AMPAHO 5 mg 30 film kapl� tablet K�sa �r�n Bilgisi
{ Ambrisentan }
1. BE�ER� TIBB� �R�N�N ADI
AMPAHO 5 mg film kapl� tablet
2. KAL�TAT�F VE KANT�TAT�F B�LE��M
Etkin madde
Her tablette:
Ambrisentan: 5 mg
Yard�mc� maddeler
Laktoz monohidrat (inek s�t�nden elde edilir): 58,5 mg Kroskarmelloz sodyum: 2,7 mg
Yard�mc� maddeler i�in B�l�m 6.1'e bak�n�z.
3. FARMAS�T�K FORMU
Film tablet.
M�stahzar pembe, yuvarlak film kapl� tabletler halindedir.
4. KL�N�K �ZELL�KLER
4.1. Terap�tik endikasyonlar
AMPAHO, WHO fonksiyonel s�n�f II ve III pulmoner arteriyel hipertansiyonlu (PAH) eri�kin hastalar�n tedavisinde, kombinasyon tedavisi dahil, endikedir. �diyopatik PAH (IPAH) ve ba� doku hastal��� ile ili�kili PAH'ta etkilili�i g�sterilmi�tir.
4.2. Pozoloji ve uygulama �ekli
Pozoloji / uygulama s�kl��� ve s�resi:Tedavi, PAH tedavisinde deneyimli bir hekim taraf�ndan ba�lat�lmal�d�r.
Ambrisentan monoterapisi:
AMPAHO ba�lang�� dozu g�nl�k 5 mg'd�r. Hastan�n klinik cevab�na ve tolerabilitesine g�re doz g�nl�k 10 mg'a kadar ��kar�labilir.
Ambrisentan�n tadalafil ile kombinasyonu:
AMPAHO'nun tadalafil ile birlikte kullan�ld��� durumlarda doz g�nl�k 10 mg'a ayarlanmal�d�r.
AMBITION �al��mas�nda, hastalara tolerabiliteye ba�l� olarak 10 mg ambrisentan uygulanmadan �nce ilk 8 haftaboyuncag�nde5mgambrisentan uygulanm��t�r (bkz.B�l�m
tadalafil uygulanm��t�r. Tolerabiliteye ba�l� olarak tadalafil dozu 4 haftadan sonra 40 mg'a ve ambrisentan dozu 8 haftadan sonra 10 mg'a art�r�lm��t�r. Hastalar�n %90'�ndan fazlas�nda ba�ar�l� olunmu�tur. Dozlar ayr�ca tolerabiliteye ba�l� olarak azalt�labilmi�tir.
S�n�rl� veriler ambrisentan tedavisinin aniden kesilmesinin PAH semptomlar�n�n tekrar
k�t�le�mesi ile ili�kili olmad���n� d���nd�rmektedir.
Siklosporin A ile birlikte kullan�mda AMPAHO' nun dozu g�nl�k 5 mg ile s�n�rland�r�lmal� ve hasta dikkatle takip edilmelidir.
Uygulama �ekli:
AMPAHO tabletler oral yolla al�n�r. Tabletler b�t�n olarak yutulmal�d�r ve a� veya tok
karn�na al�nabilir. Tabletler b�l�nmemeli, ezilmemeli veya �i�nenmemelidir.
�zel pop�lasyonlara ili�kin ek bilgiler:
B�brek yetmezli�i:
B�brek yetmezli�i olan hastalarda dozun ayarlanmas� gerekli de�ildir (bkz. B�l�m 5.2). �iddetli b�brek yetmezli�i (kreatinin klirensi <30 ml/dak) olan hastalarda AMPAHO ile ilgili veriler s�n�rl�d�r; bu hasta pop�lasyonunda tedavi dikkatli bir �ekilde ba�lat�lmal� ve doz 10 mg AMPAHO'ya ��kar�ld���nda �zel dikkat g�sterilmelidir.
Karaci�er yetmezli�i:
Ambrisentan �iddetli karaci�er yetmezli�i olan (sirozun e�lik etti�i veya etmedi�i) bireylerde incelenmemi�tir. Ambrisentan metabolizmas�n�n temel yolaklar� glukuronidasyon ve daha sonra safradaki eliminasyon ile oksidasyon oldu�undan, karaci�er yetmezli�inin ambrisentan maruziyetini (Cmaks ve EAA) art�rmas� beklenebilir. Bu nedenle, �iddetli karaci�er yetmezli�i veya klinik a��dan anlaml� �ekilde y�ksek hepatik aminotransferaz de�erleri (normal �st s�n�r�n 3 kat�ndan y�ksek (>3xULN)) olan hastalarda AMPAHO tedavisine ba�lanmamal�d�r (bkz. B�l�m 4.3 ve 4.4).
Pediyatrik pop�lasyon:
18 ya��n alt�ndaki hastalarda g�venlilik ve etkilili�ine ili�kin veri bulunmad���ndan, bu hasta grubunda AMPAHO kullan�m� �nerilmemektedir (bkz.B�l�m 5.3).
Geriyatrik pop�lasyon:
65 ya� ve �zerindeki hastalarda dozun ayarlanmas� gerekli de�ildir (bkz. B�l�m 5.2).
4.3. Kontrendikasyonlar
Etkin madde
Hamile hastalarda (bkz. B�l�m 4.6),
G�venilir do�um kontrol y�ntemlerinden birini uygulamayan �ocuk do�urma potansiyeli bulunan kad�nlarda (bkz. B�l�m 4.4 ve 4.6),
Emzirme d�nemindeki kad�nlarda (bkz. B�l�m 4.6),
Siroz ile birlikte veya tek ba��na �iddetli karaci�er yetmezli�i olan hastalarda (bkz. B�l�m 4.2),
4.4.),
Sekonder pulmoner hipertansiyonla birlikte olsun ya da olmas�n idiyopatik pulmoner fibroz (IPF) hastalar�nda kullan�lmas� kontrendikedir (bkz. B�l�m 5.1).
4.4. �zel kullan�m uyar�lar� ve �nlemleri
Ambrisentan, WHO fonksiyonel s�n�f I olarak klasifiye edilen PAH hastal���nda yarar/risk dengesinin belirlenmesine yetecek say�da hasta �zerinde incelenmemi�tir.
WHO fonksiyonel s�n�f IV olarak klasifiye edilen PAH hastalar�nda monoterapi olarak ambrisentan�n etkilili�i belirlenmemi�tir. Klinik durumun k�t�le�mesi halinde, hastal���n �iddetli evresinde �nerilen tedavi (�rne�in, epoprostenol) d���n�lmelidir.
Karaci�er fonksiyonu:
Karaci�er fonksiyon anomalileri PAH ile ili�kilendirilmi�tir. Ambrisentan ile; potansiyel olarak tedavi ile ili�kili olarak altta yatan otoimm�n hepatitin alevlenmesi ihtimali, hepatik hasar ve hepatik enzim y�kselmeleri dahil olmak �zere, otoimm�n hepatit ile uyumlu vakalar g�zlenmi�tir (bkz B�l�m 4.8 ve 5.1). Bu nedenle, hepatik aminotransferazlar (ALT ve AST) ambrisentan tedavisine ba�lamadan �nce de�erlendirilmelidir ve ba�lang�� ALT ve/veya AST de�erleri normal �st limitin 3 kat�ndan y�ksek olan hastalarda AMPAHO tedavisine ba�lanmamal�d�r (bkz. B�l�m 4.3).
Hastalar klinik olarak hepatik hasar bulgular� a��s�ndan g�zlenmelidir ve ALT ile AST'nin ayl�k takibi �nerilmektedir. Hastalarda inat��, a��klanamayan, klinik a��dan anlaml� ALT ve/veya AST art��� geli�irse veya ALT ve/veya AST art���na hepatik hasar (�rne�in, sar�l�k) bulgu ve semptomlar� e�lik ediyorsa, ambrisentan tedavisi sonland�r�lmal�d�r.
