ALUNBRIG 30 mg film kapl� tablet (28 tablet) K�sa �r�n Bilgisi
{ Brigatinib }
1. BE�ER� TIBB� �R�N�N ADI
ALUNBR�G 30 mg film kapl� tablet Sitotoksik.
2. KAL�TAT�F VE KANT�TAT�F B�LE��M
Etkin madde
Brigatinib 30 mg
Yard�mc� maddeler
Laktoz monohidrat (inek s�t�) 56,06 mg Yard�mc� maddeler i�in b�l�m 6.1'e bak�n�z.
3. FARMAS�T�K FORMU
Film kapl� tablet
Bir y�z�nde “U3” bask�l� di�er y�z� d�z, yakla��k 7 mm �ap�nda, beyaz-beyaz�ms�, yuvarlak film kapl� tabletler.
4. KL�N�K �ZELL�KLER
4.1. Terap�tik endikasyonlar
�nceden ALK-Hedefli Tedavi Almam�� �leri Evre ALK-Pozitif K���k H�creli D��� Akci�er Kanseri (KHDAK)
ALUNBR�G (brigatinib)'in, anaplastik lenfoma kinaz (ALK)-pozitifli�i (standardize FISH veya RT-PCR veya yeni nesil dizileme y�ntemleri ile tespit edilen rearanjman/f�zyon varl���; veya imm�nhistokimya ALK pozitifli�i) saptanan, ilerlemi� k���k h�creli d��� akci�er kanseri (KHDAK) olan hastalar�n tedavisinde progresyona kadar kullan�m� endikedir.
�nceden Krizotinib Tedavisi Alm�� ALK-Pozitif �leri Evre veya Metastatik K���k H�creli D��� Akci�er Kanseri (KHDAK)
ALUNBR�G (brigatinib)'in, krizotinib ile �nceden tedavi edilmi� anaplastik lenfoma kinaz (ALK)- pozitifli�i (standardize FISH veya RT-PCR veya yeni nesil dizileme y�ntemleri ile tespit edilen rearanjman/f�zyon varl���; veya imm�nhistokimya ALK pozitifli�i) saptanan, ileri evre veya metastatik k���k h�creli d��� akci�er kanseri (KHDAK) olan hastalar�n tedavisinde progresyona kadar kullan�m� endikedir.
4.2. Pozoloji ve uygulama �ekli
ALUNBR�G ile tedavi antikanser t�bbi �r�nlerinin kullan�m� konusunda deneyimli bir uzman hekim taraf�ndan ba�lat�lmal� ve takip edilmelidir.
ALUNBR�G tedavisine ba�lamadan �nce ALK-pozitif KHDAK durumu belirlenmelidir. ALK- pozitif KHDAK hastalar�n�n se�imi i�in, valide edilmi� bir ALK testi gereklidir (bkz. b�l�m 5.1.). ALK-pozitif KHDAK de�erlendirmesi, kullan�lmakta olan spesifik teknoloji konusunda yetkinli�i kan�tlanm�� laboratuvarlar taraf�ndan ger�ekle�tirilmelidir.
Pozoloji/uygulama s�kl��� ve s�resi:
ALUNBR�G'in tavsiye edilen ba�lang�� dozu ilk 7 g�n i�in g�nde bir defa 90 mg, daha sonra g�nde bir defa 180 mg'd�r.
ALUNBR�G advers reaksiyonlar d���ndaki nedenlerle 14 g�n veya daha uzun s�re kesilirse; tedaviye, daha �nce tolere edilen doza y�kseltilmeden �nce 7 g�n boyunca g�nde bir kez 90 mg'da devam edilmelidir.
Bir dozun atlanmas� veya doz al�m� sonras�nda kusma meydana gelmesi durumunda, ilave bir doz uygulanmamal� ve bir sonraki doz planlanan zamanda al�nmal�d�r.
Tedavi, klinik yarar sa�land��� s�rece devam etmelidir.
Doz ayarlamas�
Bireysel g�venlilik ve tolerabiliteye ba�l� olarak dozun kesilmesi ve/veya azalt�lmas� gerekebilir. ALUNBR�G doz azaltma seviyeleri Tablo 1'de �zetlenmektedir.
Tablo 1: Tavsiye Edilen ALUNBR�G Doz Azaltma Seviyeleri
Doz | Doz Azaltma Seviyeleri | ||
Birinci | �kinci | ���nc� | |
G�nde bir defa 90 mg (ilk 7 g�n) | G�nde bir defa 60 mg'a azalt�l�r | Kal�c� olarak kesilir | Ge�erli de�ildir. |
G�nde bir defa 180 mg | G�nde bir defa 120 mg'a azalt�l�r. | G�nde bir defa 90 mg'a azalt�l�r. | G�nde bir defa 60 mg'a azalt�l�r. |
Hastan�n g�nde bir defa 60 mg'l�k dozu tolere edememesi durumunda, ALUNBR�G kal�c� olarak kesilmelidir.
Advers reaksiyonlar�n y�netimi i�in ALUNBR�G doz modifikasyonlar�na ili�kin tavsiyeler Tablo 2'de �zetlenmektedir.
Tablo 2: Advers Reaksiyonlar i�in Tavsiye Edilen ALUNBR�G Doz Modifikasyonlar�
Advers reaksiyon | �iddeti* | Doz modifikasyonu |
�nterstisyel Akci�er Hastal��� (ILD)/ pn�moni | 1. derece | |
2. derece |
Tedavinin ilk 7 g�n� i�erisinde olay meydana gelirse; ALUNBR�G ba�lang�� seviyesine iyile�me olana kadar durdurulmal�, daha sonra ayn� dozda devam ettirilmelidir ve g�nl�k 180 mg'a art�r�lmamal�d�r.
�lk 7 g�nl�k tedaviden sonra ILD/pn�monit meydana gelirse, ALUNBR�G ba�lang�� seviyesine iyile�me olana kadar durdurulmal�d�r ve daha sonra ayn� dozda devam ettirilmelidir.
ILD/pn�monitin yeniden meydana gelmesi durumunda, ALUNBR�G kal�c� olarak kesilmelidir.
Tedavinin ilk 7 g�n� i�erisinde ILD/pn�monit meydana gelirse, ALUNBR�G ba�lang�� seviyesine iyile�me olana kadar durdurulmal�, daha sonra Tablo 1'de a��klanan bir sonraki d���k dozda devam ettirilmelidir ve g�nl�k 180 mg'a art�r�lmamal�d�r.
�lk 7 g�nl�k tedaviden sonra ILD/pn�monit meydana gelirse, ALUNBR�G ba�lang�� seviyesine iyile�me olana kadar
|
| durdurulmal�d�r. ALUNBR�G, Tablo 1'de a��kland��� �zere, bir sonraki d���k dozda devam ettirilmelidir. |
3. veya 4. derece | ||
Hipertansiyon | 3. derece hipertansiyon (Sistolik kan bas�nc� (SKB) ≥160 mmHg veya Diastolik kan bas�nc� (DKB) ≥100 mmHg, t�bbi m�dahale gerektirir, birden fazla antihipertansif t�bbi �r�n veya daha �nce kullan�landan daha yo�un tedavi gerektirir) | |
4. derece hipertansiyon (ya�am� tehdit edici sonu�lar� olan, acil m�dahale gerektiren) | ||
Bradikardi (Dakikada 60'dan d���k kalp at�m h�z�) | Semptomatik bradikardi | kalp h�z� sa�land���nda, Tablo 1'e g�re bir sonraki daha d���k doz seviyesinde devam ettirilmelidir. |
Ya�am� tehdit edici sonu�lar� olan bradikardi, acil m�dahale gerektiren | ||
CPK y�ksekli�i | Derece ≥2 kas a�r�s� veya zay�fl��� ile 3. veya 4. derece CPK y�ksekli�i (>5 x normalin �st s�n�r� (N�S)) | olana kadar durdurulmal�, daha sonra Tablo 1'e g�re bir sonraki daha d���k doz seviyesinde devam ettirilmelidir. |
Lipaz veya amilaz y�ksekli�i | 3. derece lipaz veya amilaz y�ksekli�i (>2 x N�S) |
ILD/pn�monitin yeniden meydana gelmesi durumunda ALUNBR�G kal�c� olarak kesilmelidir.
ALUNBR�G kal�c� olarak kesilmelidir.
Hipertansiyon derece ≤1'e (SKB <140 mmHg ve DKB <90 mmHg) kadar iyile�me g�sterene kadar ALUNBBR�G tedavisi durdurulmal�, daha sonra ayn� dozda devam ettirilmelidir.
3.derece hipertansiyonun tekrarlamas� durumunda; ALUNBR�G hipertansiyon derece ≤ 1'e kadar iyile�me g�sterene kadar durdurulmal�, daha sonra Tablo 1'e g�re bir sonraki daha d���k doz seviyesinde devam edilmeli veya kal�c� olarak kesilmelidir.
Hipertansiyon derece ≤ 1'e (SKB <140 mmHg ve DKB <90 mmHg) kadar iyile�me g�sterene kadar ALUNBR�G tedavisi durdurulmal�, daha sonra Tablo 1'e g�re bir sonraki daha d���k doz seviyesinde devam edilmeli veya kal�c� olarak kesilmelidir.
4.derece hipertansiyonun yeniden meydana gelmesi durumunda; ALUNBR�G kal�c� olarak kesilmelidir.
Asemptomatik bradikardi sa�lanana kadar veya dakikada 60 kalp at�m h�z� veya daha y�ksek dinlenme kalp h�z� sa�lanana kadar ALUNBR�G durdurulmal�d�r.
Bradikardiye neden oldu�u bilinen e� zamanl� kullan�lan bir t�bbi �r�n�n tan�mlanmas� ve kesilmesi veya dozunun ayarlanmas� durumunda; ALUNBR�G asemptomatik bradikardi sa�land���nda veya dakikada 60 kalp at�m h�z� veya daha y�ksek dinlenme kalp h�z� sa�land���nda, ayn� dozda devam ettirilmelidir.
E�er bradikardiye neden oldu�u bilinen e� zamanl� kullan�lan herhangi bir ila� belirlenmemi� veya katk� sa�layan e� zamanl� kullan�lan ila�lar�n kullan�m�na son verilmemi� veya doz ayarlamas� yap�lmam��sa, ALUNBR�G asemptomatik bradikardi veya dakikada 60 kalp at�m h�z� veya daha y�ksek bir dinlenme
Katk� sa�layan e� zamanl� kullan�lan bir t�bbi �r�n�n tan�mlanmas� ve kesilmesi veya dozunun ayarlanmas� durumunda; ALUNBR�G klinik olarak gerekli s�kl�kta takip ile, asemptomatik bradikardi veya dakikada 60 kalp at�m h�z� veya daha y�ksek dinlenme kalp h�z� sa�land���nda, Tablo 1'e g�re bir sonraki daha d���k doz seviyesinde devam ettirilmelidir.
Katk� sa�layan e� zamanl� kullan�lan herhangi bir t�bbi �r�n�n tan�mlanmamas� durumunda, ALUNBR�G tedavisi kal�c� olarak kesilmelidir.
N�ksetme durumunda; ALUNBR�G tedavisi kal�c� olarak kesilmelidir.
ALUNBR�G derece ≤ 1 (≤ 2.5 × N�S) CPK y�ksekli�ine veya ba�lang�� seviyesine iyile�me olana kadar durdurulmal�, daha sonra ayn� dozda devam ettirilmelidir.
3. veya 4. derece CPK y�ksekli�inin Derece ≥ 2 kas a�r�s� veya zay�fl��� ile tekrarlamas� durumunda; ALUNBR�G derece ≤ 1 (≤ 2,5 × N�S) CPK y�ksekli�ine veya ba�lang�� seviyesine iyile�me
ALUNBR�G derece ≤ 1 (≤1,5 × N�S) veya ba�lang�� seviyesine iyile�me olana kadar durdurulmal�, daha sonra ayn� dozda devam ettirilmelidir.
