, uzun süreli güneş maruziyetinden kaçınmaları tavsiye edilmelidir. Hastalara ayrıca potansiyel güneş yanığından korunmaya yardımcı olması için geniş spektrumlu Ultraviyole A (UVA) / Ultraviyole B (UVB) güneş kremi ve dudak merhemi (SPF ≥ 50) kullanmaları da tavsiye edilmelidir.
Çocuk doğurma potansiyeli olan kadınlar
ALECENSA gebe bir kadına uygulandığında fetüse zarar verebilir. ALECENSA kullanan çocuk doğurma potansiyeline sahip kadın hastalar tedavi sırasında ve son ALECENSA dozunu takiben en az 3 ay boyunca yüksek etkililiğe sahip doğum kontrol yöntemleri kullanmalıdır (bkz. Bölüm 4.5, 4.6 ve 5.3).
Yardımcı maddeler
ALECENSA kapsüller, laktoz monohidrat içerir. Nadir kalıtımsal galaktoz intoleransı, Lapp laktaz yetmezliği ya da glikoz-galaktoz malabsorpsiyon problemi olan hastaların bu ilacı kullanmamaları gerekir.
Günlük önerilen ALECENSA dozu (1200 mg) 48 mg sodyum ihtiva eder. Bu durum, kontrollü sodyum diyetinde olan hastalar için göz önünde bulundurulmalıdır.
Bu tıbbi ürün az miktarda (her “doz†da 100 mg dan az) etanol (alkol) içerir (baskı mürekkebinde % 21 oranında bulunan (0,49 mikrogram) dehidre etil alkol baskı prosesi sırasında tamamen uzaklaştırılmaktadır).
4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri
Diğer ilaçların alektinib üzerindeki etkileri:
In vitro çalışmalara göre, CYP3A4, alektinib ve majör aktif metaboliti M4'ün metabolizmasında yer alan ana enzimdir ve CYP3A total hepatik metabolizmaya %40 - %50 katkıda bulunur. M4'ün, ALK'ye karşı in vitro olarak benzer potense ve aktiviteye sahip olduğu gösterilmiştir.
CYP3A indükleyicileri
Güçlü bir CYP3A indükleyicisi olan rifampisinden günde bir kez 600 mg'lık çoklu oral dozların 600 mg alektinib tekli oral dozu ile birlikte uygulanması, alektinibin Cve EAAdeğerlerini sırasıyla %51 ve %73 azaltmış ve M4'ün Cve EAAdeğerlerini sırasıyla 2,20 ve 1,79 kat artırmıştır. Alektinib ve M4'e birlikte maruziyette etki küçük olup, Cve EAAdeğerleri sırasıyla %4 ve %18 azalmıştır. Alektinib ve M4'e birlikte maruziyet üzerindeki etkiler esas alındığında, ALECENSA CYP3A indükleyicileri ile birlikte uygulandığında doz ayarlaması gerekmemektedir. Eşzamanlı olarak güçlü CYP3A indükleyicilerini (karbamazepin, fenobarbital, fenitoin, rifabutin, rifampisin ve sarı kantaron (Hypericum perforatum) vb.) kullanan hastalar için uygun izlem önerilir.
CYP3A inhibitörleri
Güçlü bir CYP3A inhibitörü olan posakonazolden günde iki kez 400 mg'lık çoklu oral dozların 300 mg alektinib tekli oral dozu ile birlikte uygulanması, alektinibin Cve EAAdeğerlerini sırasıyla 1,18 ve 1,75 kat artırmış ve M4'ün Cve EAAdeğerlerini sırasıyla
%71 ve %25 azaltmıştır. Alektinib ve M4'e birlikte maruziyette etki küçük olup, Cdeğeri
%7 azalırken, EAAdeğeri 1,36 kat artmıştır. Alektinib ve M4'e birlikte maruziyet üzerindeki etkiler esas alındığında, ALECENSA CYP3A inhibitörleri ile birlikte uygulandığında doz ayarlaması gerekmemektedir. Eşzamanlı olarak güçlü CYP3A inhibitörlerini (ritonavir, sakinavir, telitromisin, ketokonazol, itrakonazol, vorikonazol, posakonazol, nefazodon, greyfurt veya turunç vb.) kullanan hastalar için uygun izlem önerilir.
Gastrik pH düzeyini artıran tıbbi ürünler
Alektinibin in vitro suda çözünebilirliği pH'a bağlı olmakla birlikte, klinik ilaç-ilaç etkileşimi çalışmasında, günde bir kere 40 mg esomeprazol (proton pompa inhibitörü), alektinib ve M4'e birlikte maruziyet üzerinde klinik öneme sahip hiçbir etki göstermemiştir. Bu nedenle, ALECENSA, proton pompa inhibitörleri veya gastrik pH'yi artıran diğer ilaçlarla (H2 reseptör antagonistleri veya antasitler gibi) eşzamanlı uygulandığında hiçbir doz ayarlaması gerekmemektedir.
Taşıyıcıların alektinibin dispozisyonuna etkisi
M4, P-gp'nin bir substratıdır. Alektinib, P-gp'yi inhibe ettiğinden, P-gp inhibitörlerinin eş zamanlı kullanımının M4 maruziyeti üzerinde bir etki göstermesi beklenmemektedir.
Alektinibin diğer tıbbi ürünler üzerindeki etkileri:
P-gp substratları
İn vitro olarak alektinib ve majör aktif metaboliti M4, dışa-atım taşıyıcısı P-glikoproteinin (P- gp) inhibitörleridir. Bu nedenle alektinib ve M4, birlikte uygulanan P-gp substratlarının plazma konsantrasyonlarını artırma potansiyeline sahip olabilir. ALECENSA, P-gp substratları (örn., digoksin, dabigatran eteksilat, topotekan, sirolimus, everolimus, nilotinib ve lapatinib) ile birlikte uygulandığında uygun izlem önerilir.
BCRP substratları
İn vitro olarak alektinib ve M4, dışa-atım taşıyıcısı Meme Kanseri Direnç Proteininin (BCRP) inhibitörleridir. Bu nedenle alektinib ve M4, birlikte uygulanan BCRP substratlarının plazma konsantrasyonlarını artırma potansiyeline sahip olabilir. ALECENSA, BCRP substratları (örn., metotreksat, mitoksantron, topotekan ve lapatinib) ile birlikte uygulandığında uygun izlem önerilir.
CYP substratları
İn vitro olarak alektinib ve M4, zayıf, zamana bağlı CYP3A4 inhibisyonu gösterir ve alektinib klinik konsantrasyonlarda CYP3A4 ve CYP2B6 için güçsüz bir indüksiyon potansiyeline sahiptir.