Karaci�er hasar� veya sar�l��a ili�kin klinik belirtilerin bulunmad��� hastalarda hepatik enzim anomalilerinin d�zelmesini takiben ambrisentan tedavisine yeniden ba�lanmas� d���n�lebilir. B�yle durumlarda bir hepatolo�a dan���lmas� �nerilmektedir.
Hemoglobin konsantrasyonu:
Hemoglobin konsantrasyonlar� ve hematokrit de�erlerindeki d����ler ambrisentan dahil endotelin resept�r antagonisti (ERA) s�n�f� ila�lar ile ili�kilendirilmi�tir ve bazen transf�zyon gerektiren anemi ile sonu�lanabilir (bkz B�l�m 4.8). Bu d����lerin b�y�k bir b�l�m� tedavinin ilk 4 haftas� s�resince saptanm�� ve hemoglobin de�erleri genelde daha sonra stabil hale gelmi�tir. Hemoglobin konsantrasyonunda ba�lang�ca g�re ortalama d���� (0,9 g/dl ila 1,2 g/dl) pivotal Faz 3 klinik �al��malar�n uzun s�reli a��k etiketli uzatma evrelerinde tedavinin 4. y�l�na dek s�rm��t�r. Pazarlama sonras� d�nemde, kan h�cresi transf�zyonu gerektiren anemi vakalar� rapor edilmi�tir (bkz. B�l�m 4.8).
Klinik a��dan anlaml� anemi tan�s� konulan hastalarda AMPAHO tedavisine ba�lanmas� �nerilmemektedir. AMPAHO tedavisine ba�lanmadan �nce, tedavi s�ras�nda (�rne�in; 1. ayda, ve 3. ayda) ve klinik uygulamaya ba�l� olarak daha sonra periyodik �ekilde hemoglobin ve/veya hematokrit d�zeylerinin �l��lmesi �nerilmektedir. Hemoglobin veya hematokrit d�zeylerinde klinik a��dan anlaml� d���� g�zlenir ve di�er nedenler elimine edilirse dozun azalt�lmas� veya tedavinin kesilmesi d���n�lmelidir. Anemi insidans�, ambrisentan ve tadalafilin monoterapi olarak verilmesinde g�r�len anemi insidans�na k�yasla (s�ras�yla %7 ve %11) ambrisentan tadalafil ile kombinasyon halinde uyguland���nda artm��t�r (%15 advers olay s�kl���).
![]()
Ambrisentan dahil ERA ile periferik �dem g�zlenmi�tir. Ambrisentan ile yap�lan klinik �al��malarda �o�u periferik �dem vakas� hafif ila orta �iddette olmu�tur. Buna kar��n 65 ya� ve �st� hastalarda periferik �dem s�kl��� ve �iddeti daha fazla g�r�lebilir. K�sa d�nem klinik �al��malarda periferik �dem 10 mg ambrisentan dozuyla daha s�k �ekilde bildirilmi�tir (bkz. B�l�m 4.8).
Ambrisentan tedavisine ba�land�ktan sonraki birka� hafta i�erisinde s�v� tutulumu vakalar�na ili�kin pazarlama sonras� raporlar al�nm�� ve baz� vakalarda s�v� dengesinin d�zenlenmesi veya dekompanse kalp yetmezli�i i�in bir di�retik kullan�m� veya hastane yat��� gerekmi�tir. Hastalarda �nceden mevcut a��r� s�v� y�k� mevcutsa, ambrisentan tedavisine ba�lanmadan �nce bu sorun klinik olarak uygun �ekilde d�zeltilmelidir.
Ambrisentan tedavisi s�ras�nda kilo art��� ile birlikte veya kilo art��� olmaks�z�n klinik olarak anlaml� s�v� tutulumu meydana gelirse, bu durumun ambrisentana veya altta yatan kalp yetmezli�ine ba�l� olup olmad���n�n ve �zel bir tedaviye ya da ambrisentan tedavisinin kesilmesine gerek olup olmad���n�n belirlenmesi i�in ek de�erlendirmeler yap�lmal�d�r.Periferik �dem insidans�, ambrisentan ve tadalafilin monoterapi olarak verilmesinde g�r�len periferik �dem insidans�na k�yasla (s�ras�yla %38 ve %28), ambrisentan tadalafil ile kombinasyon halinde uyguland���nda artm��t�r (%45 advers olay s�kl���). Periferik �dem en fazla tedavinin ba�lat�ld��� ilk ay i�inde g�zlenmi�tir.
�ocuk do�urma potansiyeli olan kad�nlar:
AMPAHO �ocuk do�urma potansiyeli olan kad�nlarda tedavi �ncesi hamilelik testi negatif ��kmad�ysa ve g�venli kontrasepsiyon uygulanm�yorsa kullan�lmamal�d�r. Hastaya uygun kontrasepsiyon ile ilgili herhangi bir ��phe olmas� durumunda kad�n do�um uzman�na dan���lmas� �nerilmektedir. Ambrisentan ile tedavi s�resince ayl�k gebelik testinin yap�lmas� �nerilmektedir (bkz.B�l�m 4.3 ve 4.6).
Pulmoner venookl�zif hastal�k:
Pulmoner venookl�zif hastal��� olan hastalarda endotelin resept�r antagonistleri gibi vazodilatat�r ila�lar ile pulmoner �dem olgular� bildirilmi�tir. Ambrisentan ile tedavi s�ras�nda PAH hastalar�nda akut pulmoner �dem geli�irse, pulmoner venookl�zif hastal�k olas�l��� d���n�lmelidir.
Di�er t�bbi �r�nlerle birlikte kullan�m�:
Ambrisentan kullanmakta olan hastalarda rifampisin tedavisi ba�lat�lmas� durumunda hastan�n yak�n takibi gerekir (bkz. B�l�m 4.5 ve 5.2).
Yard�mc� maddeler
AMPAHO laktoz monohidrat i�ermektedir. Bu nedenle; nadir kal�t�msal galaktoz intolerans�, Lapp laktaz yetmezli�i ya da glukoz-galaktoz malabsorpsiyon problemi olan hastalar�n bu ilac� kullanmamalar� gerekir.
Bir tablet AMPAHO i�eri�inde 23 mg'dan daha az sodyum bulunmaktad�r. Yani esas�nda
sodyum i�ermez.
4.5. Di�er t�bbi �r�nler ile etkile�imler ve di�er etkile�im �ekilleri
konsantrasyonlarda Faz I veya II ila� metabolize edici enzimleri inhibe etmez veya ind�klemez. Bu durum, ambrisentan�n bu yolaklar ile metabolize edilen t�bbi �r�nlerin profilini de�i�tirme potansiyelinin d���k oldu�unu d���nd�rmektedir.
Ambrisentan�n CYP3A4 aktivitesini ind�kleme potansiyeli sa�l�kl� g�n�ll�lerde ara�t�r�lm�� ve ambrisentan�n CYP3A4 izoenzimi �zerinde ind�kleyici etkiye sahip olmad���n� d���nd�ren bulgular elde edilmi�tir.
Siklosporin A:
Sa�l�kl� g�n�ll�lerde, kararl� durumda ambrisentan ve siklosporin A'n�n e� zamanl� uygulanmas� ambrisentan maruziyetini 2 kat art�rm��t�r. Bunun nedeni ambrisentan farmakokineti�inde etkili ta��y�c�lar�n ve metabolik enzimlerin siklosporin A taraf�ndan inhibisyonu olabilir. Bu nedenle, ambrisentan dozu siklosporin A ile birlikte kullan�ld���nda g�nde 5 mg ile s�n�rland�r�lmal�d�r (bkz. B�l�m 4.2). Ambrisentan�n �oklu dozlar� siklosporin A maruziyeti �zerinde etkili olmam��t�r ve bu nedenle siklosporin A'n�n dozunun ayarlanmas� gerekli de�ildir.