3. derece lipaz ve amilaz y�ksekli�inin tekrarlamas� durumunda;
|
| ALUNBR�G derece ≤ 1 (≤1,5 × N�S) veya ba�lang�� seviyesine iyile�me olana kadar durdurulmal�, daha sonra Tablo 1'e g�re bir sonraki daha d���k doz seviyesinde devam ettirilmelidir. |
4. derece Lipaz veya amilaz y�ksekli�i (>5,0 x N�S) | ||
Hepatotoksisite | Bilirubin ≤ 2 x N�S ile aspartat transferaz (AST) veya alanin aminotransferaz (ALT)'da Derece ≥3 y�kselme (>5,0 x N�S) | |
Kolestaz veya hemoliz yoklu�unda e�zamanl� total bilirubin y�kselmesi > 2 x N�S ile ALT veya AST'de Derece ≥2 y�kselme (>3 x N�S) | ||
Hiperglisemi | Derece 3 (250 mg/dL veya 13.9 mmol/L'den daha y�ksek) veya daha y�ksek | ALUNBR�G Tablo 1'e g�re bir sonraki daha d���k dozda devam ettirilebilir veya kal�c� olarak kesilebilir. |
G�rme bozuklu�u | Derece 2 veya 3 | |
Derece 4 | ||
Di�er advers reaksiyonlar | Derece 3 | |
| Derece 4 |
ALUNBR�G derece ≤ 1'e (≤1,5 × N�S) kadar iyile�me olana kadar durdurulmal�, daha sonra Tablo 1'e g�re bir sonraki daha d���k doz seviyesinde devam ettirilmelidir.
ALUNBR�G ba�lang�� seviyesine ya da ≤ 3x N�S'ye iyile�me olana kadar durdurulmal�, daha sonra Tablo 1'e g�re bir sonraki daha d���k doz seviyesinde devam ettirilmelidir.
ALUNBR�G kal�c� olarak kesilmelidir.
Optimum t�bbi y�netim ile yeterli hiperglisemik kontrol�n sa�lanamamas� durumunda; ALUNBR�G yeterli hiperglisemik kontrol elde edilene kadar durdurulmal�d�r. �yile�me oldu�unda,
ALUNBR�G derece 1'e veya ba�lang�� seviyesine iyile�me olana kadar durdurulmal�, daha sonra Tablo 1'e g�re bir sonraki daha d���k doz seviyesinde devam ettirilmelidir.
ALUNBR�G kal�c� olarak kesilmelidir.
ALUNBR�G ba�lang�� seviyesine iyile�me olana kadar durdurulmal�, daha sonra ayn� doz seviyesinde devam ettirilmelidir.
Derece 3 olay�n�n tekrarlamas� durumunda; ALUNBR�G ba�lang�� seviyesine iyile�me olana kadar durdurulmal�, daha sonra Tablo 1'e g�re daha d���k doz seviyesinde devam ettirilmeli veya kal�c� olarak kesilmelidir.
ALUNBR�G ba�lang�� seviyesine iyile�me olana kadar durdurulmal�, daha sonra Tablo 1'e g�re bir sonraki daha d���k doz seviyesinde devam ettirilmelidir.
Derece 4 olay�n�n tekrarlamas� durumunda; ALUNBR�G ba�lang�� seviyesine iyile�me olana kadar durdurulmal�, daha sonra Tablo 1'e g�re bir sonraki daha d���k doz seviyesinde devam ettirilmeli veya kal�c� olarak kesilmelidir.
*Ulusal Kanser Enstit�s� Advers Olay Ortak Terminoloji Kriterleri Versiyon 4.0'a (NCI CTCAE v4) g�re derecelendirilmi�tir.
Uygulama �ekli:
ALUNBR�G oral kullan�m i�indir. Tabletler b�t�n olarak suyla yutulmal�d�r. ALUNBR�G a� ya da tok olarak al�nabilir.
Greyfurt veya greyfurt suyu brigatinibin plazma konsantrasyonlar�n� art�raca�� i�in t�ketilmesinden ka��n�lmal�d�r (bkz. b�l�m 4.5.).
�zel pop�lasyonlara ili�kin ek bilgiler:
B�brek yetmezli�i:
Hafif veya orta dereceli b�brek yetmezli�i olan hastalar i�in ALUNBR�G doz ayarlamas� gerekli de�ildir (hesaplanan glomer�ler filtrasyon h�z� (eGFR) ≥30 mL/dk). �iddetli b�brek yetmezli�i (eGFR) <30 mL/dk) olan hastalar i�in; ilk 7 g�n boyunca g�nde bir defa 60 mg'l�k azalt�lm��
ba�lang�� dozu, daha sonra g�nde bir defa 90 mg tavsiye edilir (bkz. b�l�m 5.2.). �iddetli b�brek yetmezli�i olan hastalar �zellikle ilk haftada interstisyel akci�er hastal��� (ILD)/pn�monit belirtisi te�kil edebilecek yeni veya k�t�le�en solunum semptomlar� (�rne�in: dispne, �ks�r�k vs.) a��s�ndan yak�ndan takip edilmelidir (bkz. b�l�m 4.4.).
Karaci�er yetmezli�i:
Hafif dereceli karaci�er yetmezli�i (Child-Pugh s�n�f A) veya orta derecede karaci�er yetmezli�i (Child-Pugh s�n�f B) olan hastalar i�in ALUNBR�G doz ayarlamas� gerekli de�ildir. �iddetli karaci�er yetmezli�i (Child-Pugh s�n�f C) olan hastalar i�in; ilk 7 g�n boyunca g�nde bir defa 60 mg'l�k azalt�lm�� ba�lang�� dozu, daha sonra g�nde bir defa 120 mg tavsiye edilir (bkz. b�l�m 5.2.)
Pediyatrik pop�lasyon:
18 ya��n alt�ndaki hastalarda ALUNBR�G'in g�venlili�i ve etkilili�i hen�z belirlenmemi�tir. Veri mevcut de�ildir.
Geriyatrik pop�lasyon:
65 ya� ve �zeri hastalarda ALUNBR�G'in g�venlili�i ve etkilili�ine ili�kin s�n�rl� veriler ya�l� hastalarda doz ayarlamas�n�n gerekli olmad���n� �ne s�rmektedir (bkz. b�l�m 4.8.). 85 ya� �zeri hastalara ili�kin veri mevcut de�ildir.
4.3. Kontrendikasyonlar
Etkin madde
4.4. �zel kullan�m uyar�lar� ve �nlemleri
Pulmoner advers reaksiyonlar
ALUNBR�G ile tedavi olan hastalarda ILD/pn�monit ile tutarl� �zelliklere sahip olanlar dahil olmak �zere; ciddi, ya�am� tehdit edici ve �l�mc�l pulmoner advers reaksiyonlar meydana gelebilir (bkz. b�l�m 4.8.).
Bir�ok pulmoner advers reaksiyon tedavinin ilk 7 g�n� i�erisinde g�zlemlenmi�tir. Derece 1-2 pulmoner advers reaksiyonlar, tedavinin kesilmesi veya doz ayarlamas� ile giderilmi�tir. Ya� art��� ve krizotinibin son dozu ve ALUNBR�G'in ilk dozu aras�ndaki daha k�sa zaman aral��� (7 g�nden az) ba��ms�z olarak bu pulmoner advers reaksiyon oran�n�n artmas� ile ili�kilendirilmi�tir. ALUNBR�G tedavisine ba�lan�rken bu fakt�rler g�z �n�nde bulundurulmal�d�r. ILD veya ila�- ind�kl� pn�monit �yk�s� olan hastalar pivotal �al��malardan ��kar�lm��t�r.
Baz� hastalar ALUNBR�G tedavisinden sonra pn�monit ge�irmi�tir.
Hastalar; �zellikle tedavinin ilk haftas�nda, yeni veya k�t�le�en solunum semptomlar� (�rn. dispne, �ks�r�k, v.b.) bak�m�ndan takip edilmelidir. K�t�le�en solunum semptomlar�na sahip herhangi bir hastadaki pn�monit bulgusu derhal incelenmelidir. Pn�monitten ��phelenilmesi durumunda; ALUNBR�G tedavisi durdurulmal� ve hasta semptomlar�n di�er nedenleri (�rn. pulmoner embolizm, t�m�r progresyonu ve enfeksiy�z pn�monit) bak�m�ndan de�erlendirilmeli ve bu do�rultuda doz ayarlamas� yap�lmal�d�r (bkz. b�l�m 4.2.).
Hipertansiyon
ALUNBR�G ile tedavi edilen hastalarda hipertansiyon meydana gelmi�tir (bkz. b�l�m 4.8.). ALUNBR�G tedavisi s�ras�nda kan bas�nc� d�zenli olarak takip edilmelidir. Hipertansiyon, kan
bas�nc�n�n kontrol edilmesine y�nelik standart k�lavuzlara g�re tedavi edilmelidir. Kalp h�z�; bradikardiye neden oldu�u bilinen bir t�bbi �r�n ile e� zamanl� kullan�mdan ka��n�lamad��� durumda, hastalarda daha s�kl�kla takip edilmelidir. �iddetli hipertansiyon (≥ derece 3) i�in; ALUNBR�G, hipertansiyon derece 1 veya ba�lang�� seviyesine iyile�ene kadar durdurulmal�d�r. Bu do�rultuda doz ayarlamas� yap�lmal�d�r (bkz. b�l�m 4.2.)
Bradikardi
ALUNBR�G ile tedavi edilen hastalarda bradikardi meydana gelmi�tir (bkz. b�l�m 4.8.). ALUNBR�G'in bradikardiye neden oldu�u bilinen di�er ajanlar ile kombinasyon halinde uygulanmas� durumunda dikkatli olunmal�d�r. Kalp h�z� ve kan bas�nc� d�zenli olarak takip edilmelidir.
Semptomatik bradikardinin meydana gelmesi durumunda; ALUNBR�G tedavisi durdurulmal� ve bradikardiye neden oldu�u bilinen e� zamanl� kullan�lan tedaviler de�erlendirilmelidir. �yile�me oldu�unda; bu do�rultuda doz ayarlamas� yap�lmal�d�r (bkz. b�l�m 4.2.). Ya�am� tehdit edici bradikardi durumunda; katk� sa�layan birlikte kullan�lan herhangi bir ilac�n tan�mlanmamas� veya tekrarlama durumunda, ALUNBR�G tedavisi kesilmelidir (bkz. b�l�m 4.2.).
G�rme bozuklu�u
ALUNBR�G ile tedavi edilen hastalarda g�rme bozuklu�u advers reaksiyonlar� meydana gelmi�tir (bkz. b�l�m 4.8.). Hastalara, g�rme semptomlar�n� raporlamas� �nerilmelidir. Yeni veya k�t�le�en ciddi g�rme semptomlar�n olmas� durumunda; g�z muayenesi ve doz azalt�lmas� de�erlendirilmelidir (bkz. b�l�m 4.2.).
Kreatin fosfokinaz (CPK) art���
ALUNBR�G ile tedavi edilen hastalarda CPK art��� meydana gelmi�tir (bkz. b�l�m 4.8). Hastalara; a��klanamayan herhangi bir kas a�r�s�, hassasl�k veya zay�fl�k durumunu raporlamalar� tavsiye edilmelidir. ALUNBR�G tedavisi s�ras�nda CPK seviyeleri d�zenli olarak takip edilmelidir. CPK art���n�n �iddetine ba�l� olarak; ALUNBR�G tedavisi durdurulmal� ve bu do�rultuda doz ayarlamas� yap�lmal�d�r (bkz. b�l�m 4.2.).