Alektinibin 600 mg'lık çoklu dozları duyarlı bir CYP3A substratı olan midazolamın (2 mg) maruziyeti üzerinde bir etki göstermemiştir. Bu nedenle birlikte uygulanan CYP3A substratları için doz ayarlaması gerekli değildir.
CYP3A4 dışında CYP2B6 ve PXR ile düzenlenen enzimlerin indüksiyonu açısından risk tamamen ekarte edilemez. Doğum kontrol haplarının eşzamanlı uygulama ile etkililikleri azalabilir.
Özel popülasyonlara iliskin ek bilgiler
Özel popülasyonlarda herhangi bir etkileşim çalışması yürütülmemiştir.
Pediyatrik popülasyon
Pediyatrik popülasyonda herhangi bir etkileşim çalışması yürütülmemiştir.
4.6. Gebelik ve laktasyon
Gebelik kategorisi: D
Çocuk doğurma potansiyeline sahip kadınlar/Doğum Kontrolü (Konstrasepsiyon)
kadın hastalar tedavi sırasında ve son ALECENSA dozunu takiben en az 3 ay boyunca yüksek etkililiğe sahip doğum kontrol yöntemleri kullanmalıdır (bkz. Bölüm 4.4 ve 4.5).
Gebelik dönemi
Gebe kadınlarda ALECENSA kullanımına ilişkin veri yoktur ya da çok sınırlıdır. Etki mekanizması temelinde, ALECENSA gebe bir kadına uygulandığında fetüse zarar verebilir. Hayvanlarda yürütülen çalışmalar üreme toksisitesini göstermiştir (bkz. Bölüm 5.3).
ALECENSA kullanırken veya son ALECENSA dozundan sonraki 3 ayda gebe kalan kadın hastalar doktorlarını aramalı ve fetüs için potansiyel zarar konusunda bilgilendirilmelidir.
ALECENSA'nın gebelik ve/veya fetus/yeni doğan üzerindezararlı farmakolojik etkileri bulunmaktadır.
Laktasyon dönemi
Alektinibin ve/veya metabolitlerinin anne sütüne geçip geçmediği ve alektinibin emzirilen bebek veya süt yapımı üzerindeki etkileriyle ilgili veri bulunmamaktadır. Yenidoğan veya bebeklere olan riski gözardı edilemez. Emziren kadınlara, ALECENSA tedavisi sırasında emzirmemeleri gerektiği uyarısı yapılmalıdır.
Üreme yeteneği / Fertilite
Hayvanlarda ALECENSA etkisini değerlendiren hiçbir fertilite çalışması gerçekleştirilmemiştir. Genel toksikoloji çalışmalarında erkek ve dişi üreme organları üzerinde hiçbir advers etki gözlenmemiştir (bkz. Bölüm 5.3).
4.7. Araç ve makine kullanımı üzerindeki etkiler
ALECENSA araç ve makine kullanma yeteneği üzerinde çok küçük bir etkiye sahiptir. Hastalar ALECENSA kullanırken semptomatik bradikardi (örn., senkop, baş dönmesi, hipotansiyon) veya görme bozuklukları yaşayabileceklerinden araç veya makine kullanılırken dikkat gösterilmelidir (bkz. Bölüm 4.8).
4.8. İstenmeyen etkiler
Güvenlilik profilinin özeti:
Aşağıda belirtilen veriler, bir randomize faz III klinik çalışma (BO28984) ve iki tek-kollu faz II klinik çalışmaya (NP28761, NP28673) katılan, ilerlemiş ALK-pozitif küçük hücreli dışı akciğer kanserli 405 hastada ALECENSA'ya maruziyeti yansıtmaktadır. Bu hastalar, önerilen doz olan günde iki kez 600 mg ile tedavi edilmişlerdir. Faz II klinik çalışmalarda (NP28761, NP28673; N=253) ALECENSA'ya ortalama maruziyet süresi 11,2 ay olmuştur. BO28984 (ALEX; N=152) çalışmasında ALECENSA'ya ortalama maruziyet süresi 28,1 ay, krizotinibe ortalama maruziyet süresi ise 10,8 ay olmuştur.
En yaygın advers reaksiyonlar (≥ %20); kabızlık, miyalji, ödem, anemi, döküntü, bilirubin seviyesinde yükselme ve bulantı olmuştur.
Advers reaksiyonların tablosu:
0 -
![]()
Tablo 3, iki faz II klinik çalışma (NP28761, NP28673) ve bir faz III klinik çalışma BO28984 (ALEX) ve pazarlama sonrası dönemde ALECENSA alan hastalarda gözlenen advers olayları listelemektedir.
Tablo 3'te listelenen advers olaylar aşağıdaki sıra kullanılarak tanımlanmış sıklık kategorileri ve sistem organ sınıfına göre sunulmaktadır: Çok yaygın (≥1/10), yaygın (≥1/100 i <1/10), yaygın olmayan (≥1/1,000 - <1/100), seyrek (≥1/10,000 - <1/1000), çok seyrek (<1/10,000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). Her bir sıklık grubunun içinde istenmeyen etkiler azalan sıklık ve şiddet sırası ile sunulmaktadır. Aynı sıklık ve şiddet gruplamasında, istenmeyen etkiler azalan ciddiyet sıralamasına göre sunulmuştur.