Rifampisin
Sa�l�kl� g�n�ll�lerde, rifampisin (organik anyon ta��y�c� polipeptit (OATP) inhibit�r�, g��l� CYP3A ve 2C19 ind�kleyicisi, P-gp ve �ridin-difosfo-glukuronosiltransferaz [UGT] ind�kleyicisi) ile e� zamanl� uygulama ilk doz ard�ndan ambrisentan maruziyetini ge�ici olarak art�rm��t�r (yakla��k 2 kat). Bununla birlikte, 8. g�ne kadar kararl� durumda rifampisin verilmesinin ambrisentan maruziyeti �zerinde klinik a��dan �nemli etkisi g�r�lmemi�tir.. Ambrisentan kullanmakta olan hastalarda rifampisin tedavisi ba�lat�lmas� durumunda hastan�n yak�n takibi gerekir (bkz. B�l�m 4.4 ve 5.2).
Fosfodiesteraz inhibit�rleri:
Sa�l�kl� g�n�ll�lerde ambrisentan�n, ister sildenafil isterse tadalafil (her ikisi de CYP3A4 substrat�d�r) olsun, bir fosfodiesteraz inhibit�r� ile birlikte verilmesi ne fosfodiesteraz inhibit�r�n�n ne de ambrisentan�n farmakokinetiklerini anlaml� bir �ekilde etkilememi�tir (bkz. B�l�m 5.2).
Di�er hedeflendirilmi� PAH tedavileri:
PAH tedavisi i�in di�er ila�larla (�rne�in, prostanoidler ve ��z�n�r guanilat siklaz stim�lat�rleri) birlikte uyguland���nda ambrisentan�n etkilili�i ve g�venlili�i PAH hastalar�nda y�r�t�len kontroll� klinik �al��malarda �zel olarak ara�t�r�lmam��t�r (bkz. B�l�m 5.1). Bilinen biyotransformasyon verileri temelinde ��z�n�r guanilat siklaz stim�lat�rleri veya prostanoidler ile spesifik ila�-ila� etkile�imleri beklenmemektedir (bkz. B�l�m 5.2). Bununla birlikte, bu ila�larla �zel ila�-ila� etkile�imi �al��malar� y�r�t�lmemi�tir. Bu nedenle, bu ila�lar�n birlikte dikkatli �ekilde kullan�lmas� �nerilmektedir.
Oral kontraseptifler:
Sa�l�kl� g�n�ll�lerde y�r�t�len bir klinik �al��mada, g�nde bir kez ambrisentan 10 mg ile sa�lanan kararl� durumda kombine oral kontraseptiflerin etinil �stradiol ve noretindron bile�enlerinin tek doz farmakokineti�i anlaml� derecede etkilenmemi�tir (bkz.B�l�m 5.2). Bu farmakokinetik �al��maya dayanarak, ambrisentan�n �strojen ya da progestojen i�eren temelli kontraseptiflerin maruziyetini anlaml� derecede etkilemesi beklenmez.
Varfarin:
farmakokineti�ini ve antikoag�lan aktivitesini etkilememi�tir (bkz. B�l�m 5.2). Varfarin ayr�ca ambrisentan farmakokineti�i �zerinde klinik a��dan anlaml� bir etkiye sahip olmam��t�r. Buna ek olarak; ambrisentan�n hastalarda haftada bir al�nan varfarin tipi antikoag�lan dozu, protrombin zaman� (PT) ve uluslararas� normalle�tirilmi� oran (INR) �zerinde genel bir etkisi g�zlenmemi�tir.
Ketokonazol
Kararl� durumda ketokonazol (g��l� bir CYP3A4 inhibit�r�) uygulanmas� ambrisentan maruziyetinde klinik a��dan anlaml� bir art��a neden olmam��t�r (bkz. B�l�m 5.2).
Ambrisentan�n ksenobiyotik ta��y�c�lar �zerindeki etkisi
�n vitro olarak; klinik a��dan uygun konsantrasyonlarda ambrisentan�n P-glikoprotein (Pgp), meme kanseri diren� proteini (BCRP), �oklu ila� direnci ile ili�kili protein 2 (MRP2), safra tuzu d��ar� at�m pompas� (BSEP), organik anyon transport polipeptitleri (OATP1B1 ve OATP1B3) ve sodyum ba��ml� taurokolat ko-transporter polipeptit (NTCP) dahil insan ta��y�c�lar�nda inhibe edici etkisi yoktur.
Ambrisentan, bir P-gp-arac�l� d��ar� ak�� substrat�d�r.
S��an hepatositlerinde ger�ekle�tirilen in vitro �al��malar ayr�ca ambrisentan�n P-gp, BSEP veya MRP2 protein ekspresyonunu ind�klemedi�ini g�stermi�tir.
Sa�l�kl� g�n�ll�lerde kararl� durumda ambrisentan uygulanmas�, bir Pgp substrat� olan digoksinin tek doz farmakokineti�i �zerinde klinik a��dan anlaml� etki g�stermemi�tir (bkz. B�l�m 5.2).
4.6. Gebelik ve laktasyon
Gebelik kategorisi: X
�ocuk do�urma potansiyeli bulunan kad�nlar/Do�um kontrol� (Kontrasepsiyon)
Tedavi �ncesi yap�lan gebelik testi negatif de�ilse ve g�venilir do�um kontrol y�ntemi uygulanm�yorsa, �ocuk do�urma potansiyeli bulunan kad�nlarda AMPAHO tedavisine ba�lanmamal�d�r. Bireysel olarak hastalarda hangi kontraseptif y�ntemin kullan�laca��na karar verilemiyorsa bir jinekolo�a dan���lmal�d�r. AMPAHO tedavisi s�ras�nda ayda bir gebelik testi yap�lmal�d�r (bkz. B�l�m 4.3).
Gebelik d�nemi
AMPAHO gebelik d�neminde kontrendikedir (bkz. B�l�m 4.3). Hayvanlarda yap�lan �al��malar ambrisentan�n teratojenik oldu�unu g�stermi�tir. �nsanlarda deneyim mevcut de�ildir.
AMPAHO kullanan kad�nlar fetal hasar riski konusunda uyar�lmal� ve gebe kald�klar� taktirde alternatif tedaviye ba�lanmal�d�r (bkz.B�l�m 4.3, 4.4 ve 5.3)
Laktasyon d�nemi
Ambrisentan�n anne s�t�ne ge�ip ge�medi�i bilinmemektedir. Hayvanlarda ambrisentan�n anne s�t�ne ge�i�i incelenmemi�tir. Bu nedenle, laktasyon s�ras�nda AMPAHO kullan�m� kontrendikedir (bkz. B�l�m 4.3).
�reme yetene�i/Fertilite
Erkek hayvanlarda, ambrisentan dahil kronik ERA uygulamas� testik�ler t�b�ler atrofi geli�imi ile ili�kilendirilmi�tir (bkz. B�l�m 5.3). ARIES-E �al��mas�nda ambrisentana uzun s�reli maruziyetin sperm say�s� �zerindeki zararl� etkisine ili�kin net bir kan�ta rastlanmasa da kronik ambrisentan uygulamas� spermatogenez belirte�lerinde de�i�iklikler ile ili�kilendirilmi�tir. Plazma inhibin-B konsantrasyonunda bir azalma ve plazma FSH konsantrasyonunda bir art�� g�zlenmi�tir. Erkek insan fertilitesi �zerindeki etki bilinmemekle birlikte, spermatogenezde bozulma g�z ard� edilemez. Klinik �al��malarda kronik ambrisentan uygulamas� plazma testosteronunda bir de�i�iklik ile ili�kilendirilmemi�tir.