Pankreatik enzimlerde art��
ALUNBR�G ile tedavi edilen hastalarda amilaz ve lipaz art��� meydana gelmi�tir (bkz. b�l�m 4.8.). ALUNBR�G tedavisi s�ras�nda lipaz ve amilaz d�zenli olarak takip edilmelidir. Laboratuvar anormalliklerinin �iddetine ba�l� olarak; ALUNBR�G durdurulmal� ve bu do�rultuda doz ayarlamas� yap�lmal�d�r (bkz. b�l�m 4.2.).
Hepatotoksisite:
ALUNBR�G tedavisi alan hastalarda hepatik enzimler (aspartat aminotransferaz, alanin aminotransferaz) ve bilirubin seviyelerinde art�� meydana gelmi�tir (bkz. b�l�m 4.8.). ALUNBR�G tedavisine ba�lanmadan �nce ve tedavinin ilk 3 ay� boyunca her 2 haftada bir karaci�er fonksiyonu (AST, ALT ve total bilirubin dahil) de�erlendirilmelidir. Daha sonra periyodik izlemeye devam edilmelidir. Laboratuvar anormalliklerinin ciddiyet durumuna g�re tedaviye ara verilmeli ve doz modifikasyonu yap�lmal�d�r (bkz. b�l�m 4.2.)
Hiperglisemi
ALUNBR�G ile tedavi edilen hastalarda serum glukoz seviyelerinde art�� meydana gelmi�tir. ALUNBR�G tedavisine ba�lamadan �nce a�l�k serum glukoz seviyesi de�erlendirilmeli ve akabinde periyodik olarak takip edilmelidir. Gerekirse, antihiperglisemik tedaviler ba�lat�lmal� veya optimize edilmelidir. Optimum t�bbi y�netim ile yeterli hiperglisemik kontrol sa�lanamamas� durumunda; ALUNBR�G yeterli hiperglisemik kontrol sa�lanana kadar durdurulmal�d�r. �yile�me oldu�unda, Tablo 1'de a��kland��� �ekilde dozun azalt�lmas� de�erlendirilebilir veya ALUNBR�G kal�c� olarak
kesilebilir.
�la�-ila� etkile�imleri
ALUNBR�G'in g��l� CYP3A inhibit�rleri ile e� zamanl� kullan�m�ndan ka��n�lmal�d�r. G��l� CYP3A inhibit�rlerinin birlikte kullan�m�n�n ka��n�lmaz oldu�u durumunda; ALUNBR�G dozu 180 mg'dan 90 mg'a veya 90 mg'dan 60 mg'a d���r�lmelidir. G��l� CYP3A inhibit�r� kesildikten sonra; ALUNBR�G tedavisine, g��l� CYP3A inhibit�r� ba�lat�lmadan �nce tolere edilen dozda devam edilmelidir.
ALUNBR�G'in g��l� ve orta dereceli CYP3A ind�kleyicileri ile e�zamanl� kullan�m�ndan ka��n�lmal�d�r (bkz. b�l�m 4.5.). Orta dereceli CYP3A ind�kleyicilerinin e�zamanl� kullan�m�ndan ka��n�lamazsa, ALUNBR�G dozu, tolere edilen mevcut ALUNBR�G dozu ile 7 g�nl�k tedaviden sonra 30 mg'l�k art��larla, orta dereceli CYP3A ind�kleyicisinin ba�lat�lmas�ndan �nce tolere edilen ALUNBR�G dozunun maksimum iki kat�na kadar art�r�labilir. Orta derecede bir CYP3A ind�kleyicisinin kesilmesinden sonra, ALUNBR�G'e orta dereceli CYP3A ind�kleyicisinin ba�lat�lmas�ndan �nce tolere edilen dozda devam edilmelidir.
Fotosensitivite ve fotodermatoz
ALUNBR�G ile tedavi edilen hastalarda g�ne� �����na kar�� ����a duyarl�l�k olu�mu�tur (bkz. b�l�m 4.8). Hastalara ALUNBR�G al�rken ve tedavinin kesilmesinden sonra en az 5 g�n boyunca g�ne�e uzun s�re maruz kalmaktan ka��nmalar� tavsiye edilmelidir. D��ar�dayken, hastalara �apka ve koruyucu giysi giymeleri ve olas� g�ne� yan���na kar�� korunmaya yard�mc� olmak i�in geni� spektrumlu Ultraviyole A (UVA)/ Ultraviyole B (UVB) g�ne� kremi ve dudak kremi (SPF ≥30) kullanmalar� tavsiye edilmelidir. �iddetli fotosensitivite reaksiyonlar� (≥ Derece 3) i�in ALUNBR�G, ba�lang�� d�zeyine geri d�nene kadar durdurulmal�d�r. Doz buna g�re de�i�tirilmelidir (bkz. b�l�m 4.2).
Fertilite
�ocuk do�urma potansiyeli olan kad�nlara ALUNBR�G ile tedavi s�ras�nda ve son dozu takiben en az 4 ay boyunca etkili, hormonal olmayan kontrasepsiyon kullan�m� tavsiye edilmelidir. �ocuk do�urma potansiyeline sahip kad�n partneri olan erkeklere, tedavi s�ras�nda ve son ALUNBR�G dozundan sonra en az 3 ay s�reyle etkili kontrasepsiyon kullan�m� tavsiye edilmelidir (bkz. b�l�m 4.6.).
Laktoz
ALUNBR�G laktoz monohidrat i�erir. Nadir kal�t�msal galaktoz intolerans�, total laktaz yetmezli�i ya da glukoz-galaktoz malabsorpsiyon problemi olan hastalar�n bu ilac� kullanmamalar� gerekir.
Sodyum
Bu t�bbi �r�n her “doz”unda 1 mmol (23 mg)'dan daha az sodyum ihtiva eder; yani asl�nda “sodyum i�ermez”.
4.5. Di�er t�bbi �r�nler ile etkile�imler ve di�er etkile�im �ekilleri
Brigatinib plazma konsantrasyonlar�n� artt�rabilen ajanlarCYP3A inhibit�rleri
�n vitro �al��malar brigatinibin CYP3A4/5'in bir substrat� oldu�unu g�stermi�tir. Sa�l�kl� g�n�ll�lerde; g��l� bir CYP3A inhibit�r� olan itrakonazol�n g�nde iki defa 200 mg'l�k �oklu dozu ile 90 mg'l�k tekli brigatinib dozunun e� zamanl� uygulanmas�, tek ba��na uygulanan 90 mg'l�k brigatinib dozuna g�re, brigatinib C'�n� % 21, EAA'u % 101 (2 kat) ve EAA'yi % 82
(<2 kat) oran�nda art�rm��t�r. Bunlarla s�n�rl� olmamak �zere belirli antiviral ajanlar (�rn. indinavir, nelfinavir, ritonavir, sakinavir), makrolid antibiyotikler (�rn. klaritromisin, telitromisin, troleandomisin), antifungal ajanlar (�rn. ketokonazol, vorikonazol) ve nefazodon dahil olmak �zere g��l� CYP3A inhibit�rlerinin ALUNBR�G ile e� zamanl� kullan�m�ndan ka��n�lmal�d�r. G��l� CYP3A inhibit�rlerinin birlikte kullan�m�n�n ka��n�lmaz oldu�u durumunda; ALUNBR�G dozu yakla��k % 50 oran�nda (yani 180 mg'dan 90 mg'a veya 90 mg'dan 60 mg'a) d���r�lmelidir. G��l� bir CYP3A inhibit�r� kesildikten sonra; ALUNBR�G tedavisine, g��l� CYP3A inhibit�r� ba�lat�lmadan �nce tolere edilen dozda devam edilmelidir.
Orta dereceli CYP3A inhibit�rleri (�rn. diltiazem ve verapamil), fizyolojik-bazl� bir farmakokinetik modelden elde edilen sim�lasyonlara dayanarak, brigatinib EAA'sini yakla��k % 40 oran�nda art�rabilir. Orta dereceli CYP3A inhibit�rleri ile kombinasyonda, ALUNBR�G i�in doz ayarlamas� gerekli de�ildir. ALUNBR�G'in orta dereceli CYP3A inhibit�rleri ile e� zamanl� uygulanmas� durumunda, hastalar yak�ndan takip edilmelidir.
Greyfurt veya greyfurt suyu da brigatinibin plazma konsantrasyonlar�n� artt�rabilir ve bu nedenle kullan�m�ndan ka��n�lmal�d�r (bkz. b�l�m 4.2.).
CYP2C8 �nhibit�rleri
�n vitro �al��malar brigatinibin CYP2C8'in bir substrat� oldu�unu g�stermi�tir. Sa�l�kl� g�n�ll�lerde; g��l� bir CYP2C8 inhibit�r� olan gemfibrozilin g�nde iki defa 600 mg'l�k �oklu dozu ile 90 mg'l�k tekli brigatinib dozunun e� zamanl� uygulanmas�, tek ba��na uygulanan 90 mg'l�k brigatinib dozuna g�re, brigatinib C'�n� % 41, EAA'u % 12 ve EAA'yi % 15 oran�nda azaltm��t�r. Gemfibrozilin brigatinibin farmakokineti�i �zerindeki etkisi klinik olarak anlaml� de�ildir ve azalm�� brigatinib maruziyetinin altta yatan mekanizmas� bilinmemektedir. G��l� CYP2C8 inhibit�rleri ile e� zamanl� kullan�m s�ras�nda doz ayarlamas� gerekli de�ildir.
P-gp ve BCRP inhibit�rleri
Brigatinib, in vitro P-glikoprotein (P-gp) ve meme kanseri diren� proteininin (BCRP) bir substrat�d�r. Brigatinibin y�ksek ��z�n�rl�k ve y�ksek ge�irgenlik g�sterdi�i g�z �n�nde bulunduruldu�unda; P-gp ve BCRP inhibisyonunun, brigatinibin sistemik maruziyetinde klinik olarak anlaml� bir de�i�ikli�e yol a�mas� beklenmemektedir. P-gp ve BCRP inhibit�rleri ile e� zamanl� kullan�m s�ras�nda ALUNBR�G dozunun ayarlanmas� gerekli de�ildir.
Brigatinib plazma konsantrasyonlar�n� azaltabilen ajanlar
CYP3A ind�kleyicileri
Sa�l�kl� g�n�ll�lerde; g��l� bir CYP3A ind�kleyicisi olan rifampisinin g�nl�k 600 mg'l�k �oklu dozu ile 180 mg'l�k tekli brigatinib dozunun e� zamanl� uygulanmas�, tek ba��na uygulanan 180 mg'l�k brigatinib dozuna g�re, brigatinib C'�n� % 60, EAA'u % 80 (5 kat) ve EAA'yi
% 80 (5 kat) oran�nda azaltm��t�r. Bunlarla s�n�rl� olmamak �zere rifampisin, karbamazepin, fenitoin, rifabutin, fenobarbital ve St. John's Wort dahil g��l� CYP3A ind�kleyicileri ile ALUNBR�G'in e� zamanl� kullan�m�ndan ka��n�lmal�d�r.
Orta dereceli CYP3A ind�kleyicileri, fizyolojik-bazl� bir farmakokinetik modelden elde edilen sim�lasyonlara dayanarak, brigatinib EAA'sini yakla��k % 50 oran�nda azaltabilir. Bunlarla s�n�rl� olmamak �zere efavirenz, modafinil, bosentan, etravirin ve nafsilin dahil olmak �zere orta dereceli CYP3A ind�kleyicileri ile ALUNBR�G'in e� zamanl� kullan�m�ndan ka��n�lmal�d�r. Orta dereceli CYP3A ind�kleyicilerinin e�zamanl� kullan�m�ndan ka��n�lamazsa, ALUNBR�G dozu, tolere edilen mevcut ALUNBR�G dozu ile 7 g�nl�k tedaviden sonra 30 mg'l�k art��larla, orta dereceli CYP3A ind�kleyicisinin ba�lat�lmas� �nce tolere edilen ALUNBR�G dozunun maksimum iki kat�na kadar
art�r�labilir. Orta derecede bir CYP3A ind�kleyicisinin kesilmesinden sonra, ALUNBR�G''e orta dereceli CYP3A ind�kleyicisinin ba�lat�lmas�ndan �nce tolere edilen dozda devam edilmelidir.