Tablo 3 ALECENSA klinik çalışmalarında (NP28761, NP28673, BO28984; N=405)ve pazarlama sonrası dönemde gözlenen advers olaylar
Sistem organ sınıfı Advers olaylar (MedDRA) | ALECENSA N=405 | ||
| Sıklık kategorisi (tüm dereceler) | Sıklık kategorisi (derece 3-4 (%) | |
Kan ve lenf sistemi hastalıkları |
|
|
|
Anemi |
| Çok yaygın | Yaygın |
Hemolitik anemi |
| Yaygın olmayan | - |
Sinir sistemi hastalıkları |
|
|
|
Disgözi |
| Yaygın | Yaygın olmayan |
Göz hastalıkları |
|
|
|
Görme bozuklukları |
| Çok yaygın | - |
Kardiyak hastalıklar |
|
|
|
Bradikardi |
| Çok yaygın | - |
Solunum, göğüs bozuklukları ve mediastinal hastalıklar |
|
|
|
İnterstisyel akciğer hastalığı / Pnömonit |
| Yaygın | Yaygın olmayan |
Gastrointestinal hastalıklar |
|
|
|
İshal |
| Çok yaygın | Yaygın |
Kusma |
| Çok yaygın | Yaygın olmayan |
Kabızlık |
| Çok yaygın | Yaygın olmayan |
Bulantı |
| Çok yaygın | Yaygın olmayan |
Stomatit |
| Yaygın | - |
Hepato-bilier hastalıklar |
|
|
|
AST seviyesinde yükselme |
| Çok yaygın | Yaygın |
ALT seviyesinde yükselme |
| Çok yaygın | Yaygın |
Bilirubin seviyesinde yükselme |
| Çok yaygın | Yaygın |
Alkalin fosfataz seviyesinde | Yaygın | ||
İlaç-bağımlı karaciğer hasarı |
| Yaygın olmayan | Yaygın olmayan |
Deri ve deri altı doku hastalıkları |
|
|
|
Döküntü |
| Çok yaygın | Yaygın |
Işığa duyarlılık |
| Yaygın | Yaygın olmayan |
Kas-iskelet bozukluklar, bağ doku ve kemik hastalıkları |
|
|
|
Miyalji |
| Çok yaygın | Yaygın |
Kan kreatinin fosfokinaz seviyesinde yükselme |
| Çok yaygın | Yaygın |
Böbrek ve idrar yolu hastalıkları |
|
|
|
Akut böbrek hasarı |
| Yaygın | Yaygın |
Kan kreatinin seviyesinde yükselme |
| Yaygın | Yaygın olmayan |
Genel bozukluklar ve uygulama bölgesine iliskin hastalıklar |
|
|
|
Ödem |
| Çok yaygın | Yaygın |
Araştırmalar |
|
|
|
Kilo artışı |
| Çok yaygın | Yaygın olmayan |
* Derece 3-4 advers olayı görülmemiştir.
** 1 adet Derece 5 olayı içerir
Seçilmiş advers ilaç reaksiyonlarına ilişkin açıklama
1 -
![]()
İnterstisyel akciğer hastalığı (ILD) / pnömonit:
ALECENSA ile tedavi edilen hastalarda şiddetli ILD/pnömonit meydana gelmiştir. Klinik çalışmalarda (NP28761, NP28673, BO28984), ALECENSA ile tedavi edilen 405 hastadan 1'i (%0,2) Derece 3 ILD yaşamıştır. Bu olay ALECENSA tedavisinin kesilmesine yol açmıştır. BO28984 faz III klinik çalışmasında, krizotinible tedavi edilen hastalarda %2 görülme oranına karşılık, ALECENSA ile Derece 3 veya 4 ILD/pnömonit vakası gözlenmemiştir. Çalışmaların hiçbirinde ölümcül ILD vakaları görülmemiştir. Hastalar pnömonite işaret eden pulmoner semptomlar açısından izlenmelidir (bkz. Bölüm 4.2 ve 4.4).
Hepatotoksisite:
Klinik çalışmalarda (NP28761, NP28673, BO28984) Derece 3-4 AST (Aspartat transaminaz)/ALT (Alanin transaminaz) artışlarına sahip iki hastada karaciğer biyopsisi ile belgelenmiş, ilaca-bağlı karaciğer hasarı görülmüştür. Ayrıca bir hastada Derece 4 ilaca-bağlı karaciğer hasarı yan etkisi bildirilmiştir. Bu vakalardan ikisi ALECENSA tedavisinin kesilmesine yol açmıştır. Klinik çalışmalarda (NP28761, NP28673, BO28984) ALECENSA ile tedavi edilen hastalarda artmış AST ve ALT düzeyleri (sırasıyla %17 ve %16) bildirilmiştir. Bu olayların çoğu Derece 1 ve 2 şiddetinde olup, Derece ≥3 olaylar hastaların sırasıyla %3,7 ve %3,7'sinde bildirilmiştir. Olaylar genellikle tedavinin ilk 3 ayı sırasında meydana gelmiş olup, çoğunlukla geçici yapıdadır ve ALECENSA tedavisinin geçici olarak kesilmesi (hastaların sırasıyla %1,5 ve %3'ü için bildirilmiş) veya dozda azaltma (sırasıyla
%2 ve %1,5) ile düzelmiştir. Hastaların %1,2'sinde AST ve %1,5'inde ALT artışları ALECENSA tedavisinin kesilmesine yol açmıştır. BO28984 faz III klinik çalışmasında, ALECENSA alan hastalarda Derece 3 veya 4 ALT veya AST yükselmelerinin her biri %5 oranında görülürken, krizotinib alan hastalarda bu oranlar sırasıyla %16 ve %11 olmuştur.
Klinik çalışmalarda (NP28761, NP28673, BO28984) ALECENSA ile tedavi edilen hastaların
%21'inde bilirubin artışları bildirilmiştir. Olayların çoğu Derece 1 ve 2 şiddetindedir; Derece 3 olaylar hastaların %3,7'sinde bildirilmiştir. Olaylar genellikle tedavinin ilk 3 ayı sırasında meydana gelmiş olup, çoğunlukla geçicidir ve doz modifikasyonu ile düzelmiştir. Hastaların
%7,7'sinde bilirubin artışları doz modifikasyonlarına yol açmış, %2'sinde ise ALECENSA tedavisinin kesilmesini gerektirmiştir. BO28984 faz III klinik çalışmasında, ALECENSA alan hastalarda Derece 3 veya 4 bilirubin artışları %3,9 oranında görülürken, krizotinib alan hastalarda Derece 3 veya 4 bilirubin artışı görülmemiştir.
Klinik çalışmalarda, ALECENSA ile tedavi edilen bir hastada (%0,2) normal alkalin fosfataz eşliğinde ALT veya AST'de normalin üst limitinin üç katı veya daha yüksek ve total bilirubinde normalin üst limitinin iki katı ya da daha yüksek eşzamanlı artışlar meydana gelmiştir.
Hastalar ALT, AST ve total bilirubin dahil karaciğer fonksiyonları için Bölüm 4.4'te belirtildiği gibi takip edilmeli ve Bölüm 4.2'de önerildiği gibi tedavi edilmelidir.
Bradikardi:
çalışmasında, ALECENSA alan hastaların %15'inde doz sonrası kalp hızı 50 atım/dakikanın
altına düşmüş, krizotinib alan hastalarda bu oran %21 olmuştur. Semptomatik bradikardi yaşayan hastalar Bölüm 4.2 ve Bölüm 4.4'te önerildiği gibi tedavi edilmelidir. ALECENSA tedavisini bırakmaya neden olan bradikardi vakaları görülmemiştir.