4.7. Ara� ve makine kullan�m� �zerindeki etkiler
Ambrisentan, ara� ve makine kullanma becerisi �zerinde hafif veya orta �iddette bir etkiye sahiptir. Hastan�n muhakeme, motor veya bili�sel yetenekler gerektiren g�revleri yerine getirme kapasitesi de�erlendirilirken hastan�n klinik durumu ve ambrisentan�n advers reaksiyon profili (hipotansiyon, ba� d�nmesi, asteni, yorgunluk gibi) ak�lda bulundurulmal�d�r (bkz. B�l�m 4.8). Hastalar, ara� veya makine kullanmadan �nce ambrisentandan nas�l etkilenebilecekleri konusunda bilgilendirilmelidir.
4.8. �stenmeyen etkiler
G�venlilik profilinin �zeti:
Ambrisentan�n g�venlili�i, monoterapi ve/veya kombinasyon halinde, PAH'� bulunan 1200'den fazla hastan�n dahil edildi�i klinik �al��malarda de�erlendirilmi�tir (bkz. B�l�m 5.1.). 12 hafta s�reli plasebo kontroll� klinik �al��ma verilerine dayanarak tan�mlanm�� olan advers reaksiyonlar ortaya ��kt�klar� sistem organ s�n�f�na ve g�r�lme s�kl�klar�na g�re a�a��da yer almaktad�r. Daha uzun s�reli plasebo kontroll� olmayan �al��malardan (ARIES-E ve AMBITION (tadalafil ile kombinasyon halinde)) elde edilen bilgiler de a�a��da sunulmaktad�r. Uzun s�reli tedavi ile veya tadalafil ile kombinasyon halinde ambrisentan i�in daha �nce bilinmeyen advers reaksiyonlar tan�mlanmam��t�r. Daha uzun g�zlem s�relerine sahip kontroll� olmayan �al��malarda belirlenen g�venlilik profilinin (ortalama g�zlem s�resi 79 hafta) k�sa s�reli �al��malarda g�zlenene benzer oldu�u bulunmu�tur. Rutin farmakovijilans verileri de ayr�ca sunulmaktad�r.
Ambrisentan ile g�zlemlenen en yayg�n advers reaksiyonlar periferal �dem, s�v� tutulmas� ve ba� a�r�s� (sin�s ba� a�r�s� ve migren dahil olmak �zere) olmu�tur. Daha y�ksek doz (10 mg) bu advers reaksiyonlar i�in daha y�ksek insidans ile ili�kili bulunmu�tur ve k�sa d�nem klinik ara�t�rmalarda periferal �dem 65 ya� ve �zeri hastalarda daha �iddetli g�r�lme e�iliminde olmu�tur(bkz. B�l�m 4.4).
S�kl�k ifadeleri �u �ekilde tan�mlanm��t�r: �ok yayg�n (≥1/10), yayg�n (≥1/100 ila <1/10), yayg�n olmayan (≥1/1.000 ila <1/100), seyrek (≥1/10.000 ila <1/1.000), �ok seyrek (<1/10.000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). Doz ile ili�kili advers reaksiyonlar i�in s�kl�k kategorisi daha y�ksek ambrisentan dozunu yans�tmaktad�r. S�kl�k kategorileri; �al��ma s�relerindeki farkl�l�klar, �nceden var olan di�er durumlar ve �al��man�n ba�lang�c�ndaki hasta �zellikleri gibi fakt�rlere a��klama getirmemektedir. Klinik ara�t�rma deneyimlerine ba�l� olarak belirlenen advers reaksiyon s�kl�k kategorileri, normal klinik pratikte ortaya ��kan advers reaksiyonlar�n s�kl���n� yans�tmayabilir. Advers reaksiyonlar, s�kl�k kategorilerine g�re listelenmi�tir.
Ambrisentan ile tedavide ARIES-E ve AMBITION �al��malar�ndan elde edilen istenmeyen etkiler:
Kan ve lenf sistemi hastal�klar�
�ok yayg�n: Anemi (hemoglobin ve/veya hematokrit seviyesinin azalmas�)
Ba����kl�k sistemi hastal�klar�
Yayg�n: A��r� duyarl�l�k reaksiyonlar� (�rne�in, anjiyo�dem, deride d�k�nt�, ka��nt�)
Sinir sistemi hastal�klar�
�ok yayg�n: Ba� a�r�s� (sin�s ba� a�r�s�, migren dahil), ba� d�nmesi
G�z hastal�klar�
Yayg�n: G�rme bozuklu�u (bulan�k g�rme, g�rmede azalma dahil)
Kardiyak hastal�klar� �ok yayg�n: Palpitasyon Yayg�n: Kalp yetmezli�i
Vask�ler hastal�klar�
Yayg�n: S�cak basmas�, hipotansiyon, senkop
Solunum, g���s bozukluklar� ve mediastinal hastal�klar�
�ok yayg�n: Dispne, nazofarenjit, nazal konjesyon Yayg�n: Epistaksis, sin�zit, rinit
Gastrointestinal hastal�klar�
�ok yayg�n: Bulant�, diyare
Yayg�n: Kar�n a�r�s�, konstipasyon, kusma
Deri ve deri alt� doku hastal�klar�
Yayg�n: Deride d�k�nt�
Genel bozukluklar ve uygulama b�lgesine ili�kin hastal�klar�
�ok yayg�n: Periferik �dem, s�v� tutulumu, yorgunluk Yayg�n: G���s a�r�s�/g���ste rahats�zl�k hissi, asteni
Ambrisentan�n tadalafil ile kombinasyonunda AMBITION �al��mas�ndan elde edilen
istenmeyen etkiler:
Kan ve lenf sistemi hastal�klar�
�ok yayg�n: Anemi (hemoglobin ve/veya hematokrit seviyesinin azalmas�)
Ba����kl�k sistemi hastal�klar�
Yayg�n: A��r� duyarl�l�k reaksiyonlar� (�rne�in, anjiyo�dem, deride d�k�nt�, ka��nt�)
Sinir sistemi hastal�klar�
�ok yayg�n: Ba� a�r�s� (sin�s ba� a�r�s�, migren dahil), ba� d�nmesi
G�z hastal�klar�
Yayg�n: G�rme bozuklu�u (bulan�k g�rme, g�rmede azalma dahil)
Kulak ve i� kulak hastal�klar�
Yayg�n: Tinnitus
Yayg�n olmayan: Ani duyma kayb�
Kardiyak hastal�klar� �ok yayg�n: Palpitasyon Yayg�n: Kalp yetmezli�i
Vask�ler hastal�klar�
�ok yayg�n: S�cak basmas� Yayg�n: Hipotansiyon, senkop
Solunum, g���s bozukluklar� ve mediastinal hastal�klar�
�ok yayg�n: Dispne, nazofarenjit, nazal konjesyon Yayg�n: Epistaksis, sin�zit, rinit
Gastrointestinal hastal�klar�
�ok yayg�n: Bulant�, diyare, kusma Yayg�n: Kar�n a�r�s�, konstipasyon
Deri ve deri alt� doku hastal�klar�
Yayg�n: Deride d�k�nt�
Genel bozukluklar ve uygulama b�lgesine ili�kin hastal�klar�
�ok yayg�n: Periferik �dem, s�v� tutulumu, yorgunluk, g���s a�r�s�/g���ste rahats�zl�k hissi Yayg�n: Asteni
Ambrisentan ile tedavide ARIES-C ve pazarlama sonras� �al��malardan elde edilen istenmeyen
etkiler:
Kan ve lenf sistemi hastal�klar�
Yayg�n: Anemi (hemoglobin ve/veya hematokrit seviyesinin azalmas�)
Ba����kl�k sistemi hastal�klar�
Yayg�n olmayan: A��r� duyarl�l�k reaksiyonlar� (�rne�in, anjiyo�dem, deride d�k�nt�, ka��nt�)
Sinir sistemi hastal�klar�
�ok yayg�n: Ba� a�r�s� (sin�s ba� a�r�s�, migren dahil) Yayg�n: Ba� d�nmesi
G�z hastal�klar�
Bilinmiyor: G�rme bozuklu�u (bulan�k g�rme, g�rmede azalma dahil)
Kardiyak hastal�klar�
Yayg�n: Kalp yetmezli�i, palpitasyon
Vask�ler hastal�klar�
Yayg�n: S�cak basmas�, hipotansiyon Yayg�n olmayan: Senkop
Solunum, g���s bozukluklar� ve mediastinal hastal�klar�
nazofarenjit, rinit, sin�zit
Gastrointestinal hastal�klar�
Yayg�n: Kar�n a�r�s�, konstipasyon, bulant�, kusma, diyare
Hepato-bilier hastal�klar�
Yayg�n: Hepatik transaminaz y�kselmesi
Yayg�n olmayan: Hepatik hasar, otoimmun hepatit (bkz. B�l�m 4.4)
Genel bozukluklar ve uygulama b�lgesine ili�kin hastal�klar�
�ok yayg�n: Periferik �dem, s�v� tutulumu
Yayg�n: Asteni, yorgunluk, g���s a�r�s�/g���ste rahats�zl�k hissi
Se�ilmi� advers reaksiyonlar�n tan�m�
Hemoglobin seviyesinde azalma:
Pazarlama sonras� d�nemde kan h�cresi transf�zyonu gerektiren anemi vakalar� raporlanm��t�r (bkz. B�l�m 4.4). Hemoglobin seviyesinde azalma (anemi) s�kl���n�n 10 mg'l�k ambrisentan dozu ile daha y�ksek oldu�u g�zlenmi�tir. 12 haftal�k plasebo kontroll� Faz 3 klinik �al��malarda, ortalama hemoglobin konsantrasyonu ambrisentan alan hasta gruplar�nda azalm�� ve bu azalma 4. hafta gibi erken bir s�rede saptanm��t�r (azalma 0,83 g/dl); ba�lang�ca g�re ortalama de�i�im sonraki 8 haftal�k s�rede stabil kalm��t�r. Ambrisentan tedavisi alan gruptaki toplam 17 hastada (%6,5) hemoglobin de�erinde ba�lang�ca g�re %15 ve daha fazla azalma g�zlenmi� ve daha sonra bu de�er normal s�n�r�n alt�na inmi�tir.