Plazma konsantrasyonlar� brigatinib ile de�i�ebilen ajanlar
CYP3A substratlar�
Hepatositlerle ger�ekle�tirilen in vitro �al��malar brigatinibin CYP3A4 ind�kleyicisi oldu�unu g�stermi�tir. Kanser hastalar�nda, g�nde 180 mg'l�k �oklu ALUNBR�G dozlar�n�n, hassas bir CYP3A substrat� olan 3 mg'l�k tek bir oral midazolam dozu ile birlikte uygulanmas�, tek ba��na uygulanan 3 mg oral midazolam dozuna g�re midazolam C'�n� %16, AUC'yi %26 ve AUC'� %30 oran�nda azaltm��t�r. Brigatinib; b�y�k �l��de CYP3A ile metabolize edilen, e� zamanl� uygulanan t�bbi �r�nlerin plazma konsantrasyonlar�n� azaltabilir. Bu nedenle, ALUNBR�G'in dar terap�tik indekse sahip CYP3A substratlar� (�rn: alfentanil, fentanil, kinidin, siklosporin, sirolimus, takrolimus) ile birlikte kullan�m�ndan ka��n�lmal�d�r zira etkinliklerinde azalma olabilir.
ALUNBR�G ayr�ca CYP3A ind�ksiyonundan sorumlu ayn� mekanizmalar (�rn. pregnane X resept�r aktivasyonu) ile di�er enzimleri ve ta��y�c�lar� (�rn. CYP2C, P-gp) da ind�kleyebilir.
Ta��y�c� substratlar
Brigatinibin P-gp substratlar� (�rn. digoksin, dabigatran, kol�isin, pravastatin), BCRP (�rn. metotreksat, rosuvastatin, s�lfasalazin), organik katyon ta��y�c�s� 1 (OCT1), �oklu ila� ve toksin ekstr�zyon proteini 1 (MATE1) ve 2K (MATE2K) substratlar� ile e� zamanl� uygulanmas�, bu substratlar�n plazma konsantrasyonlar�n� artt�rabilir. ALUNBR�G dar terap�tik indekse sahip ta��y�c� substratlarla (�rn: digoksin, dabigatran, metotreksat) birlikte kullan�l�rken hastalar yak�ndan takip edilmelidir.
4.6. Gebelik ve laktasyon
Genel tavsiyeGebelik kategorisi: C
�ocuk do�urma potansiyeli bulunan kad�nlar/Kad�n ve Erkeklerde Do�um kontrol� (kontrasepsiyon)
ALUNBR�G ile tedavi edilen, �ocuk do�urma potansiyeli bulunan kad�nlar�n hamile kalmamalar� ve ALUNBR�G ile tedavi edilen erkeklerin ise tedavi s�ras�nda �ocuk sahibi olmamalar� tavsiye edilmelidir. �reme potansiyeli olan kad�nlara ALUNBR�G ile tedavi s�ras�nda ve son dozu takiben en az 4 ay boyunca etkili, hormonal olmayan kontrasepsiyon kullanmalar� tavsiye edilmelidir. �reme potansiyeline sahip kad�n partneri olan erkeklere, tedavi s�ras�nda ve son ALUNBR�G dozundan sonra en az 3 ay s�reyle etkili kontrasepsiyon kullan�m� �nerilmelidir.
Gebelik d�nemi
ALUNBR�G gebe bir kad�na uyguland���nda �l�mc�l sonu�lar do�urabilir. Hayvanlar �zerinde yap�lan �al��malar, �reme toksisitesini g�stermektedir (bkz. b�l�m 5.3.). ALUNBR�G'in gebe kad�nlardaki kullan�m�na ili�kin klinik veri bulunmamaktad�r. Annenin klinik durumu tedavi gerektirmiyorsa; ALUNBR�G gebelik d�nemi s�ras�nda kullan�lmamal�d�r. ALUNBR�G gebelik s�ras�nda kullan�l�rsa veya hasta bu t�bbi �r�n� al�rken hamile kal�rsa; hastaya potansiyel olarak fet�se zarar verebilece�i bilgisi verilmelidir.
Laktasyon d�nemi
ALUNBR�G'in anne s�t�ne ge�ip ge�medi�i bilinmemektedir. Mevcut veriler ilac�n anne s�t�ne ge�me potansiyelini hari� tutamaz. ALUNBR�G tedavisi s�ras�nda emzirme durdurulmal�d�r.
�reme yetene�i/Fertilite
ALUNBR�G'in insan fertilitesi �zerindeki etkilerine ili�kin veri mevcut de�ildir. Hayvanlar �zerinde yap�lan �al��malar esas al�nd���nda; ALUNBR�G erkeklerde fertilitede azalmaya neden olabilir (bkz. b�l�m 5.3.). Bu bulgular�n insan fertilitesine y�nelik klinik anlam� bilinmemektedir.
4.7. Ara� ve makine kullan�m� �zerindeki etkiler
ALUNBR�G ara� ve makine kullanma yetene�i �zerinde min�r bir etkiye sahiptir. Ancak; hastalar ALUNBR�G'i kullan�rken g�rme bozuklu�u, ba� d�nmesi veya yorgunluk ya�ayabilece�i i�in, ara� veya makine kullan�rken dikkatli olmal�d�rlar.
4.8. �stenmeyen etkiler
G�venlilik profilinin �zeti
Tavsiye edilen doz rejiminde ALUNBR�G ile tedavi edilen hastalarda raporlanan en yayg�n advers reaksiyonlar (≥ %25); AST art���, CPK art���, hiperglisemi, lipaz art���, hiperins�linemi, diyare, ALT art���, amilaz art���, anemi, mide bulant�s�, bitkinlik, hipofosfatemi, lenfosit say�s�nda azalma, �ks�r�k, alkalin fosfataz art���, d�k�nt�, APTT art���, miyalji, ba� a�r�s�, hipertansiyon, akyuvar say�s�nda azalma, dispne ve kusmad�r.
Neoplazma progresyonu ile ili�kili olaylar d���nda; tavsiye edilen doz rejiminde ALUNBR�G ile tedavi edilen hastalarda rapor edilen en yayg�n ciddi advers reaksiyonlar (≥ %2) pn�moni, pn�monit, dispne ve pireksi olmu�tur.
Advers reaksiyonlar�n tablo halinde listesi:
A�a��da tan�mlanan veriler �� klinik �al��mada tavsiye edilen doz rejiminde ALUNBR�G'e maruziyeti yans�tmaktad�r: Daha �nce bir ALK-inhibit�r� ile tedavi g�rmemi� ileri evre ALK- pozitif KHDAK hastalar�nda bir Faz 3 �al��ma (ALTA 1L) (N=136), daha �nce krizotinib tedavisine progresyon g�stermi� olup ALUNBR�G'le tedavi edilen ALK-pozitif KHDAK hastalar�nda bir faz 2 �al��ma (N=110) ve ilerlemi� malignansili hastalarda bir faz 1/2 doz eskalasyon/ekspansiyon �al��mas� (N=28). Bu �al��malarda, tavsiye edilen doz rejiminde ALUNBR�G kullanan hastalarda medyan maruziyet s�resi 21,8 ay olmu�tur.
Rapor edilen advers reaksiyonlar Tablo 3'te sunulmaktad�r ve Sistem Organ S�n�f�na, tavsiye edilen terime ve g�r�lme s�kl���na g�re listelenmi�tir. S�kl�k kategorileri: (�ok yayg�n ≥1/10, yayg�n (≥1/100 ila <1/10), yayg�n olmayan ≥1/1.000 ila <1/100, seyrek ≥1/10.000 ila <1/1.000; bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). Her bir s�kl�k grubu i�inde istenmeyen etkiler azalan �iddete g�re verilmi�tir.
Tablo 3: ALUNBR�G ile 180 mg rejiminde tedavi edilen hastalarda (N= 274) rapor edilen advers reaksiyonlar (Advers Olaylar i�in Ortak Terminoloji Kriterleri (CTCAE) versiyon 4.03'e g�re)
Sistem Organ S�n�f� | S�kl�k Kategorisi | Advers reaksiyonlar T�m dereceler | Advers reaksiyonlar Derece 3 – 4 |
Enfeksiyonlar ve enfestasyonlar | �ok yayg�n | Pn�moni �st solunum yolu enfeksiyonu |
|
Yayg�n |
| Pn�moni | |
Kan ve lenfatik sistem bozukluklar� | �ok yayg�n | Anemi Lenfosit say�s�nda azalma APTT art��� Akyuvar say�s�nda azalma N�trofil say�s�nda azalma | Lenfosit say�s�nda azalma |
Yayg�n | Platelet say�s�nda azalma | APTT art��� Anemi | |
Yayg�n olmayan |
| N�trofil say�s�nda azalma | |
Metabolizma ve beslenme hastal�klar� | �ok yayg�n | Hiperglisemi Hiperins�linemi Hipofosfatemi Hipomagnezemi Hiperkalsemi Hiponatremi Hipokalemi ��tah azalmas� |
|
Yayg�n |
| Hipofosfatemi Hiperglisemi Hiponatremi Hipokalemi ��tah azalmas� | |
Psikiyatrik hastal�klar | Yayg�n: | Uykusuzluk |
|
Sinir sistemi hastal�klar� | �ok yayg�n: | Ba� a�r�s� Periferal n�ropati Ba� d�nmesi |
|
Yayg�n | Haf�za bozuklu�u Disguzi | Ba� a�r�s� Periferal n�ropati | |
Yayg�n olmayan |
| Ba� d�nmesi | |
G�rme bozukluklar� | �ok yayg�n | G�rme bozuklu�u |
|
Yayg�n |
| G�rme bozuklu�u | |
Kardiyak bozukluklar | Yayg�n | Bradikardi Elektrokardiyogram QT uzamas� Ta�ikardi Palpitasyonlar | Elektrokardiyogram QT uzamas� |
Yayg�n olmayan |
| Bradikardi | |
Vask�ler bozukluklar | �ok yayg�n | Hipertansiyon | Hipertansiyon |
Solunum, g���s bozukluklar� ve mediyastinal hastal�klar | �ok yayg�n | �ks�r�k Dispne |
|
Yayg�n | Pn�monit | Pn�monit Dispne | |
Gastointestinal hastal�klar | �ok yayg�n | Lipaz art��� Diyare Amilaz art��� Bulant� Kusma Kar�n a�r�s� Konstipasyon | Lipaz art��� |
|
| Stomatit |
|
Yayg�n | A��z kurulu�u Dispepsi Flatulans | Amilaz art��� Bulant� Kar�n a�r�s� Diyare | |
Yayg�n olmayan | Pankreatit | Kusma Stomatit Dispepsi Pankreatit | |
Hepatobiliyer bozukluklar | �ok yayg�n | AST art��� ALT art��� Alkalin fosfataz art��� |
|
Yayg�n | Kan laktat dehidrogenaz art��� Hiperbilirubinemi | ALT art��� AST art��� Alkalin fosfataz art��� | |
Yayg�n olmayan |
| Hiperbilirubinemi | |
Deri ve deri alt� dokusu hastal�klar� | �ok yayg�n | D�k�nt� Prurit |
|
Yayg�n | Cilt kurulu�u Fotosensitivite reaksiyonu | D�k�nt� Fotosensitivite reaksiyonu | |
Yayg�n olmayan |
| Cilt kurulu�u Prurit | |
Kas-iskelet sistemi ve ba� dokusu hastal�klar� | �ok yayg�n | Kan CPK art��� Miyalji Artralji | Kan CPK art��� |
Yayg�n | Kas-iskelet g���s a�r�s� Kollar ve bacaklarda a�r� Kas-iskelet sisteminde kat�l�k |
| |
Yayg�n olmayan |
| Kollar ve bacaklarda a�r� Kas-iskelet g���s a�r�s� Miyalji | |
B�brek ve idrar yolu hastal�klar� | �ok yayg�n | Kan kreatinin art��� |
|
Genel bozukluklar ve uygulama yerine ili�kin durumlar | �ok yayg�n | Bitkinlik �dem Pireksi |
|
Yayg�n | Kardiyak olmayan g���s a�r�s� G���ste rahats�zl�k A�r� | Bitkinlik | |
Yayg�n olmayan |
| Pireksi �dem Kardiyak olmayan g���s a�r�s� | |
Ara�t�rmalar | Yayg�n | Kan kolesterol seviyesinde art�� Kilo kayb� |
|
Yayg�n olmayan |
| Kilo kayb� | |
bozuklu�u, vitr�z y�rt�lma, vitroz flat�r, ge�ici amorozu i�erir.