Şiddetli miyalji vakaları ve Kreatin fosfokinaz (CPK) yükselmesi:
Klinik çalışmalarda (NP28761, NP28673, BO28984), ALECENSA ile tedavi edilen hastalarda miyalji olayları (%23), kas iskelet ağrıları (%0,5) ve artralji (%19) dahil miyalji vakaları (%35) bildirilmiştir. Olayların çoğu Derece 1 veya 2'dir, Dörthasta (%1) Derece 3 olay yaşamıştır. Bu advers olaylar nedeniyle ALECENSA tedavisi doz modifikasyonları sadece iki hastada (%0,5) gerekmiştir; miyaljiye bağlı ALECENSA tedavisi bırakma vakası gözlenmemiştir. Klinik çalışmalardan (NP28761, NP28673, BO28984) elde edilen CPK laboratuvar verilerine göre CPK yükselmesi, ALECENSA ile tedavi edilen 363 hastanın
%48'İnde görülmüştür. Derece 3 yükselmelerin sıklığı %4,2'dir. Klinik çalışmalarda (NP28761, NP28673, BO28984), Derece 3 CPK yükselmesine kadar geçen medyan zaman 14 gündür. Hastaların %3,5'inde CPK yükselmesi sonucu doz modifikasyonu gerekmiş; CPK yükselmesine bağlı olarak tedavinin kesildiği hasta olmamıştır. BO28984 klinik çalışmasında ALECENSA kolunda bir hastada (%0,7) ve krizotinib kolunda iki hastada (%1,3) şiddetli artralji bildirilmiştir. ALECENSA alan hastaların %3,9'unda ve krizotinib alan hastaların
%3,3'ünde Derece 3 CPK yükselmesi bildirilmiştir. Hemolitik anemi:
Pazarlama sonrası dönemde, anemi şiddeti Derece 1 ila Derece 3 arasında değişen, hemolitik anemi vakaları bildirilmiştir. Alectinib ile sonucu bilinen ve alınan aksiyonun bilindiği 30 vakadan, çoğunluğu (%66,7) iyileşmiş veya alektinib doz ayarlamasını takiben iyileşme göstermiştir; %10'u herhangi bir doz ayarlaması olmaksızın iyileşmiştir. Klinik çalışmalar içerisinde (NP28761, NP28673, BO28984, MO29750, BO39694, BO29554 kohort A,
YO29449), ALECENSA ile tedavi edilen 716 hastadan 2'si (%0,3), hemolitik anemiyi düşündüren ciddi olmayan Derece 1 durum yaşamıştır. Vakalardan biri ALECENSA tedavisinin kesilmesini gerektirmiştir. Klinik çalışmalarda veya pazarlama sonrası izlemde Derece 4 veya Derece 5 (ölümcül) hemolitik anemi vakası gözlenmemiştir (bkz. Bölüm 4.2 ve 4.4).
Gastrointestinal etkiler:
Kabızlık (%38), bulantı (%20), diyare (%19) ve kusma (%14) en yaygın bildirilen gastrointestinal (GI) reaksiyonlardır. Bu olayların çoğu hafif veya orta şiddettedir; Derece 3 olaylar diyare (%1), bulantı (%0,5), kusma (%0,2) ve kabızlık (%0,2) için bildirilmiştir. Bu olaylar ALECENSA tedavisinin kesilmesine yol açmamıştır. Klinik çalışmalarda (NP28761, NP28673, BO28984), kabızlık, bulantı, diyare ve/veya kusma olayları için başlangıca kadar geçen medyan süre 22 gün olmuştur. Olayların sıklığı tedavinin ilk ayından sonra azalmıştır. Faz III BO28984 klinik çalışmasında ALECENSA kolunda bir hastada (%0,7) Derece 3 ve 4 bulantı, diyare ve kabızlık bildirilirken; krizotinib kolunda da bir hastada Derece 3 ve 4 bulantı, diyare ve kusma görülme sıklıkları sırasıyla %3,3, %2 ve %3,3 olmuştur.
Şüpheli advers reaksiyonların raporlanması
Ruhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması,ilacınyarar/riskdengesinin sürekli olarak izlenmesine
Farmakovijilans Merkezi'ne (TÜFAM) bildirmeleri gerekmektedir (www.titck.gov.tr: e- posta: tufam@titck.gov.tr, tel: 0 800 314 00 08; faks: 0 312 218 35 99).
4.9. Doz aşımı ve tedavisi
Doz aşımı yaşayan hastalar yakından takip edilmeli ve destekleyici bakım uygulanmalıdır. ALECENSA ile doz aşımı için spesifik bir antidot mevcut değildir.
5. FARMAKOLOJİK ÖZELLİKLER
5.1. Farmakodinamik özellikler
Farmakoterapötik grup: Protein kinaz inhibitörleri, anaplastik lenfoma kinaz (ALK) inhibitörleri
ATC kodu: L01ED03
Etki mekanizması:
Alektinib, yüksek düzeyde selektif ve potent ALK ve RET tirozin kinaz inhibitörüdür. Klinik öncesi çalışmalarda, ALK tirozin kinaz aktivitesinin inhibisyonu, STAT 3 ve PI3K/AKT dahil hücre içi sinyal yolaklarının blokajı ve tümör hücre ölümü (apoptoz) indüksiyonuna yol açmıştır.
Alektinib, krizotinib direncinden sorumlu mutasyonlar dahil, ALK enziminin mutant formlarına karşı in vitro ve in vivo aktivite göstermiştir. Alektinibin ana metaboliti (M4) in vitro olarak benzer potens ve aktivite göstermiştir.
5.3. Klinik öncesi güvenlilik verileri
e göre, alektinib, her ikisi de kan beyin bariyerinde eflüks taşıyıcı olan p- glikoprotein veya BCRP'nin substratı değildir. Bu nedenle, merkezi sinir sistemine dağılıp yerleşebilmektedir.
Klinik etkililik ve güvenlilik:
ALK-pozitif küçük hücreli dışı akciğer kanseri
Daha önce tedavi almamış hastalar
ALECENSA'nın güvenliliği ve etkililiği, daha önce tedavi almamış olan ALK-pozitif KHDAK hastalarında global randomize Faz III açık etiketli klinik çalışmada (BO28984, ALEX) araştırılmıştır. Randomizasyon öncesinde Ventana anti-ALK (D5F3) immunohistokimyası (IHC) ile tüm hastalardan alınan doku örneklerinin ALK protein ekspresyonu pozitifliği için merkezi olarak test edilmesi gerekmiştir.