��pheli advers reaksiyonlar�n raporlanmas�:
Ruhsatland�rma sonras� ��pheli ila� advers reaksiyonlar�n�n raporlanmas� b�y�k �nem ta��maktad�r. Raporlama yap�lmas�, ilac�n yarar/risk dengesinin s�rekli olarak izlenmesine olanak sa�lar. Sa�l�k mesle�i mensuplar�n�n herhangi bir ��pheli advers reaksiyonu T�rkiye Farmakovijilans Merkezi (T�FAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e-posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks: 0 312 218 35 99).
4.9. Doz a��m� ve tedavisi
G�nl�k 10 mg'�n �st�nde ambrisentandozlar�iletedaviedilenPAH hastalar� ile ilgili deneyim
mevcut de�ildir. Sa�l�kl� g�n�ll�lerde tek dozluk 50 ve 100 mg'l�k uygulamalar (�nerilen maksimum dozun 5 ila 10 kat�) ba� a�r�s�, s�cak basmas�, ba� d�nmesi, bulant� ve nazal konjesyon ile ili�kili olarak bulunmu�tur.
Etki mekanizmas� nedeniyle, AMPAHO doz a��m� potansiyel olarak hipotansiyona yol a�abilir (bkz. B�l�m 5.3). Belirgin hipotansiyon vakalar�nda aktif kardiyovask�ler deste�e ihtiya� duyulabilir. Bilinen spesifik bir antidotu yoktur.
5. FARMAKOLOJ�K �ZELL�KLER
5.1. Farmakodinamik �zellikler
Farmakoterap�tik grup: Pulmoner arteriyel hipertansiyon i�in antihipertansifler
ATC kodu: C02KX02
Etki mekanizmas�:
Ambrisentan; oral uygulamaya y�nelik, endotelin A (ET) resept�r� i�in selektif bir propanoik
asit s�n�f� ERA'd�r. Endotelin, PAH patofizyolojisinde �nemli bir role sahiptir.
Ambrisentan g��l� (Ki 0,016 nM) ve y�ksek oranda se�ici bir ETA antagonistidir (ETB ile kar��la�t�r�ld���nda ETA se�icili�i yakla��k 4.000 kat daha fazlad�r).
Ambrisentan, temelde vask�ler d�z kas h�creleri ve kalp kas� h�crelerinde bulunan ETA resept�r alt tipini bloke etmektedir. Bu blokaj, vazokonstriksiyon ve d�z kas h�cre proliferasyonuna neden olan ikinci haberci sistemlerin endotelin arac�l� aktivasyonunu �nler.
Ambrisentan�n ET'ye k�yasla ET resept�r� selektivitesinin ET resept�r� arac�l� vazodilat�r nitrik oksit ve prostasiklin �retimini korumas� beklenmektedir.
Klinik etkililik ve g�venlik:
�ki randomize, �ift k�r, �ok merkezli, plasebo kontroll� Faz 3 pivotal �al��ma (ARIES-1 ve 2) yap�lm��t�r. ARIES-1 �al��mas�nda 201 hasta yer alm�� ve ambrisentan 5 mg ve 10 mg ile plasebo kar��la�t�r�lm��t�r. ARIES-2 �al��mas�nda 192 hasta yer alm�� ve ambrisentan 2,5 mg ve 5 mg ile plasebo kar��la�t�r�lm��t�r. Her iki �al��mada ambrisentan hastalar�n digoksin, antikoag�lanlar, di�retikler, oksijen ve vazodilat�rler (kalsiyum kanal blokerleri, ACE inhibit�rleri) kombinasyonunu i�erebilen destekleyici/temel ilaca eklenmi�tir. Dahil edilen hastalarda ba� doku hastal�klar� ile ili�kili PAH veya �PAH mevcuttur (PAH-CTD). Hastalar�n �o�unda WHO fonksiyonel s�n�f II (% 38,4) veya s�n�f III (% 55) semptomlar� mevcuttur. �nceden mevcut karaci�er hastal��� (siroz veya klinik a��dan anlaml� �ekilde artan aminotransferaz) ve PAH i�in di�er hedefe y�nelik tedavilerin (�rne�in, prostanoidler) kullan�ld��� hastalar �al��maya dahil edilmemi�tir. Bu �al��malarda hemodinamik parametreler de�erlendirilmemi�tir.
Faz 3 �al��malar i�in tan�mlanan primer sonlanma noktas�, ba�lang�ca g�re 6 dakikal�k y�r�me mesafesinde (6DYM) 12. haftada g�r�len de�i�iklik ile de�erlendirilen egzersiz kapasitesindeki d�zelme olmu�tur. Her iki �al��ma da ambrisentan tedavisi t�m ambrisentan dozlar� i�in 6DYM'de anlaml� d�zelme sa�lam��t�r.
d�zelme 51,4 m olmu�tur (% 95 GA: 26,6 ila 76,2; p <0,001).
Faz 3 �al��malar�n �nceden tan�mlanm�� bir birle�ik analizi (ARIES-C) yap�lm��t�r. 6DYM'de plaseboya g�re ayarlanm�� ortalama d�zelme 5 mg dozu i�in 44,6 m (% 95 GA: 24,3 ile 64,9; p<0,001) ve 10 mg dozu i�in 52,5 m olmu�tur (% 95 GA: 28,8 ila 76,2; p<0,001).