I���a duyarl�l�k reaksiyonu, polimorf ���k er�psiyonu, solar dermatiti i�erir.
Se�ili advers reaksiyonlar�n tan�m�
Pulmoner advers reaksiyonlar
ALTA 1L'de; hastalar�n % 2,9'unda tedavinin ba�lang�c�nda (8 g�n i�inde) herhangi bir Derece ILD/pn�monit, hastalar�n % 2,2'sinde Derece 3-4 ILD/pn�monit g�r�lm��t�r. �l�mc�l ILD/pn�monit mevcut de�ildir. �lave olarak, hastalar�n % 3,7'sinde pn�monit tedavinin ilerleyen zamanlar�nda g�r�lm��t�r.
ALTA'da, hastalar�n % 6,4'�nde tedavinin ba�lar�nda (9 g�n i�inde, medyan ba�lang��: 2 g�n) ILD/pn�monit, pn�moni ve dispne dahil olmak �zere, herhangi bir derecede pulmoner advers reaksiyonlar g�r�lm��t�r; hastalar�n % 2,7'sinde Derece 3-4 pulmoner advers reaksiyon ve 1 hastada (% 0,5) �l�mc�l pn�moni g�r�lm��t�r. Derece 1-2 pulmoner advers reaksiyonlar� takiben; ALUNBR�G ile tedavi kesilmi�tir ve daha sonra yeniden ba�lat�lm�� veya doz azalt�lm��t�r. �� �l�mc�l vaka (hipoksi, akut solunum g��l��� sendromu ve pn�moni) dahil olmak �zere, erken pulmoner advers reaksiyonlar hastalarda (N= 137) (�al��ma 101)bir doz y�kseltme �al��mas�nda da meydana gelmi�tir. �lave olarak, ALTA'da hastalar�n % 2,3'�nde tedavinin ilerleyen zamanlar�nda pn�monit g�r�lm��t�r, 2 hastada Derece 3 pn�monit g�r�lm��t�r (bkz. b�l�m 4.2 ve 4.4.).
Ya�l� hastalar
Erken pulmoner advers reaksiyon, 65 ya� alt� hastalar�n %3,1'inde, 65 ya� ve �zeri hastalar�n ise
%10,1'inde rapor edilmi�tir.
Hipertansiyon
ALUNBR�G ile 180 mg rejiminde tedavi edilen hastalar�n % 30'unda hipertansiyon bildirilmi�tir olup; bu hastalar�n % 11'i Derece 3 hipertansiyona sahiptir. 180 mg rejiminde % 1,5 oran�nda hipertansiyon i�in doz azalt�lmas� ger�ekle�tirilmi�tir. T�m hastalarda ortalama sistolik ve diyastolik kan bas�nc� zamanla artm��t�r (bkz. b�l�m 4.2 ve 4.4.).
Bradikardi
ALUNBR�G ile 180 mg rejiminde tedavi edilen hastalar�n % 8,4'�nde bradikardi rapor edilmi�tir.
180 mg tedavi rejiminde hastalar�n % 8,4'�nde dakikada 50 kalp at�m h�z�ndan az kalp at�m h�z� rapor edilmi�tir (bkz. b�l�m 4.2. ve 4.4.).
G�rme bozuklu�u
ALUNBR�G ile 180 mg rejiminde tedavi edilen hastalar�n % 14'�nde g�rme bozuklu�u advers reaksiyonlar� rapor edilmi� olup; bunlardan 3 tanesi, mak�ler �dem ve katarakt dahil olmak �zere Derece 3 advers reaksiyon (% 1,1) olarak rapor edilmi�tir.
180 mg rejiminde iki hastada (% 0,7) g�rme bozuklu�u i�in doz azalt�lmas� ger�ekle�tirilmi�tir (bkz. b�l�m 4.2 ve 4.4.).
Periferal n�ropati
180 mg rejiminde tedavi edilen hastalar�n % 20'sinde periferal n�ropati advers reaksiyonlar� rapor edilmi�tir. Hastalar�n % 33'�nde t�m periferal n�ropati advers reaksiyonlar� ortadan kalkm��t�r. Periferal n�ropati advers reaksiyonlar�n�n medyan s�resi 6.6 ay ve maksimum s�re 28,9 ayd�r.
Kreatin fosfokinaz (CPK) art���
ALTA 1L ve ALTA'da, ALUNBR�G ile 180 mg rejiminde tedavi edilen hastalar�n % 64'�nde CPK art��� rapor edilmi�tir. Derece 3-4 CPK art��� insidans� % 18 olmu�tur. CPK art���n�n ba�lang�c�na kadar ge�en medyan s�re 28 g�n olmu�tur.
180 mg rejiminde hastalar�n % 10'unda CPK art��� i�in doz azalt�lmas� ger�ekle�tirilmi�tir (bkz. b�l�m 4.2. ve 4.4.).
Pankreatik enzimlerin art���
ALUNBR�G ile 180 mg rejiminde tedavi edilen hastalar�n s�ras�yla % 47 ve % 54'�nde amilaz ve lipaz art��lar� rapor edilmi�tir. Derece 3 ve 4 art��lar� i�in amilaz ve lipaz insidans� s�ras�yla % 7,7 ve % 15 olmu�tur. Amilaz ve lipaz art���n�n ba�lang�c�na kadar ge�en medyan s�re s�ras�yla 16 g�n ve 29 g�n olmu�tur.
180 mg rejiminde hastalar�n s�ras�yla % 4,7 ve % 2,9'unda lipaz ve amilaz art��� i�in doz azalt�lmas� ger�ekle�tirilmi�tir (bkz. b�l�m 4.2. ve 4.4.).
Hepatik enzimlerin art���
ALUNBR�G ile 180 mg rejiminde tedavi edilen hastalar�n s�ras�yla % 49 ve % 68'inde ALT ve AST art��lar� rapor edilmi�tir. Derece 3 ve 4 art��lar� i�in, insidanslar ALT ve AST i�in s�ras�yla % 4,7 ve % 3,6 olmu�tur.
180 mg rejiminde hastalar�n s�ras�yla % 0,7 ve % 1,1'inde ALT ve AST art��� i�in doz azalt�lmas� ger�ekle�tirilmi�tir (bkz. b�l�m 4.2. ve 4.4.).
Hiperglisemi
Hastalar�n % 61'inde hiperglisemi g�r�lm��t�r. Hastalar�n % 6,6's�nda Derece 3 hiperglisemi meydana gelmi�tir.
Hi�bir hastada hiperglisemi nedeniyle doz azalt�lmas� ger�ekle�tirilmemi�tir.
Fotosensitivite ve fotodermatoz
ALUNBR�G ile farkl� doz rejimlerinde tedavi edilen 804 hastadan al�nan verilerle yedi klinik �al��man�n havuzlanm�� analizi, hastalar�n %5,8'inde fotosensitivite ve fotodermatozun rapor edildi�ini ve hastalar�n %0,7'sinde Derece 3-4 meydana geldi�ini g�stermi�tir. Hastalar�n %0,4'�nde doz azalt�lm��t�r (bkz. b�l�m 4.2 ve 4.4).
��pheli advers reaksiyonlar�n raporlanmas�
Ruhsatland�rma sonras� ��pheli ila� advers reaksiyonlar�n�n raporlanmas� b�y�k �nem ta��maktad�r. Raporlama yap�lmas�, ilac�n yarar/risk dengesinin s�rekli olarak izlenmesine olanak sa�lar. Sa�l�k
mesle�i mensuplar�n�n herhangi bir ��pheli advers reaksiyonu T�rkiye Farmakovijilans Merkezi (T�FAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e-posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks: 0 312 218 35 99).
4.9. Doz a��m� ve tedavisi
ALUNBR�G'in doz a��m�na ili�kin spesifik herhangi bir antidot mevcut de�ildir. Doz a��m� meydana gelmesi durumunda; hastalar advers reaksiyonlar (bkz. b�l�m 4.8) bak�m�ndan takip edilir ve uygun bir destek tedavisi sa�lan�r.
5. FARMAKOLOJ�K �ZELL�KLER
5.1. Farmakodinamik �zellikler
Farmakoterap�tik grup: Antineoplastik ila�, protein kinaz inhibit�rleri ATC kodu: L01ED04
Etki mekanizmas�
Brigatinib, ins�lin-benzeri b�y�me fakt�r� 1 resept�r� (IGF-1R) ve ALK'y�, c-ros onkogen 1(ROS1), hedefleyen bir tirozin kinaz inhibit�r�d�r. Brigatinib, in vitro ve in vivo tayinlerde ALK'nin otofosforilasyonunu ve a�a�� y�nl� sinyal proteini STAT3'�n ALK arac�l� fosforilasyonunu inhibe etmi�tir.
Brigatinib, farelerde EML4-ALK ve NPM-ALK f�zyon proteinlerini eksprese eden h�cre hatlar�n�n in vitro proliferasyonunu inhibe etmi�tir ve EML4-ALK-pozitif KHDAK ksenograft b�y�mesinin doza ba�l� inhibisyonunu g�stermi�tir.
Brigatinib; G1202R ve L1196M dahil, ALK inhibit�rlerine diren� ile ili�kilendirilen EML4- ALK'n�n mutant formlar�n� eksprese eden h�crelerin canl�l���n� in vitro ve in vivoda inhibe etmi�tir.
Kardiyak elektrofizyoloji
�al��ma 101'de, ilerlemi� malignansili 123 hastada 30 mg ila 240 mg'l�k g�nl�k brigatinib dozu ile ALUNBR�G'in QT aral���n� uzatma potansiyeli de�erlendirilmi�tir. Ba�lang�� noktas�ndan maksimum ortalama QTcF (Friedericia y�ntemi ile d�zeltilmi� QT) de�i�imi 10 milisaniyeden k�sa olmu�tur. Bir maruziyet-QT analizi, konsantrasyona ba�l� herhangi bir QTc aral��� uzamas� olmad���n� �ne s�rm��t�r.
Klinik etkililik ve g�venlilik
ALTA 1L
ALUNBR�G'in g�venlili�i ve etkilili�i; daha �nceden ALK-hedefli bir tedavi almam�� ve ilerlemi� ALK-pozitif KHDAK'l� 275 yeti�kin hastada ger�ekle�tirilen randomize (1:1), a��k etiketli, �ok merkezli bir �al��mada (ALTA 1L) de�erlendirilmi�tir.