Faz III çalışmaya dahil edilen toplam 303 hastadan, 151'i krizotinib koluna ve 152'si günde iki kez 600 mg'lık önerilen dozda oral olarak ALECENSA alan ALECENSA koluna randomize edilmiştir.
noktası, RECIST 1.1 kullanılarak araştırmacı değerlendirmesi uyarınca, krizotinib ile
karşılaştırıldığında ALECENSA'nın üstünlüğünü progresyonsuz sağkalıma (PFS) dayalı olarak göstermektir. ALECENSA için başlangıçtaki demografik ve hastalık özellikleri, medyan yaş 58 (krizotinib için 54), % 55 kadın (krizotinib için % 58), % 55 Asya dışı (krizotinib için % 54), % 61'i sigara içmemis (krizotinib için % 65), % 93 ECOG PS 0 veya 1 (krizotinib için% 93), % 97 Derece IV hastalığı olan (krizotinib için % 96), % 90 adenokarsinoma histolojisi (krizotinib için % 94), başlangıçta MSS metastazı olan % 40 (krizotinib için 38 %) ve daha MSS radyasyonuna maruz kalmış olan %17 (krizotinib için
%14) olmuştur.
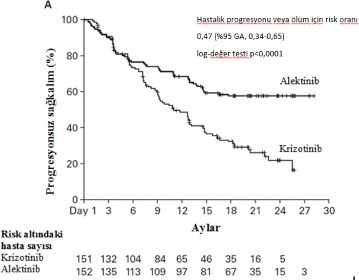
Çalışma, primer analizde araştırmacı tarafından değerlendirildiği üzere, PFS'de istatistiksel olarak önemli bir iyileşme göstererek, primer sonlanım noktasını karşılamıştır. Etkililik verileri Tablo 4'te özetlenmekte ve araştırmacı tarafından değerlendirilen PFS için Kaplan- Meier eğrileri Şekil 1'de gösterilmektedir.
Tablo 4 BO28984 (ALEX) çalışmasının etkililik sonuçları özeti
| Krizotinib N=151 | ALECENSA N=152 |
Medyan takip süresi (Ay) | 17,6 (0,3 – 27 aralığı) | 18,6 (0,5 – 29 aralığı) |
Primer etkililik parametreleri |
102 (68%) 11,1 [9,1; 13,1] |
62 (41%) NE [17,7; NE] |
PFS (INV) Vaka yaşayan hastalar n (%) Medyan (ay) [95% GA] | ||
HR [95% GA] Katmanlı log-sıra p-değeri |
0,47 [0,34, 0,65] p <0,0001 | |
Sekonder etkililik parametreleri |
92 (61%) 10,4 [7,7; 14,6] |
63 (41%) 25,7 [19,9; NE] |
PFS (IRC)* Vaka yaşayan hastalar n (%) Medyan (ay) [95% GA] | ||
HR [95% GA] Katmanlı log-sıra p-değeri |
0,5 [0,36; 0,7] p < 0.0001 | |
SSS progresyonuna kadar geçen zaman (IRC)*, ** Vaka yaşayan hastalar n (%) |
68 (45%) |
18 (12%) |
Sebep-spesifik HR [95% GA] Katmanlı log-sıra p-değeri | 0,16 [0,1; 0,28] p < 0,0001 | |
SSS progresyonu 12 aylık toplam insidans (IRC) % (95% GA) | 41,4% [33,2; 49,4] | 9,4% [5,4; 14,7] |
ORR (INV)*, *** Yanıt verenler n (%) [95% GA] |
114 (75,5%) [67,8; 82,1] |
126 (82,9%) [76,; 88,5] |
Genel sağkalım* Vaka yaşayan hastalar n (%) Medyan (ay) [95% GA] |
40 (27%) NE [NE; NE] |
35 (23%) NE [NE; NE] |
HR [95% GA] |
0,76 [0,48; 1,2] | |
Cevap süresi (INV) Medyan (ay) 95 % GA | N=114 11,1 [7,9; 13] | N=126 NE [NE; NE] |
Temelde ölçülebilir SSS metastazları olan hastalar için SSS-ORR SSS yanıt verenler n (%) [95% GA]
SSS-CR n (%)
SSS-DOR, Medyan (ay) 95% GA | N=22 11 (50%) [28,2; 71,8]
1 (5%)
5,5 [2,1; 17,3] | N=21 17 (81%) [58,1; 94,6]
8 (38%)
17,3 [14,8; NE] |
Temelde ölçülebilir ve ölçülemeyen SSS metastazları için SSS-ORR (IRC) SSS yanıt verenler n (%) [95% GA]
SSS-CR n (%)
SSS-DOR, Medyan (ay) 95% GA | N=58
15 (25,9%) [15,3; 39]
5 (9%)
3,7 [3,2; 6,8] | N=64
38 (59,4%) [46,4; 71,5] 29 (45%) NE [17,3; NE] |
* Hiyerarşik testlerin parçası olan anahtar ikincil sonlanım noktaları
** SSS progresyonunun yarışmalı risk analizi, yarışmalı olaylar olarak sistemik progresyon ve ölüm
*** Krizotinib kolunda 2 hasta ve alektinib kolunda 6 hasta tam cevap gösterdi.
GA: Güven aralığı; SSS: Santral sinir sistemi; CR: tam yanıt; DOR: yanıt süresi; HR: risk oranı; IRC: Bağımsız inceleme komitesi; INV: araştırıcı; NE: tahmin edilebilir değil; ORR: objektif yanıt oranı; PFS: progresyonsuz sağkalım
PFS faydasının büyüklüğü, başlangıçta merkezi sinir sistemi metastazı olan hastalar (HR = 0,4, 95% GA: 0,25-0,64, ALECENSA için medyan PFS = NE, 95% GA: 9,2-NE, krizotinib
için medyan PFS = 7,4 ay, 95% GA: 6,6-9,6) ve başlangıçta merkezi sinir sistemi metastazı olmayan hastalar (HR = 0,51, 95% GA: 0,33-0,8, ALECENSA için medyan PFS = NE, 95% GA: NE, NE, krizotinib için medyan PFS = 14,8 ay, 95% GA:10,8-20,3) için tutarlı olmuş ve her iki alt grupta da ALECENSA'nın krizotinib üzerindeki faydasını göstermiştir.
Şekil 1: BO28984 (ALEX) çalışmasında araştırıcı tarafından değerlendirilen progresyonsuz sağkalım için Kaplan-Meier eğrisi
Daha önce krizotinib ile tedavi edilen hastalar
Krizotinib ile önceden tedavi edilen ALK-pozitif KHDAK hastalarda ALECENSA güvenliliği ve etkililiği iki Faz I/II klinik çalışmasında (NP28673 ve NP28761) incelenmiştir.