ARIES-2 �al��mas�nda; ambrisentan tedavisi (kombine doz grubu), plasebo ile kar��la�t�r�ld���nda, PAH'�n klinik k�t�le�mesini anlaml� �ekilde geciktirmi�tir (p<0,001) ve risk oran� (HR) %80'lik d���� oldu�unu g�stermi�tir (%95 GA; %47 ila 92). �l��m kriterleri; �l�m, akci�er transplantasyonu, PAH nedeniyle hospitalizasyon, atriyal septostomi, PAH tedavisinde kullan�lan di�er ajanlar�n tedaviye eklenmesi ve tedaviyi erken b�rakma kriterlerini i�ermi�tir. Plasebo ile kar��la�t�r�ld���nda, kombine doz tedavi grubunda SF-36 Sa�l�k Anketinin fiziksel fonksiyon �l�e�inde istatistiksel olarak anlaml� �ekilde y�kselme ortaya ��kt��� g�zlenmi�tir (-0,20±8,14'e kar��l�k 3,41±6,96, p=0,005). Ambrisentan tedavisi ile 12. haftada Borg Dispne �ndeksinde (BDI) istatistiksel olarak anlaml� �ekilde iyile�me ortaya ��km��t�r (plaseboya g�re d�zeltilmi� BDI; -1,1 [%95 GA: -1,8 ila -0,4; p=0,019; kombine doz grubu]).
Uzun s�reli veriler:
ARIES 1 ve 2 �al��malar�na dahil edilen hastalar, bu �al��malar� takip eden uzun s�reli, a��k etiketli bir uzatma �al��mas�na (ARIES- E) devam etmek �zere uygun bulunmu�tur (n=383).
Kombine ortalama maruziyet 145 ± 80 hafta ve maksimum maruziyet yakla��k 295 haftaolmu�tur. Bu �al��man�n temel birincil sonlanma noktalar�, serum LFT'leri de dahil olmak �zere uzun s�reli ambrisentan maruziyeti ile ili�kili advers olaylar�n insidans� ve �iddeti olmu�tur. Uzun vadeli ambrisentan maruziyeti ile g�zlemlenen g�venlilik bulgular�, genellikle 12 haftal�k plasebo kontroll� �al��malarda g�zlemlenenlerle uyumlu olmu�tur.
Ambrisentan kullanan deneklerde g�zlenen sa�kal�m oranlar� (kombine ambrisentan dozu
grubu) bir y�ll�k s�rede % 93, iki y�ll�k s�rede % 85 ve 3. Y�ll�k s�rede ise %79 olmu�tur.
A��k bir �al��mada (AMB222), ambrisentan aminotransferaz anomalileri nedeniyle daha �nce di�er ERA tedavisinin kesildi�i hastalarda, serum aminotransferaz konsantrasyonlar�nda art���n insidans�n�n de�erlendirilmesi i�in 36 hastada incelenmi�tir. Ambrisentan ile ortalama 53 haftal�k tedavi s�ras�nda dahil edilen hastalar�n hi�birisinde tedavinin kesilmesini gerektirecek derecede do�rulanm�� serum ALT>3xULN bulgusu g�zlenmemi�tir. Bu s�re zarf�nda hastalar�n % 50'sinde 5 mg ambrisentan dozunun 10 mg dozuna artt�r�lmas� gerekmi�tir.
T�m Faz 2 ve 3 �al��malarda (ilgili a��k ek �al��malar dahil) >3xULN serum aminotransferaz anomalisinin k�m�latif insidans� ortalama 79,5 haftal�k maruziyet s�resinde 17/483 g�n�ll� olmu�tur. Bu, ambrisentan i�in 100 hasta y�l� ba��na 2,3 olay oran�na kar��l�k gelmektedir. A��k etiketli uzun s�reli ARIES-E uzatma �al��mas�nda, ambrisentan ile tedavi edilen hastalarda
>3xULN serum aminotransferaz y�kselmesi olu�umunun 2 y�ll�k riski %3,9 olmu�tur.
Di�er klinik bilgiler:
Bir Faz 2 �al��mada (n=29) (AMB220), PAH hastalar�nda 12 hafta sonra hemodinamik parametrelerde bir d�zelme g�zlenmi�tir. Ambrisentan tedavisi ortalama kardiyak endekste bir art��a, ortalama pulmoner arter bas�nc�nda bir d����e ve ortalama pulmoner vask�ler diren�te bir d����e neden olmu�tur.
hafta s�reli plasebo kontroll� klinik �al��malarda, ba�lang��tan tedavinin sonuna kadar sistolik ve diyastolik kan bas�n�lar�ndaki ortalama azalma s�ras�yla 3 mm Hg ve 4,2 mm Hg olmu�tur. Sistolik ve diyastolik kan bas�n�lar�ndaki ortalama azalmalar, uzun vadeli a��k etiketli ARIES- E �al��mas�nda, ambrisentan ile tedavide 4 y�la kadar devam etmi�tir.
Sa�l�kl� g�n�ll�lerde ger�ekle�tirilmi� olan bir ila�-ila� etkile�imi �al��mas�nda, ambrisentan ya da sildenafilin farmakokinetik �zelliklerinde klinik olarak anlaml� bir de�i�iklik olmad��� ve kombinasyon tedavisinin iyi tolere edildi�i g�zlenmi�tir. ARIES-E �al��mas�nda 22 hasta (%5,7), AMB222 �al��mas�nda ise 17 hasta (%47) ambrisentan ile birlikte sildenafil kullanm��t�r. Bu hasta pop�lasyonunda g�venlili�e dair ek kayg�lar ortaya ��kmam��t�r.
Tadalafil ile kombinasyon halinde klinik etkililik:
Ambrisentan ve tadalafil ba�lang�� kombinasyonunun tek ba��na ambrisentan veya tadalafil monoterapisine kar�� etkilili�ini de�erlendirmek �zere daha �nce tedavi g�rmemi�, s�ras�yla 2:1:1 oran�nda randomize edilmi� 500 PAH hastas�nda �ok merkezli, �ift k�r, aktif komparat�rl�, olay y�nlendirmeli bir Faz 3 sonu� �al��mas� (AMB112565/AMBITION) y�r�t�lm��t�r. Hi�bir hasta tek ba��na plasebo kullanmam��t�r. Birincil analiz birle�tirilmi� monoterapi gruplar�na kar�� kombinasyon grubudur. Ayr� ayr� monoterapi gruplar�na kar�� kombinasyon tedavisi grubuna ili�kin destekleyici kar��la�t�rmalar da yap�lm��t�r. Ciddi anemi, s�v� tutulumu veya seyrek retinal hastal�klar� olan hastalar ara�t�r�c� kriterlerine g�re hari� tutulmu�tur. Ba�lang��ta ALT ve AST de�erleri >2xULN olan hastalar da hari� tutulmu�tur.
Ba�lang��ta, hastalar�n %96's� daha �nce PAH'a �zg� tedavi g�rmemi� olup, tan�dan �al��maya giri�e kadar ge�en medyan s�re 22 g�nd�r. Hastalar ambrisentan 5 mg ve tadalafil 20 mg dozuna ba�lat�lm�� ve tolerabilite sorunlar� ya�amad�klar� s�rece 4. haftada 40 mg tadalafil ve
8. haftada 10 mg ambrisentana titre edilmi�tir. Kombinasyon tedavisi i�in medyan �ift k�r
tedavi s�resi 1,5 y�ldan fazla olmu�tur.
Birincil sonlan�m noktas� a�a��daki �ekilde tan�mlanan ilk klinik ba�ar�s�zl�k olay�na kadar
ge�en s�re olmu�tur:
�l�m veya
PAH'da k�t�le�me nedeniyle hastaneye yat�r�lma,
Hastal�k progresyonu,
Tatmin edici olmayan uzun vadeli klinik yan�t.
T�m hastalar i�in ortalama ya� 54 olmu�tur (SD 15; aral�k 18-75 ya�). Ba�lang��ta hastalar�n WHO FC de�eri II (%31) ve FC III (%69) olmu�tur. �diyopatik veya kal�t�msal PAH �al��ma pop�lasyonundaki en yayg�n etiyoloji olup (%56), bunu ba� dokusu bozukluklar�na ba�l� PAH (%37), ila�lar ve toksinler ile ili�kili PAH (%3), d�zeltilmi� basit konjenital kalp hastal��� ile ili�kili PAH (%2) ve HIV ile ili�kili PAH (%2)izlemi�tir. WHO FC II ve III durumuna sahip hastalar ba�lang��ta ortalama 353 metrelik 6DYM'ye sahip olmu�tur.