Uygunluk kriterleri; lokal bir bak�m standard� testine dayal� olarak belgelenen bir ALK d�zenlemesine sahip ve ECOG performans skoru 0-2 olan hastalar�n dahil edilmesine izin vermi�tir. Hastalar�n lokal olarak ilerlemi� veya metastatik ortamda �nceden 1 taneye kadar kemoterapi rejimi almas�na izin vermi�tir. Leptomeningeal metastazlar da dahil olmak �zere tedavi edilmi� veya tedavi edilmemi� merkezi sinir sistemi (CNS) metastazlar� olan n�rolojik olarak stabil hastalar uygundur. Pulmoner interstisyel hastal���, ilaca ba�l� pn�monit veya radyasyon pn�moniti �yk�s� olan hastalar �al��ma d��� b�rak�lm��t�r.
Hastalar; g�nde bir kez 90 mg'l�k dozda 7 g�nl�k bir tedavi sonras� g�nde bir kez 180 mg (N=137) ALUNBR�G veya oral olarak g�nde iki defa krizotinib 250 mg (N= 138) almak �zere 1:1 oran�nda randomize edilmi�tir. Randomizasyon beyin metastaz� (var, yok) ve lokal olarak ilerlemi� veya metastatik hastal�k i�in �nceden kemoterapi kullan�m� (evet, hay�r) ile katmanlara ayr�lm��t�r.
Hastal�k progresyonu ya�ayan krizotinib kolundaki hastalara ALUNBR�G ile tedavi g�rmeleri i�in �apraz ge�i� �nerildi. Krizotinib koluna randomize edilen ve son analiz zaman�na kadar �al��ma tedavisini b�rakan 121 hastan�n tamam� aras�nda, 99 (%82) hastaya m�teakip ALK tirozin kinaz inhibit�rleri (TKI'ler) verildi. Krizotinib koluna randomize edilen seksen (%66) hasta, daha sonra ALUNBR�G tedavisi ald�; buna, �al��mada �apraz ge�i� yapan 65 (%54) hasta da dahildir.
Maj�r sonu� �l��m�, �ift K�r Hakemli Ba��ms�z �nceleme Komitesi (BIRC) taraf�ndan de�erlendirilen Solid T�m�rlerdeki Yan�t De�erlendirme Kriteri'ne (RECIST v1.1) g�re progresyonsuz sa�kal�m (PFS) olmu�tur. BIRC taraf�ndan de�erlendirilen ilave sonu� �l��mleri, do�rulanm�� objektif yan�t oran�n� (ORR), yan�t s�resini (DOR), yan�t verilene kadar ge�en s�reyi, hastal�k kontrol oran�n� (DCR), intrakraniyal ORR'yi, intrakraniyal PFS'yi ve intrakraniyal DOR'u i�ermi�tir. Ara�t�rmac� taraf�ndan de�erlendirilen sonu�lar aras�nda PFS ve genel sa�kal�m yer alm��t�r.
ALTA 1L'deki temel demografik �zellikler ve hastal�k �zellikleri �u �ekildedir: medyan ya� 59 (% 32'si 65 ya� ve �zeri olmak �zere 27 –89 aras�), % 59 beyaz �rk ve % 39 Asyal�, % 55 kad�n, % 39 ECOG PS 0 ve % 56 ECOG PS 1, % 58 hi� sigara kullanmam��, % 93 hastal�k evresi IV, % 96 adenokarsinoma histolojisine sahip, % 30 ba�lang��ta CNS metastaz�na sahip, % 14 daha �nce beyin i�in radyoterapi alm�� ve % 27 daha �nce kemoterapi alm��. Ekstratorasik metastaz b�lgelerine beyin (hastalar�n % 30'u), kemik (hastalar�n % 31'i) ve karaci�er (hastalar�n % 20'si) dahildir. Medyan ba��l doz yo�unlu�u ALUNBR�G i�in %97, krizotinib i�inse %99 idi.
ALUNBR�G kolunda 11 ayl�k medyan takip s�resinde ger�ekle�tirilen primer analizde; ALTA 1L �al��mas�, BIRC taraf�ndan PFS'de istatistiksel olarak anlaml� bir iyile�me g�steren primer sonlanma noktas�n� kar��lam��t�r.
ALUNBR�G kolunda 24,9 ayl�k medyan takip s�resinde 28 Haziran 2019'da protokole g�re belirlenmi� bir ara analiz ger�ekle�tirilmi�tir. ITT pop�lasyonunda BIRC'ye g�re medyan PFS, ALUNBR�G kolunda 24 ay ve krizotinib kolunda 11 ayd� (HR =0,49 [%95 GA (0,35, 0,68)], p
<0,0001).
ALUNBR�G kolunda medyan 40.4 ayl�k takip s�resinde ger�ekle�tirilen son hasta son temas tarihi olan 29 Ocak 2021 ile protokolde belirtilen nihai analizin sonu�lar� a�a��da sunulmu�tur.
Tablo 4: ALTA 1L'deki Etkililik Sonu�lar� (ITT Pop�lasyonu)
Etkililik Parametreleri | ALUNBR�G N = 137 | Krizotinib N = 138 |
Medyan takip s�resi (ay) | 40,4 (aral�k: 0–52,4) | 15,2 (aral�k: 0,1–51,7) |
Birincil etkililik parametreleri | ||
PFS (BIRC) | ||
Olayl� Hasta Say�s�, n (%) | 73 (%53,3) | 93 (%67,4) |
�lerleyen Hastal�k, n (%) | 66 (%48,2)b | 88 (%63,8)c |
�l�m, n (%) | 7 (%5,1) | 5 (%3,6) |
Medyan (ay cinsinden) (%95 CI) | 24,0 (18,5, 43,2) | 11,1 (9,1, 13) |
Tehlike oran� (%95 CI) | 0,48 (0,35, 0,66) | |
Log-rank p-de�eri | <0,0001 | |
�kincil etkililik parametreleri | ||
Do�rulanm�� Objektif Yan�t Oran� (BIRC) | ||
Yan�tlayanlar, n (%) (%95 CI) | 102 (%74,5) (66,3, 81,5) | 86 (%62,3) (53,7, 70,4) |
p-de�eri | 0,033 | |
Tam Yan�t, % | %24,1 | %13 |
K�smi Yan�t, % | %50,4 | %49,3 |
Do�rulanm�� Yan�t�n S�resi (BIRC) | ||
Ortanca (ay) (%95 CI) | 33,2 (22,1, NE) | 13,8 (10,4, 22,1) |
Genel Sa�kal�m | ||
Olay Say�s�, n (%) | 41 (%29,9) | 51 (%37) |
Medyan (ay cinsinden) (%95 CI) | NE (NE, NE) | NE (NE, NE) |
Tehlike oran� (%95 CI) | 0,81 (0,53, 1,22) | |
G�nl�k s�ralamas� p de�eri | 0,3311 | |
36 ayda Genel Sa�kal�m | %70,7 | %67,5 |
�ekil 1: ALTA 1L'de BIRC Taraf�ndan Progresyonsuz Sa�kal�m�n Kaplan-Meier �izimi
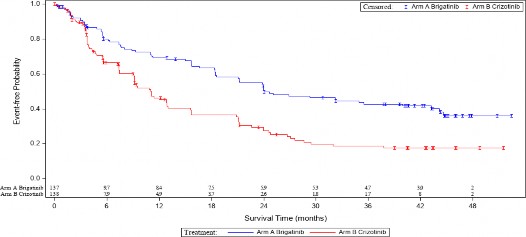
Bu �ekildeki sonu�lar, son hasta son temas tarihi olan 29 Ocak 2021 ile yap�lan son etkililik analizine dayanmaktad�r.
Ba�lang��ta herhangi bir beyin metastaz� olan hastalarda ve �l��lebilir beyin metastazlar� (en uzun �ap ≥ 10 mm) olan hastalarda RECIST v1.1'e g�re intrakraniyal etkilili�im BIRC de�erlendirmesi Tablo 5'te �zetlenmi�tir.
Tablo 5: ALTA 1L'deki Hastalarda BIRC-De�erlendirmeli �ntrakraniyal Etkililik
Etkililik Parametreleri | Ba�lang��ta �l��lebilir Beyin Metastazlar� Olan Hastalar | |
ALUNBR�G N= 18 | Krizotinib N= 23 | |
Do�rulanm�� �ntrakraniyal Objektif Yan�t Oran� | ||
Yan�tlayan, n (%) (% 95 CI) | 14 (% 77,8) (52,4, 93,6) | 6 (% 26,1) (10,2, 48,4) |
p-de�eri | 0.0014 | |
Tam yan�t %'si | % 27,8 | %0 |
K�smi yan�t %'si | % 50 | % 26,1 |
Do�rulanm�� �ntrakraniyal Yan�t S�resi | ||
Medyan (ay) (% 95 CI) | 27,9 (5,7, NE) | 9,2 (3,9, 9,2) |
| Ba�lang��ta Herhangi bir Beyin Metastaz� Olan Hastalar | |
ALUNBR�G N= 47 | ALUNBR�G N= 49 | |
Do�rulanm�� �ntrakraniyal Objektif Yan�t Oran� | ||
Yan�tlayan, n (%) (% 95 CI) | 31 (% 66) (50,7, 79,1) | 7 (%14,3) (5,9, 27,2) |
p-de�eri | <0.0001 | |
Tam yan�t (%) | % 44,7 | % 2 |
K�smi yan�t (%) | % 21,3 | % 12,2 |
Do�rulanm�� �ntrakraniyal Yan�t S�resi | ||
Medyan (ay) (% 95 CI) | 27,1 (16,9, 42,8) | 9,2 (3,9, NE) |
�nkraniyal PFS | ||
Olaylar� olan hasta say�s�, n (%) | 27 (%57,4) | 35 (%71,4) |
Progresif hastal�k, n (%) | 27 (%57,4) | 32 (%65,3) |
�l�m, n (%) | 0 (%0) | 3 (%6,1) |
Medyan (ay) (% 95 CI) | 24 (12,9, 30,8) | 5,5 (3,7, 7,5) |
Tehlike oran� (% 95 CI) | 0,29 (0,17, 0,51) | |
Log-s�ra p-de�eri | < 0,0001 | |
CI= G�ven aral���; NE= Tahmin edilebilir de�ildir.
Bu tablodaki sonu�lar, son hasta son temas tarihi olan 29 Ocak 2021 ile yap�lan son etkililik analizine dayanmaktad�r. Lokal olarak ilerlemi� veya metastatik hastal�k i�in �nceden kemoterapi varl��� ile s�ras�yla log-rank testi ve Cochran Mantel-Haenszel testi ile katmanlara ayr�lm��t�r.
≥ % 20 intrakraniyal hedef lezyon �ap� b�y�mesi veya hedef olmayan intrakraniyal lezyonlar�n kesin progresyonu) veya �l�m veya sans�rleme tarihine kadar �l��lm��t�r.
ALTA
ALUNBR�G'in g�venlili�i ve etkilili�i; krizotinib tedavisine progresyon g�steren, lokal olarak ilerlemi� veya metastatik ALK-pozitif KHDAK'l� 222 yeti�kin hastada ger�ekle�tirilen randomize (1:1), a��k etiketli, �ok merkezli bir �al��mada (ALTA) de�erlendirilmi�tir.
Uygunluk kriterleri; valide edilmi� bir teste dayal� olarak belgelenen bir ALK d�zenlemesine sahip �ncesinde kemoterapi alm��, ECOG Performans Durumu 0-2 olan ve hastalar�n dahil edilmesine izin vermi�tir. �lave olarak, merkezi sinir sistemi (CNS) metastaz� olan hastalar, n�rolojik olarak stabil olmalar� ve kortikosteroid dozunda art�� gerektirmedikleri s�rece, dahil edilmi�tir. Pulmoner interstiyal hastal�k veya ila� ile ili�kili pn�moni ge�mi�i olan hastalar hari� tutulmu�tur. Pulmoner interstisyel hastal��� veya ilaca ba�l� pn�monit �yk�s� olan hastalar �al��ma d��� b�rak�lm��t�r.