Çalışma NP28673:
NP28673 çalışması, daha önce krizotinib ile tedavi edilirken progresyon gösteren ALK-pozitif ileri evre KHDAK hastalarında gerçekleştirilen Faz I/II tek kollu, uluslararası, çok merkezli bir çalışmadır. Krizotinibe ek olarak, hastalara önceden kemoterapi uygulanmış olabilir. Toplam 138 hasta çalışmanın Faz II kısmında yer almıştır ve önerilen dozda oral yolla günde iki kez 600 mg ALECENSA almıştır.
Birincil sonlanım noktası, genel popülasyonda (sitotoksik kemoterapi tedavilerine önceden maruz kalan/kalmayan) Solid Tümörlerde Yanıt Değerlendirme Kriteri (RECIST) kriter versiyonu 1.1 kullanılarak, merkezi Bağımsız İnceleme Komitesi (IRC) değerlendirmesine göre Objektif Yanıt Oranı (ORR) ile ALECENSA etkililiğini değerlendirmektir. Eş zamanlı birincil sonlanım noktası, önceden sitotoksik kemoterapi maruziyeti olan hastalarda RECIST
1.1 kullanılarak merkezi IRC değerlendirmesiyle ORR'yi incelemektir. Tahmini ORR için önceden belirlenmiş güven sınırı % 35'ten daha düşükse, istatistiksel olarak anlamlı bir sonuç elde edilir.
Hasta demografileri, ALK-pozitif KDHAK popülasyonu ile uyumludur. Genel çalışma popülasyonunun demografik özellikleri %67 beyaz, %26 Asyalı, %56 kadındır ve ortalama yaş 52'dir. Hastaların çoğunluğu hiç sigara kullanmamıştır (%70). Başlangıçta hastalarda ECOG (Doğu Ortak Onkoloji Grubu) performansı hastaların %90,6'sında 0 veya 1 ve
%9,4'inde 2'dir. Çalışmaya giriş sırasında, hastaların %99'u evre IV hastalığa sahiptir,
%61'inde beyin metastazı vardır ve %96'sında tümörler adenokarsinoma olarak sınıflandırılmıştır. Çalışmadayeralanhastaların%20'sinde hastalık daha önce sadece
krizotinib tedavisi sırasında ve %80'inde daha önce krizotinib ve en az bir kemoterapi tedavisi sırasında ilerlemiştir.
Çalışma NP28761:
Çalışma NP28761, daha önce krizotinib ile tedavi edilirken progresyon gösteren ALK pozitif ileri evre KHDAK hastalarında gerçekleştirilen Faz I/II tek kollu çok merkezli bir çalışmadır. Krizotinibe ek olarak, hastalara önceden kemoterapi uygulanmış olabilir. Toplam 87 hasta çalışmanın Faz II kısmında yer almıştır ve önerilen dozda günde iki kez 600 mg oral ALECENSA almıştır.
Birincil sonlanım noktası, RECIST kriter versiyonu 1.1 kullanılarak, merkezi Bağımsız İnceleme Komitesi (IRC) değerlendirmesine göre Objektif Yanıt Oranıyla (ORR) ALECENSA etkililiğini değerlendirmektir. %35'lik önceden belirtilmiş eşik üzerindeki hesaplanmış ORR alt güven sınırı istatistiksel olarak anlamlı bir bulgu sağlayacaktır.
Hasta demografileri KHDAK ALK-pozitif popülasyon ile uyumludur. Genel çalışma popülasyonunun demografik özellikleri %84 beyaz, %8 Asyalı, %55 kadındır ve ortalama yaş 54'tür. Hastaların çoğunluğu hiç sigara kullanmamıştır (%62). Başlangıçta hastalarda ECOG (Doğu Ortak Onkoloji Grubu) performansı durumu hastaların %89,7'sinde 0 ve 1 ve
%10,3'ünde 2'dir. Çalışmaya giriş sırasında, hastaların %99'u evre IV hastalığa sahiptir,
%60'ında beyin metastazı vardır ve %94'ünde tümörler adenokarsinoma olarak sınıflandırılmıştır. Çalışmada yer alan hastalardan %26'sında daha önce hastalık sadece krizotinib tedavisi sırasında, %74'ünde ise daha önce krizotinib ve en az bir kemoterapi tedavisi sırasında ilerlemiştir.
NP28673 ve NP28761 çalışmalarının etkililik sonuçları Tablo 5'te ve Merkezi Sinir Sistemi (SSS) sonlanım noktalarının havuzlanmış analizi Tablo 6'da özetlenmiştir.
Tablo 5 NP28673 ve NP28761 çalışmalarının etkililik sonuçları
| NP28673 ALECENSA 600 mg Günde 2 kez | NP28761 ALECENSA 600 mg Günde 2 kez |
Medyan (ay) | 21 (1 – 30 aralığı) | 17 (1 – 29 aralığı) |
Birincil etkililik parametreleri |
|
N  67 35 (%52,2) [%39,7, %64,6] |
ORR (IRC) RE populasyonu Yanıt verenler N (%) [%95 GA] | N=122 62 (%50,8) [%41,6, %60] | |
ORR (IRC) önceden kemoterapi tedavisi Alan hastalar Yanıt verenler N (%) [%95 GA] | N = 96
43 (%44,8) [%34,6, %55,3] | |
İkincil etkililik parametreleri |
N  62 |
N  35 20 (%57,1) |
DOR (IRC) |
[%95 GA] | [11,2; 24,9] | [6,9; NE] |
PFS (IRC) Vaka sayısı N (%) Medyan (ay) [%95 GA] | N = 138 98 (%71) 8,9 [5,6; 12,8] | N  87 58 (%66,7) 8,2 [6,3; 12,6] |
GA: Güven aralığı, DOR: Yanıt süresi, IRC: Bağımsız inceleme komitesi, NE: Tahmin edilemez, ORR: Objektif Yanıt Oranı, PFS: Progresyonsuz Sağkalım, RE: Değerlendirilebilir yanıt
NP28673 ve NP28761 çalışmaları için ORR bulguları, özellikle bazı alt gruplardaki az sayıda hasta dikkate alındığında, yaş, cinsiyet, ırk, ECOG performans durumu, Merkezi Sinir Sistemi (SSS) metastazı ve önceden kemoterapi kullanımı gibi başlangıç hasta özellikleri alt grupları arasında uyumludur.