Sonu� sonlan�m noktalar�
Kombinasyon tedavisi ile tedavi, birle�tirilmi� monoterapi grubuna k�yasla, nihai de�erlendirme vizitine kadar bile�ik klinik ba�ar�s�zl�k sonlan�m noktas�nda %50 risk azalmas� (tehlike oran� [HR] 0,502; %95 GA: 0,348 ila 0,724; p=0,0002) ile sonu�lanm��t�r (�ekil 1 ve
Tablo 1). Tedavi etkisi kombinasyontedavisindehastaneye
yat�r�lmalarda %63 azalmadan
sonlan�m noktas� �zerindeki etkilili�i her bir monoterapiye k�yasla ve ya�, etnik k�ken, co�rafik b�lge, etiyoloji (iPAH/hPAH ve PAH-CTD) alt gruplar� aras�nda tutarl� olmu�tur. Etki hem FC II hem de FC III hastalar� i�in anlaml� olmu�tur.
�ekil 1
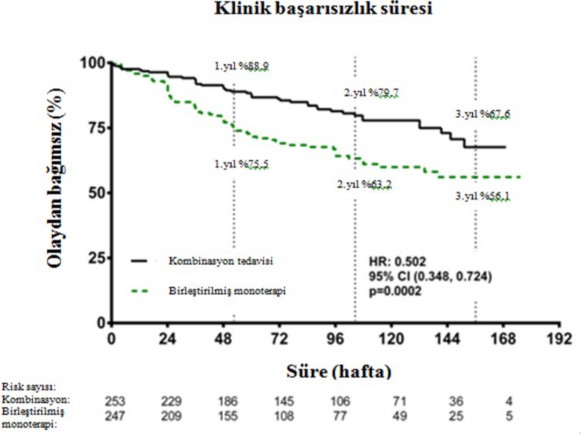
Tablo 1
| Ambrisentan + Tadalafil (N=253) | Monoterapi Birle�tirilmi� (N=247) | Ambrisentan monoterapi (N=126) | Tadalafil monoterapi (N=121) |
�lk Klinik Ba�ar�s�zl�k Olay�na Kadar Ge�en S�re (Karara Ba�lanm��) | ||||
Klinik ba�ar�s�zl�k, say� (%) | 46 (%18) | 77 (%31) | 43 (34) | 34 (28) |
Tehlike oran� (%95 GA) |
| 0,502 (0,348, 0,724) | 0,477 (0,314, 0,723) | 0,528 (0,338, 0,827) |
P-de�eri, Log-s�ra testi |
| 0,0002 | 0,0004 | 0,0045 |
�lk Klinik Ba�ar�s�zl�k Olay� Olarak Bile�en (Karara Ba�lanm��) | ||||
�l�m (t�m nedenler) | 9 (%4) | 8 (%3) | 2 (2) | 6 (5) |
A��rla�an PAH nedeniyle hastaneye yat�� | 10 (%4) | 30 (%12) | 18 (14) | 12 (10) |
Hastal�k progresyonu | 10 (%4) | 16 (%6) | 12 (10) | 4 (3) |
Tatmin edici olmayan uzun vadeli klinik yan�t | 17 (%7) | 23 (%9) | 11 (9) | 12 (10) |
o�rulama Kodu: 1ZW56M0FyZmxXRG83Q3NRRG83ZmxXQ3NR | ||||
�lk hastaneye yat��, say� (%) | 19 (%8) | 44 (%18) | 27 (%21) | 17 (%14) |
Tehlike oran� (%95 GA) |
| 0,372 | 0,323 | 0,442 |
P-de�eri, Log-s�ra testi |
| 0,0002 | < 0,0001 | 0,0124 |
�kincil sonlan�m noktalar�
�kincil sonlan�m noktalar� test edilmi�tir:
Tablo 2
�kincil sonlan�m noktalar� (ba�lang��tan 24. haftaya kadar de�i�im | Ambrisentan + Tadalafil | Monoterapi Birle�tirilmi� | Farkl�l�k ve G�ven Aral��� | p de�eri |
NT-proBNP (% azalma) | -67,2 | -50,4 | % farkl�l�k -33,8: %95 GA: -44,8, -20,7 | p<0,0001 |
24. haftada tatmin edici bir klinik yan�ta ula�an g�n�ll�lerin %'si | 39 | 29 | Olas�l�klar oran� 1,56; %95 GA: 1,05, 2,32 | p= 0,026 |
6DYM (metre, medyan de�i�im | 49 | 23,8 | 22,75 m; %95 GA: 12,00, 33,50 | p<0,0001 |
�diyopatik pulmoner fibroz:
%11'inde sekonder pulmoner hipertansiyon (WHO grup 3) bulunan 492 idiyopatik pulmoner fibroz (IPF) hastas� (ambrisentan N=329, plasebo N=163) ile yap�lan �al��ma, primer etkililik sonlan�m noktas�na ula��lamayaca�� belirlendi�inden, �al��ma erken sonland�r�lm��t�r (ARTEMIS-IPF �al��mas�).
Ambrisentan grubunda 90 (%27), plasebo grubunda 28 (%17) IPF progesyonu (solunum nedenli hastaneye yat�� dahil) ya da �l�m vakas� g�zlenmi�tir. Bu nedenle ambrisentan sekonder pulmoner hipertansiyon olsun ya da olmas�n IPF hastalar�nda kontrendikedir (bkz. B�l�m 4.3).
5.2. Farmakokinetik �zellikler
Genel �zelliklerEmilim:
Ambrisentan insanlarda h�zl� bir �ekilde emilmektedir. Oral uygulamadan sonra ambrisentan�n maksimum plazma konsantrasyonlar� (C) tipik �ekilde a�l�k ve tokluk ko�ullar�nda dozlamadan yakla��k 1,5 saat sonra meydana gelmektedir. C ve plazma konsantrasyonu zaman e�risi alt�ndaki alan (EAA) terap�tik doz aral���nda doza orant�sal olarak artmaktad�r. Kararl� duruma genelde 4 g�nl�k tekrarl� dozlama sonunda eri�ilmektedir.
Sa�l�kl� g�n�ll�lere a�l�k ko�ullar�nda ve y�ksek oranda ya� i�eren bir ���nle ambrisentan uygulan�m�n� i�eren bir g�da etkisi �al��mas� C de�erinin % 12 d��erken EAA'n�n de�i�medi�ini g�stermi�tir. Pik konsantrasyondaki bu d���� klinik a��dan anlaml� olmad���ndan ambrisentan a� veya tok karn�na al�nabilir.
Da��l�m:
Ambrisentan plazma proteinine y�ksek oranda ba�lanmaktad�r. Ambrisentan�n in vitro plazma proteinine ba�lanma oran� ortalama % 98,8 olup, 0,2 - 20 mikrogram/ml aral���nda konsantrasyondan ba��ms�zd�r. Ambrisentan temelde alb�mine (%96,5) ve daha d���k d�zeyde alfa-asit glikoproteine ba�lanmaktad�r.
Ambrisentan�n eritrositlere da��l�m� d���k olup ortalama kan:plazma oran� erkekler ve kad�nlarda s�ras�yla 0,57 ve 0,61'dir.
Biyotransformasyon:
Ambrisentan, bir non-s�lfonamid (propionik asit) ERA'd�r.