Hastalar; g�nde bir kez 90 mg (90 mg'l�k rejim, N=112) veya g�nde bir kez 90 mg'l�k dozda 7 g�nl�k bir tedavi sonras� g�nde bir kez 180 mg (180 mg'l�k rejim, N=110) ALUNBR�G almak �zere 1:1 oran�nda randomize edilmi�tir. Medyan takip s�resi 22,9 ay olmu�tur. Randomizasyon beyin metastaz� (var, yok) ve krizotinib tedavisine en iyi cevap (tam veya k�smi yan�t, di�er herhangi bir yan�t/bilinmeyen) ile katmanlara ayr�lm��t�r.
Maj�r sonu� �l��m�, ara�t�rmac� taraf�ndan de�erlendirilen Solid T�m�rlerdeki Yan�t De�erlendirme Kriteri'ne (RECIST v1.1) g�re objektif yan�t oran�n� (ORR) do�rulam��t�r. �lave sonu� �l��mleri; Ba��ms�z �nceleme Komitesi (IRC) taraf�ndan de�erlendirilen do�rulanm�� ORR'yi, yan�t verilene kadar ge�en s�reyi; progresyonsuz sa�kal�m� (PFS); yan�t s�resini (DOR); genel sa�kal�m� ve IRC taraf�ndan de�erlendirilen intrakraniyal ORR, intrakraniyal DOR'yi i�ermi�tir.
ALTA'daki temel demografik �zellikler ve hastal�k �zellikleri �u �ekildedir: medyan ya� 54 (% 23'� 65 ya� ve �zeri olmak �zere 18 –82 aras�), % 67 beyaz �rk ve % 31 Asyal�, % 57 kad�n, % 36 ECOG PS 0 ve % 57 ECOG PS 1, % 7 ECOG PS2, % 60 hi� sigara kullanmam��, % 35 daha evvel sigara kullanm��, %5 mevcut durumda sigara i�en, % 98 Evre IV, % 97 adenokarsinom ve % 74 daha �nce kemoterapi alm��. Ekstratorasik metastazlar�n en yayg�n oldu�u b�lgelere % 69 beyin (% 62'si daha �nce beynine radyasyon alm��), % 39 kemik ve % 26 karaci�er dahildir.
ALTA analizinden elde edilen etkililik sonu�lar� Tablo 6'da �zetlenmektedir ve ara�t�rmac� taraf�ndan de�erlendirilen PFS i�in Kaplan-Meier (KM) e�risi �ekil 2'de g�sterilmektedir.
Tablo 6: ALTA'daki Etkililik Sonu�lar� (ITT Pop�lasyonu)
Etkililik parametreleri | Ara�t�rmac� De�erlendirmesi | IRC De�erlendirmesi | ||
90 mg'l�k rejim* N= 112 | 180 mg'l�k rejim N= 110 | 90 mg'l�k rejim* N= 112 | 180 mg'l�k rejim N= 110 | |
Objektif yan�t oran� | ||||
(%) | % 46 | % 56 | % 51 | % 56 |
CI | (35, 57) | (45, 67) | (41, 61) | (47, 66) |
Yan�t s�resi | ||||
Medyan (ay) | 1,8 | 1,9 | 1,8 | 1,9 |
Yan�t s�resi | ||||
Medyan (ay) | 12,0 | 13,8 | 16,4 | 15,7 |
% 95 g�ven aral��� | (9,2, 17,7) | (10,2, 19,3) | (7,4, 24,9) | (12,8, 21,8) |
Progresyonsuz sa�kal�m | ||||
Medyan (ay) | 9,2 | 15,6 | 9,2 | 16,7 |
% 95 g�ven aral��� | (7,4, 11,1) | (11,1, 21) | (7,4, 12,8) | (11,6, 21,4) |
Genel sa�kal�m | ||||
Medyan (ay) | 29,5 | 34,1 | NA | NA |
%95 g�ven aral��� | (18,2, NE) | (27,7, NE) | NA | NA |
12 ayl�k sa�kal�m olas�l��� (%) | % 70,3 | % 80,1 | NA | NA |
CI= G�ven aral���; NE = tahmin edilebilir de�ildir; NA= Uygulanamaz
*G�nde bir kez 90 mg'l�k rejim
�ekil 2: Ara�t�rmac�n�n De�erlendirdi�i Sistemik Progresyonsuz Sa�kal�m: Tedavi Kolu ile ITT Pop�lasyonu (ALTA)
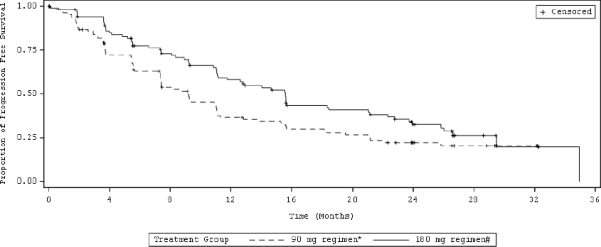
K�saltmalar: ITT = Tedavi ama�l�
Not: Progresyonsuz sa�kal�m tedavinin ba�lang�c�ndan, ilk olarak hangisinin meydana geldi�ine bak�lmaks�z�n hastal�k progresyonunun ilk olarak kan�tland��� veya �l�me kadar olan tarihe kadar ge�en s�re olarak belirlenmi�tir.
*90 mg g�nl�k tedavi rejimi
ALTA'da ba�lang��taki �l��lebilir beyin metastazlar� (en uzun �ap ≥10 mm) olan hastalardaki intrakraniyal ORR ve intrakraniyal yan�t s�resinin IRC de�erlendirmeleri Tablo 7'de �zetlenmektedir.
Tablo 7: Ba�lang��ta �l��lebilir Beyin Metastaz� olan Hastalardaki �ntrakraniyal Etkililik
IRC ile de�erlendirilen etkililik parametresi | Ba�lang��taki �l��lebilir Beyin Metastaz�na Sahip Hastalar | |
90 mg'l�k rejim* (N=26) | 180 mg'l�k rejim (N=18) | |
�ntrakraniyal objektif yan�t oran� | ||
(%) | % 50 | % 67 |
%95 g�ven aral��� | (30, 70) | (41, 87) |
�ntrakraniyal hastal�k kontrol oran� | ||
(%) | % 85 | % 83 |
%95 g�ven aral��� | (65, 96) | (59, 96) |
�ntrakraniyal yan�t s�reklili�i | ||
Medyan (ay) | 9,4 | 16,6 |
%95 g�ven aral��� | (3,7, 24,9) | (3,7, NE) |
%CI = G�ven aral���; NE = tahmin edilebilir de�ildir
*G�nde bir kez 90 mg'l�k rejim
Ba�lang��ta beyin metastaz� olan hastalarda, intrakraniyal hastal�k kontrol oran� 90 mg kolunda (N= 81) % 77.8 (% 95 CI: 67,2 – 86,3) ve 180 mg kolunda (N= 74) % 85,1 (% 95 CI: 75-92,3) olmu�tur.
�al��ma 101
Ayr� bir doz bulma �al��mas�nda, ALK-pozitif KHDAK'� olan ve krizotinibde progresyon g�steren 25 hastaya g�nde bir defa 90 mg'l�k dozda 7 g�nl�k bir tedavi sonras� g�nde 1 defa 180 mg ALUNBR�G uygulanm��t�r. Bu hastalar�n 19'u ara�t�rmac� de�erlendirmesi ile do�rulanm�� objektif bir yan�t g�stermi�tir (% 76; % 95 CI: 55, 91) ve yan�t veren 19 hasta aras�nda yan�t�n KM tahmini medyan s�resi 26,1 ay (% 91 CI: 7,9, 26,1) olmu�tur. KM medyan PFS 16,3 ayd�r (% 95 CI: 9,2,
NE) ve 12 ayl�k genel sa�kal�m olas�l��� % 84.0 (% 95 CI: 62,8, 93,7) olmu�tur.
Pediyatrik pop�lasyon
Avrupa �la� Ajans�, akci�er karsinomas�nda (k���k h�creli ve k���k h�creli d��� karsinoma) pediyatrik pop�lasyonun t�m alt gruplar�nda ALUNBR�G ile ger�ekle�tirilen �al��malar�n sonu�lar�n� sunma y�k�ml�l���nden muaf tutmu�tur (pediyatrik kullan�ma ili�kin bilgi i�in b�l�m 4.2'ye bak�n�z).
5.2. Farmakokinetik �zellikler
Genel �zelliklerEmilim:
�al��ma 101'de, hastalara brigatinibin tekli oral doz (30 – 240 mg) uygulamas�n� takiben pik konsantrasyonu i�in medyan s�re (T), dozlama sonras� 1-4 saat aral���nda de�i�mi�tir. Tekli bir dozdan sonra ve kararl� durumda, sistemik maruziyet 60 mg – 240 mg/g�n doz aral��� boyunca doz orant�sald�r. Tekrarl� dozlama sonras�nda makul bir ak�m�lasyon g�zlenmi�tir (geometrik ortalama ak�m�lasyon oran�: 1,9 – 2,4). 90 mg ve 180 mg'l�k g�nl�k dozlarda brigatinibin geometrik ortalama kararl� durum C'� s�ras�yla 552 ve 1452 ng/mL'dir ve ilgili EAAs�ras�yla 8,165 ve 20,276 h.ng/mL'dir. Brigatinib P-gp ve BCRP ta��y�c� proteinlerinin bir substrat�d�r.
Y�ksek-ya�l� ���n sonras� brigatinib uygulanan sa�l�kl� g�n�ll�lerde; bir gece boyunca a�l�k sonras� Cve EAA ile kar��la�t�r�ld���nda; EAA �zerinde bir etkisi olmaks�z�n brigatinib C'�
% 13'e indirgenmi�tir. Brigatinib yemeklerle birlikte veya yemekten ba��ms�z olarak uygulanabilir.
Da��l�m:
Brigatinib insan plazma proteinlerine orta derecede (% 91) ba�lanm��t�r ve ba�lanma konsantrasyona ba�l� de�ildir. Kan-plazma konsantrasyonu oran� 0,69'dur. Hastalara g�nde bir defa 180 mg brigatinib uygulanmas�n� takiben; kararl� durumda brigatinib da��l�m�n�n g�r�n�r geometrik hacim ortalamas� (VF) 307 L olmu�tur ve bu durum dokulara orta derecede bir da��l�m oldu�unu g�stermektedir.
Biyotransformasyon:
�n vitro �al��malar; brigatinibin primer olarak CYP2C8 ve CYP3A4 ile, ve �ok daha d���k bir d�zeyde CYP3A5 ile metabolize oldu�unu g�stermi�tir.
Oral olarak 180 mg'l�k tekli [C]-brigatinib dozunun sa�l�kl� g�n�ll�lere uygulanmas�n� takiben; N-demetilasyon ve sistein konjugasyonu iki maj�r metabolik klerens yolu olmu�tur. Kombine edilmi� idrar ve d��k�da, radyoaktif dozun % 48, % 27 ve % 9,1'i s�ras�yla de�i�memi� brigatinib, N-desmetik brigatinib (AP26123) ve brigatinib sistein konjugat� olarak at�lm��t�r. De�i�memi� brigatinib, AP26123 (% 3,5) ile birlikte dola��mdaki maj�r radyoaktif bile�enlerdir (% 92), ayr�ca primer metabolit in vitroda da g�zlenmi�tir. Hastalarda; kararl� durumda, AP26123'�n plazma EAA's� brigatinib maruziyetinin % 10'undan az olmu�tur. In vitro kinaz ve h�cresel tayinlerde, AP26123 metaboliti brigatinibden yakla��k 3-kat daha d���k bir potens ile ALK'y� inhibe etmi�tir.
Eliminasyon:
G�nde bir defa 180 mg brigatinib verilen hastalarda; kararl� durumda brigatinibin g�r�n�r geometrik ortalama oral klerensi (CL/F), 8,9 L/saat ve medyan plazma eliminasyon yar�lanma s�resi 24 saattir.