Tablo 6 NP28673 ve NP28761 çalışmalarının SSS sonlanım noktalarının toplu analiz özeti
SSS parametreleri (NP28673 ve NP28761) | Alektinib 600 mg günde iki kez |
Başlangıçta ölçülebilir SSS lezyonları olan hastalar SSS ORR (IRC) Yanıt verenler (%) [%95 GA] Tam yanıt Kısmi yanıt
SSS DOR (IRC) Vaka sayısı (%) Medyan (ay) [%95 GA] | N= 50
32 (%64) [49,2; 77,1] 11 (%22) 21(%42)
N=32 18 (%56,3) 11,1 [7,6; NE] |
GA: Güven aralığı, DOR: Yanıt süresi, IRC: Bağımsız inceleme komitesi, ORR: Objektif Yanıt Oranı, NE: Tahmin edilemez
Pediyatrik popülasyon
Avrupa İlaç Ajansı, akciğer karsinomunda (küçük hücreli ve küçük hücreli dışı karsinom) pediyatrik popülasyonun tüm alt gruplarında ALECENSA ile yapılan çalışmaların sonuçlarını sunma yükümlülüğünü kaldırmıştır (pediyatrik kullanım ile ilgili bilgi için bkz. bölüm 4.2).
5.2. Farmakokinetik özellikler
Genel özelliklerAlektinib ve ana aktif metabolitinin (M4) farmakokinetik parametreleri ALK-pozitif KHDAK hastaları ve sağlıklı deneklerde belirlenmiştir. Alektinib için geometrik ortalama (varyasyon katsayısı %) kararlı hal C, Cve EAAsırasıyla 665 ng/mL (%44,3), 572 ng/mL (%47,8) ve 7430 ng*h/mL'dir (%45,7). M4 için geometrik ortalama kararlı hal C, Cve EAAsırasıyla 246 ng/mL (%45,4), 222 ng/mL (%46,6) ve 2810 ng*h/mL'dir (%45,9).
Emilim:
ALK-pozitif KHDAK hastalarda tokluk durumunda günde iki kez 600 mg oral uygulamayı takiben alektinib hızla emilerek yaklaşık 4 ila 6 saat sonra Tdeğerine ulaşmaktadır.
Alektinib kararlı haline günde iki kez 600 mg sürekli dozla 7. günde ulaşılmaktadır. Günde iki kez 600 mg dozda kullanımı için popülasyon PK analizine göre tahmini geometrik ortalama birikim oranı 6'dır.
Alektinib mutlak biyoyararlanımı sağlıklı deneklerde tokluk durumunda %36,9'dur (%90 GA:
%33,9, %40,3).
Bol yağlı, yüksek kalorili yemek ile 600 mg tek oral uygulamayı takiben kombine alektinib ve M4 maruziyeti, açlık koşullarına kıyasla 3 kat artmıştır.
Dağılım:
Alektinib ve ana metaboliti M4 ilaç konsantrasyonundan bağımsız olarak insan plazma proteinlerine yüksek oranda bağlanır (>%99). Alektinib ve M4'ün ortalama in vitro insan kanı-plazma konsantrasyon oranları klinik olarak önemli konsantrasyonlarda sırasıyla 2,64 ve 2,5'tir.
IV uygulamasını takiben alektinibin kararlı haldeki (V) geometrik ortalama dağılım hacmi 475 L olup, dokulara aşırı dağılımı göstermektedir.
İn vitro verilere göre, alektinib bir P-gp substratı değildir. Alektinib ve M4 BCRP veya organik anyon taşıyıcı polipeptit (OATP) 1B1/B3 substratları değildir.
Biyotransformasyon:
In vitro metabolizma çalışmalarına göre, CYP3A4, alektinib ve ana metaboliti M4'ün metabolizmasına aracılık eden ana CYP izozimidir ve insan hepatositlerindeki alektinib metabolizmasına %40-50 katkıda bulunduğu tahmin edilmektedir. İnsan kütle denge çalışmalarından elde edilen sonuçlara göre, alektinib ve M4 plazma dolaşan ana parçalar olup alektinib ve M4 plazmadaki toplam radyoaktivitenin %76 kadarından sorumludur. Kararlı halde geometrik ortalama Metabolit/Ana madde oranı 0,399'dur.
Metabolit M1b sağlıklı gönüllülerde insan plazmasında ve in vitro minör bir metabolit olarak tespit edilmiştir. Metabolit M1b ve minör izomeri M1a'nın oluşumu muhtemelen CYP izoenzimleri (CYP3A dışındaki izoenzimler dahil) ve aldehit dehidrogenaz (ALDH) enzimlerinin bir kombinasyonu ile katalizlenir.
İn vitro çalışmalar alektinibin veya majör aktif metabolitinin (M4) klinik açıdan ilgili konsantrasyonlarda CYP1A2, CYP2B6, CYP2C9, CYP2C19 veya CYP2D6'yı inhibe ettiğini göstermemektedir. Alektinib in vitro olarak klinik açıdan ilgili konsantrasyonlarda OATP1B1/OATP1B3, OAT1, OAT3 veya OCT2'yi inhibe etmemiştir.
Eliminasyon:
Sağlıklı deneklere oral olarak uygulanan C etiketli alektinibin tek doz uygulanmasından
atılım vardır (ortalama geri kazanım yüzde 0,46). Dozun %84'ü değişmemiş alektinib, %5,8'i ise M4 olarak feçeste atılmıştır.
Bir popülasyon PK analizine göre, alektinibin belirgin klerensi (KL/F) 81,9 L/saattir. Alektinib için her bir eliminasyon yarı ömür tahmininin geometrik ortalaması 32,5 saattir. M4 için karşılık gelen değerler sırasıyla 217 L/saat ve 30,7 saattir.
Doğrusallık / Doğrusal olmayan durum:
Popülasyon PK analizi tokluk koşullarında 300 ila 900 mg doz aralığında alektinib doz orantısallığını desteklemektedir.
Özel popülasyonlara ilişkin ek bilgiler:
Böbrek yetmezliği:
Alektinib ve aktif metaboliti M4 ihmal edilebilir oranlarda idrarda değişmeden atılmaktadır (dozun < %0,2'si). Bir popülasyon farmakokinetik analizine bağlı olarak, alektinib ve M4 maruziyetleri hafif ve orta derecede böbrek yetmezliği olan ve normal renal fonksiyonlu hastalarda benzerdir. Şiddetli böbrek yetmezliği olan hastalarda farmakokinetik çalışma yapılmamıştır.
Karaciğer yetmezliği:
Alektinib eliminasyonu öncelikle karaciğerdeki metabolizma aracılığıyla gerçekleştiğinden, karaciğer yetmezliği alektinib ve/veya ana metaboliti M4'ün plazma konsantrasyonunu artırabilir. Bir popülasyon farmakokinetik analizine dayalı olarak, alektinib ve M4 maruziyetleri hafif karaciğer yetmezliği olan hastalarda ve normal hepatik fonksiyonlu hastalarda benzerdir.