Brigatinibin primer at�l�m yolu d��k� yoluylad�r. 6 sa�l�kl� erkek g�n�ll�ye [C] brigatinibin tekli 180 mg oral dozunun uygulanmas�n� takiben; uygulanan dozun % 65'i d��k�da ve % 25'i idrarda saptanm��t�r. De�i�memi� brigatinib d��k� ve idrarda toplam radyoaktivitenin s�ras�yla % 41 ve % 86's�n� temsil etmi�tir, geri kalan k�s�m metabolitler olmu�tur.
Hastalardaki karakteristik �zellikler
Karaci�er yetmezli�i:
Brigatinibin farmakokineti�i; normal karaci�er fonksiyonuna sahip sa�l�kl� g�n�ll�lerde (N=9), hafif karaci�er yetmezli�i olan (Child-Pugh s�n�f A, N=6), orta derecede karaci�er yetmezli�i olan (Child-Pugh s�n�f B, N=6) veya �iddetli karaci�er yetmezli�i olan (Child-Pugh s�n�f C, N=6) hastalarda karakterize edilmi�tir. Brigatinib farmakokineti�i; normal karaci�er fonksiyonuna sahip sa�l�kl� g�n�ll�ler ve hafif (Child-Pugh s�n�f A) veya orta dereceli (Child-Pugh s�n�f B) karaci�er yetmezli�i olan hastalar aras�nda benzerdir. Normal karaci�er fonksiyonuna sahip sa�l�kl� g�n�ll�ler ile kar��la�t�r�ld���nda, �iddetli karaci�er yetmezli�i (Child-Pugh s�n�f C) olan hastalarda ba�lanmam�� EAA% 37 daha y�ksek olmu�tur (bkz. b�l�m 4.2.).
B�brek yetmezli�i:
Brigatinib farmakokineti�i; pop�lasyon farmakokinetik analizleri esas al�nd���nda, normal b�brek fonksiyonuna sahip hastalar ve hafif – orta dereceli b�brek yetmezli�i (eGFR ≥ 30 mL/dk) olan hastalarda benzerdir. Bir farmakokinetik �al��mada; normal b�brek fonksiyonuna sahip hastalar (eGFR ≥ 90 mL/dk, N=8) ile kar��la�t�r�ld���nda, �iddetli b�brek yetmezli�i (eGFR <30 mL/dk, N=6) olan hastalarda ba�lanmam�� EAA% 94 daha y�ksek olmu�tur (bkz. b�l�m 4.2.).
Irk ve cinsiyet:
Pop�lasyon farmakokinetik analizleri; �rk veya cinsiyetin brigatinib farmakokinetikleri �zerinde
herhangi bir etkisi olmad���n� g�stermi�tir.
Ya�, v�cut a��rl��� ve alb�min konsantrasyonlar�:
Pop�lasyon farmakokinetik analizleri; v�cut a��rl���, ya� ve alb�min konsantrasyonunun brigatinib farmakokinetikleri �zerinde klinik olarak anlaml� bir etkisi olmad���n� g�stermi�tir.
5.3. Klinik �ncesi g�venlilik verileri
Brigatinib ile ger�ekle�tirilen g�venlilik farmakoloji �al��malar�nda pulmoner etkiler (solunum h�z�nda de�i�im; insan C'�n�n 1-2 kat�), kardiyovask�ler etkiler (kalp at�m h�z�nda ve kan bas�nc�nda de�i�im; insan C'�n�n 0,5 kat�) ve renal etkiler (renal fonksiyonda azalma; insan C'�n�n 1-2,5 kat�) i�in potansiyel tespit edilmi�; ancak QT uzamas� veya n�rofonksiyonal etki potansiyeli g�r�lmemi�tir.
Klinik kullan�mla ili�kili olabilecek �ekilde klinik maruziyet seviyelerine benzer maruziyet seviyelerinde hayvanlarda g�r�len advers reaksiyonlar gastrointestinal sistem, kemik ili�i, g�zler, testisler, karaci�er, b�brek, kemik ve kalple ilgilidir. Bu etkiler genellikle dozlaman�n yap�lmad��� iyile�me periyodu boyunca geri d�n���ml�d�r; ancak, g�zler ve testislerdeki etkiler iyile�menin olmamas� nedeniyle belirgin istisnalard�r. Tekrarl� doz toksisite �al��malar�nda, maymunlarda insan EAA's�n�n 0,2 kat� ve �zerinde akci�er de�i�imleri (k�p�ks� alveolar makrofajlar) g�r�lm��t�r; ancak bunlar minimal d�zeyde olup normal maymunlarda arka plan bulgular� olarak rapor edilenlere benzer �zellik g�stermi�tir ve bu maymunlarda solunum rahats�zl���na dair herhangi bir klinik kan�t bulunmam��t�r.
Brigatinib ile karsinojenisite �al��malar� ger�ekle�tirilmemi�tir.
Brigatinib, bakteriyel ters mutasyon (Ames) veya memeli h�cre kromozomal aberasyon testlerinde in vitroda mutajenik de�ildir; ancak s��an kemik ili�i mikronukleus testinde mikronukleus say�s�n� k���k miktarda artt�rm��t�r. Mikronukleus ind�ksiyonunun mekanizmas� anormal kromozom segragasyonudur (aneugenisite) ve kromozomlar �zerinde klastojenik bir etkisi yoktur. Bu etki g�nl�k 180 mg dozda insan maruziyetinin yakla��k 5 kat�nda g�zlemlenmi�tir.
Brigatinib erkek fertilitesini bozabilir. Tekrarl�-doz hayvan �al��malar�nda testik�ler toksisite g�zlenmi�tir. S��anlardaki bulgular; d���k a��rl�kl� testisler, seminal vezik�ller ve prostat bezini ve testik�ler t�b�ler dejenerasyonu i�ermektedir; bu etkiler, iyile�me d�nemi boyunca geri d�n���ml� olmam��t�r. Maymunlardaki bulgular; mikroskobik hipospermatogenez bulgusu ile birlikte testislerin boyutunda k���lmeyi i�ermektedir; bu etkiler, iyile�me d�nemi boyunca geri d�n���ml� olmu�tur. Genel olarak; s��an ve maymunlarda erkek �reme organlar� �zerindeki bu etkiler, g�nde bir kez 180 mg'l�k doz alan hastalarda g�zlenen EAA de�erinin 0,2 kat� ve �zeri maruziyetlerde meydana gelmi�tir. S��an ve maymunlardaki genel toksikoloji �al��malar�nda di�i �reme organlar� �zerinde belirgin bir advers etki g�zlenmemi�tir.
Organogenez s�ras�nda gebe s��anlara g�nl�k brigatinib dozlar�n�n verildi�i bir embryo-f�tal geli�im �al��mas�nda; g�nde bir kez 180 mg dozda insan maruziyetindeki EEA de�erinin yakla��k 0,7 kat� gibi d���k dozlarda doza ba�l� iskelet anormallikleri g�zlenmi�tir. Bulgular aras�nda embriyo-letalite, f�tal geli�imde yava�lama ve iskelet de�i�imleri yer almaktad�r.
6. FARMAS�T�K �ZELL�KLER
6.1. Yard�mc� maddelerin listesi
�ekirdek tablet
Laktoz monohidrat (inek s�t�) Mikrokristalin sel�loz (PH-102) Sodyum ni�asta glikolat (tip A) Hidrofobik kolloidal silika Magnezyum stearat
Tablet kaplama (Opadry II Beyaz) Talk
Polietilen glikol (Makrogol) Polivinil alkol
Titanyum dioksit
6.2. Ge�imsizlikler
Ge�erli de�ildir.
6.3. Raf �mr�
36 ay
6.4. Saklamaya y�nelik �zel tedbirler
30C'nin alt�ndaki oda s�cakl���nda ve orijinal ambalaj�nda ���ktan koruyarak saklay�n�z. �ocuklar�n g�remeyece�i ve eri�emeyece�i yerlerde saklay�n�z.
6.5. Ambalaj�n niteli�i ve i�eri�i
28 film kapl� tablet, �s�l yap��mal� ka��t lamine folyo kaplamal� �effaf termoform poli-kloro-tri- fluoro-etilen (PCTFE) blister ambalajda kullanma talimat� ile birlikte karton kutuda takdim edilir.
6.6. Be�eri t�bbi �r�nden arta kalan maddelerin imhas� ve di�er �zel �nlemler
Kullan�lmam�� olan t�bbi �r�nler ya da at�k materyaller “T�bbi At�klar�n Kontrol� Y�netmeli�i” ve “Ambalaj At�klar�n�n Kontrol� Y�netmeli�i”ne uygun olarak imha edilmelidir.
Kullan�lmam�� olan �r�nler ya da at�k materyaller, yerel d�zenlemelere uygun olarak at�lmal�d�r.
Sitotoksik ve sitostatik be�eri t�bbi �r�nlerin kullan�mlar� sonucu bo�alan i� ambalajlar�n�n at�klar� TEHL�KEL� ATIKTIR ve bu at�klar�n y�netimi 2/4/2015 tarihli ve 29314 say�l� Resmî Gazetede yay�mlanan At�k Y�netimi Y�netmeli�i'ne g�re yap�l�r.
 Artrit
Artrit, olduk�a yayg�n bir hastal�kt�r ancak iyi anla��lamam��t�r. Asl�nda �artrit� tek bir hastal���n ad� de�ildir; eklem a�r�s� veya eklem hastal�klar�n� adland�rman�n gayri resmi yoludur.
Artrit
Artrit, olduk�a yayg�n bir hastal�kt�r ancak iyi anla��lamam��t�r. Asl�nda �artrit� tek bir hastal���n ad� de�ildir; eklem a�r�s� veya eklem hastal�klar�n� adland�rman�n gayri resmi yoludur. |
 A��r� Alkol Kullan�m�, Alkolizm
Alkol ba��ml�l���, alkol kullan�m� ve alkol sorunlar� aras�ndaki fark� a��klamak g��t�r.
�rne�in, ge�mi�te alkol kullanm�� olan bir kimsenin mutlaka alkol ba��ml�s� olmas�
gerekmez.
A��r� Alkol Kullan�m�, Alkolizm
Alkol ba��ml�l���, alkol kullan�m� ve alkol sorunlar� aras�ndaki fark� a��klamak g��t�r.
�rne�in, ge�mi�te alkol kullanm�� olan bir kimsenin mutlaka alkol ba��ml�s� olmas�
gerekmez. |
 |
Do�um Sonras� Depresyonu Do�um sonras� depresyonu, do�umdan sonra her on kad�ndan biri taraf�ndan tecr�be edilen stresli bir durumdur. |
 |
Grip, So�uk Alg�nl��� ve �ks�r�k Grip ve so�uk alg�nl��� (nezle) semptomlar� aras�ndaki fark� bilmek �nemlidir. So�uk alg�nl��� gripten daha hafif belirtiler g�steren bir solunum yolu hastal���d�r. |
 |
S�rt A�r�s� S�rt a�r�s� birden bire ortaya ��k�p �iddetli (akut) olabilir veya zamanla geli�ip daha uzun s�reli sorunlara (kronik) neden olabilir. |
�LA� GENEL B�LG�LER�
Takeda �la�lar� ve Ticaret Ltd.�ti.
| Sat�� Fiyat� | 20324.42 TL [ 1 Dec 2025 ] |
| �nceki Sat�� Fiyat� | 20324.42 TL [ 24 Nov 2025 ] |
| Original / Jenerik | Original �la� |
| Re�ete Durumu | K�rm�z� Re�eteli bir ila�d�r. |
| Barkodu | 8699456090110 |
| Etkin Madde | Brigatinib |
| �thal ( ref. �lke : Avusturya ) ve Be�eri bir ila�d�r. |