Şiddetli karaciğer yetmezliği (Child Pugh C) olan hastalara 300 mg'lık bir doz uygulanmasından sonra, eşleştirilmiş sağlıklı bireylerdeki parametrelerle karşılaştırıldığında, Caynı olmakla birlikte, EAAsağlıklı bireylere kıyasla 2,2 kat fazla bulunmuştur. M4'ün Cve EAAseviyeleri sağlıklı bireylere göre sırasıyla %39 ve %34 daha düşüktür ve bu durum alektinib ve M4'ün kombine maruziyetinin şiddetli karaciğer bozukluğu olan hastalarda sağlıklı bireylere göre 1,8 kat fazla olduğunu göstermektedir.
Karaciğer yetmezliği çalışmasına aynı zamanda orta derecede karaciğer yetmezliği (Child Pugh C) olan hasta grubu dahil edilmiş ve eşleştirilmiş sağlıklı bireyler ile karşılaştırıldığında bu grupta makul derecede daha yüksek alektinib maruziyeti görülmüştür. Bununla birlikte, Child Pugh B grubundaki bireylerde genel olarak anormal bilirubin, albumin veya protrombin zamanı görülmediğinden, bu grubun tam olarak azalmış metabolik kapasitesi olan orta derecede karaciğer bozukluğu olan bireyleri tam olarak temsil edemediğini göstermektedir.
Yaş, vücut ağırlığı, ırk ve cinsiyetin etkileri:
Yaş, vücut ağırlığı, ırk ve cinsiyet alektinibin ve M4'ün sistemik maruziyeti üzerinde klinik olarak anlamlı bir etkiye sahip değildir. Klinik çalışmalara kaydedilen hastalar için vücut ağırlıkları 36,9-123 kg aralığındadır. Aşırı vücut ağırlığına (>130 kg) sahip hastalara ilişkin veri yoktur (bkz. Bölüm 4.2).
5.3. Klinik öncesi güvenlilik verileri
e göre, alektinib, her ikisi de kan beyin bariyerinde eflüks taşıyıcı olan p- glikoprotein veya BCRP'nin substratı değildir. Bu nedenle, merkezi sinir sistemine dağılıp yerleşebilmektedir.6. FARMASÖTİK ÖZELLİKLER
6.1. Yardımcı maddelerin listesi
Kapsül içeriği:
Laktoz Monohidrat (inek sütünden elde edilmektedir) Hidroksipropilselülöz
Sodyum Lauril Sülfat Karboksimetilselüloz Kalsiyum Magnezyum Stearat
Kapsül kabuğu:
Karragenan Potasyum Klorür
Titanyum Dioksit (E171) Karnauba Mumu
Mısır Nişastası Hipromeloz
Baskı mürekkebi:
Kırmızı demir oksit (E172) Sarı demir oksit (E172)
FD&C Mavi No.2 alüminyum lake (E132) Karnauba Mumu
Beyaz şellak (Lak böceğinin (Laccifer Lacca Kerr) lak salgı maddesinden elde edilmektedir) Gliseril monooleat
1-bütanol Dehidre etil alkol
6.2. Geçimsizlikler
Uygulanabilir değildir.
6.3. Raf ömrü
60 ay.
6.4. Saklamaya yönelik özel tedbirler
30°C altındaki oda sıcaklığında saklayınız.
Orijinal ambalajında, ışıktan ve nemden koruyarak saklanmalıdır.
6.5. Ambalajın niteliği ve içeriği
8 sert kapsül/blister ihtiva eden alüminyum/alüminyum perfore blisterler
Çoklu ambalajda 224 adet sert kapsül (56'lık 4 paket)
6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler
Kullanılmamış olan ürünler ya da atık materyaller, “Tıbbi Atıkların Kontrolü Yönetmeliği'' ve “Ambalaj Atıklarının Kontrolü Yönetmeliğiâ€ne uygun olarak imha edilmelidir.
 Aşırı Alkol Kullanımı, Alkolizm
Alkol bağımlılığı, alkol kullanımı ve alkol sorunları arasındaki farkı açıklamak güçtür.
Örneğin, geçmişte alkol kullanmış olan bir kimsenin mutlaka alkol bağımlısı olması
gerekmez.
Aşırı Alkol Kullanımı, Alkolizm
Alkol bağımlılığı, alkol kullanımı ve alkol sorunları arasındaki farkı açıklamak güçtür.
Örneğin, geçmişte alkol kullanmış olan bir kimsenin mutlaka alkol bağımlısı olması
gerekmez. |
 En Yaygın Alerji Türleri
Bağışıklık sistemi, polen, arı zehiri veya evcil hayvan gibi yabancı bir maddeye veya çoğu insanda reaksiyona neden olmayan bir yiyeceğe tepki gösterdiğinde alerjiler meydana gelir.
En Yaygın Alerji Türleri
Bağışıklık sistemi, polen, arı zehiri veya evcil hayvan gibi yabancı bir maddeye veya çoğu insanda reaksiyona neden olmayan bir yiyeceğe tepki gösterdiğinde alerjiler meydana gelir. |
 |
Pankreas Kanseri Pankreas karnın alt kısmında yatay şekilde bulunan bir organdır. Sindirime yardımcı olan enzimleri ve kan şekerini yönetmeye yardımcı olan hormonları vücuda dağıtmakla görevlidir. |
 |
Kalp Krizi Kalbe giden kan akışı durduğunda kalp krizi meydana gelir. |
 |
Parkinson Hastalığı Hastalık ilk kez 1817 de İngiliz doktor James Parkinson tarafından tanımlanmış ve Dr. Parkinson hastalığı “sallayıcı felç” olarak kaleme almış. |
İLAÇ GENEL BİLGİLERİ
Roche Müstahzarları Sanayi A.Ş.
| Geri Ödeme Kodu | A16802 |
| Satış Fiyatı | 92430.85 TL [ 17 Mar 2025 ] |
| Önceki Satış Fiyatı | 92430.85 TL [ 7 Mar 2025 ] |
| Original / Jenerik | Original İlaç |
| Reçete Durumu | Normal Reçeteli bir ilaçdır. |
| Barkodu | 8699505153407 |
| Etkin Madde | Alektinib Hidroklorür |
| ATC Kodu | L01ED03 |
| Birim Miktar | 150 |
| Birim Cinsi | MG |
| Ambalaj Miktarı | 224 |
| Antineoplastik ve İmmünomodülatör Ajanlar |
| İthal ( ref. ülke : Isvicre ) ve Beşeri bir ilaçdır. |