Ambrisentan, bir�ok UGT izoenzim (UGT1A9S, UGT2B7S ve UGT1A3S) arac�l���yla ambrisentan glukuronidi (%13) olu�turmak �zere glukuronizayona maruz kalmaktad�r. Ambrisentan ayr�ca ba�ta CYP3A4 ve daha d���k d�zeyde CYP3A5 ve CYP2C19 arac�l��� ile 4-hidroksimetil ambrisentan (%21) olu�turmak �zere oksidatif metabolizmaya maruz kalmakta ve bu �r�n ilave glukuronizasyon sonucu 4-hidroksimetil ambrisentan glukuronide (%5) d�n��mektedir. 4-hidroksimetil ambrisentan�n insan endotelin resept�r� i�in ba�lanma afinitesi ambrisentandan 65 kat daha d���kt�r. Bu nedenle, plazmada g�zlenen konsantrasyonlarda (ana bile�ik ambrisentana g�re yakla��k %4) 4-hidroksimetil ambrisentan�n ambrsisentan�n farmakolojik aktivitesine katk�da bulunmas� beklenmemektedir.
�n vitro veriler, 300 µM konsantrasyona kadar ambrisentan�n UGT1A1, UGT1A6, UGT1A9, UGT2B7 veya sitokrom P450 enzimleri 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 ve 3A4'� belirgin olarak inhibe etmedi�ini g�stermi�tir. Ek olarak, insan ta��y�c� genleri ile transfekte edilmi� h�cre dizilerinin kullan�ld��� in vitro �al��malar 100 µM konsantrasyona kadar ambrisentan�n P-glikoproteini (Pgp), meme kanseri resept�r protein (BCRP), �oklu-ila� diren� protein isoform-2 (MRP2) ya da safra tuzu d��a at�m pompas�n� (BSEP) inhibe etmedi�ini g�stermi�tir. Ambrisentan in vitro olarak OATP1B1, OATP1B3 ve NTCP'yi s�ras�yla 47 µM, 45 µM, ve yakla��k olarak 100 µM IC50 de�erleri ile zay�f olarak inhibe etmi�tir. S��an ve insan hepatositlerinde yap�lan in vitro �al��malarda NTCP, OATP, BSEP ve MRP2'nin ambrisentan inhibisyonuna y�nelik bir kan�t g�r�lmemi�tir. Ayr�ca ambrisentan, s��an hepatositlerinde MRP2, Pgp veya BSEP protein ekspresyonunu ind�klememi�tir. �n vitro verilere dayanarak, ambrisentan�n klinik olarak uygun konsantrasyonlarda BSEP, BCRP, Pgp, MRP2, OATP1B1/3 veya NTCP yolu ile ta��ma veya sitokrom P450 enzimleri 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 ve 3A4 veya UGT1A1, UGT1A6, UGT1A9, UGT2B7 �zerinde bir etkisinin olmas� beklenmemektedir..
Kararl� durumda ambrisentan�n (g�nde bir kez 10 mg) PT ve INR ile �l��len tek doz varfarinin (25 mg) farmakokineti�i ve farmakodinami�i �zerindeki etkileri 20 sa�l�kl� g�n�ll�de ara�t�r�lm��t�r. Ambrisentan, varfarinin farmakokineti�i veya farmakodinami�inde klinik a��dan anlaml� etki g�stermemi�tir. Benzer �ekilde varfarin ile birlikte uygulanmas� ambrisentan�n farmakokineti�ini etkilememi�tir (bkz. B�l�m 4.5).Yedi g�nl�k sildenafil dozaj�n�n (g�nde �� kez 20 mg) tek doz ambrisentan�n farmakokineti�i ve yedi g�nl�k ambrisentan dozaj�n�n (g�nde bir kez 10 mg) tek doz sildenafilin farmakokineti�i �zerindeki etkisi 19 sa�l�kl� g�n�ll�de ara�t�r�lm��t�r. Ambrisentan ile birlikteuygulanmas�n� takiben sildenafil C de�erindeki %13'l�k art�� d���nda sildenafil, N-desmetil-sildenafil ve
Kararl� durumda ambrisentan�n (g�nde bir kez 10 mg) tek doz tadalafil farmakokineti�i �zerinde ve kararl� durumda tadalafilin (g�nde bir kez 40 mg) tek doz ambrisentan farmakokineti�i �zerindeki etkileri 23 sa�l�kl� g�n�ll�de ara�t�r�lm��t�r. Tadalafil farmakokineti�i �zerinde ambrisentan�n klinik a��dan anlaml� etkisi olmam��t�r. Benzer olarak, tadalafilin birlikte uygulanmas� ambrisentan farmakokineti�ini etkilememi�tir (bkz. B�l�m 4.5)
Tekrarl� doz ketokonazol (g�nde bir kez 400 mg) uygulanmas�n�n tek doz 10 mg ambrisentan�n farmakokineti�i �zerindeki etkileri 16 sa�l�kl� g�n�ll�de ara�t�r�lm��t�r. EAA(0-inf) ve Cmaks ile �l��len ambrisentan maruziyeti s�ras�yla %35 ve %20 artm��t�r. Maruziyetteki bu de�i�ikli�in klinik a��dan anlaml� olmad��� d���n�ld���nden AMPAHO ile birlikte ketokonazol uygulanabilir.
Tekrarl� doz siklosporin A (g�nde iki kez 100-150 mg) uygulamas�n�n ambrisentan�n (g�nde bir kez 5 mg) kararl� durum farmakokineti�i �zerine etkisi ve tekrarl� doz ambrisentan (g�nde bir kez 5 mg) uygulamas�n�n siklosporin A'n�n (g�nde iki kez 100-150 mg) kararl� durum farmakokineti�i �zerine etkisi sa�l�kl� g�n�ll�lerde ara�t�r�lm��t�r. �oklu doz siklosporin A varl���nda ambrisentan�n Cmaks ve EAA(0-ᴛ)'s� artm��t�r (s�ras�yla %48 ve %121). Bu de�i�iklikler nedeniyle, siklosporin A ile birlikte uyguland���nda ambrisentan�n dozu g�nde bir kez 5 mg ile s�n�rland�r�lmal�d�r (bkz. B�l�m 4.2). Buna kar��n, ambrisentan�n �oklu doz uygulamas�n�n siklosporin A maruziyeti �zerinde klinik a��dan anlaml� bir etkisi olmam��t�r ve bu nedenle siklosporin A'n�n dozunun ayarlanmas�na gerek yoktur.
Akut ve tekrarl� doz rifampisin (g�nde bir kez 600 mg) uygulamas�n�n ambrisentan�n (g�nde bir kez 10 mg) kararl� durum farmakokineti�i �zerine etkisi sa�l�kl� g�n�ll�lerde ara�t�r�lm��t�r. Rifampisinin ilk dozlar�n� takiben, ambrisentan EAA(0-ᴛ)'s�nda ge�ici art�� (rifampisinin birinci ve ikinci dozunu takiben s�ras�yla %121 ve %116) g�zlemlenmi�tir; bunun nedeni muhtemelen rifampisinle ilgili OATP inhibisyonudur. Buna kar��n, �oklu doz rifampisin uygulamas�n� takiben 8. g�nde ambrisentan maruziyetinde klinik olarak anlaml� bir etki olmam��t�r. Ambrisentan tedavisi alan hastalarda rifampisin tedavisi ba�land���nda yak�ndan takip gereklidir (bkz. B�l�m 4.4 ve 4.5).
Tekrarl� doz �eklinde uygulanan ambrisentan (10 mg) uygulanmas�n�n tek doz �eklinde uygulanan digoksinin farmakokineti�i �zerindeki etkileri 15 sa�l�kl� g�n�ll�de ara�t�r�lm��t�r. �oklu doz ambrisentan digoksin EAA ve en d���k konsantrasyonlarda hafif bir art��a ve digoksin C de�erinde %29 art��a neden olmu�tur. Digoksin maruziyetinde �oklu doz ambrisentan varl���nda g�zlenen art�� klinik a��dan anlaml� olarak de�erlendirilmemi� olup, digoksin dozunda ayarlama gerekli de�ildir (bkz. B�l�m 4.5).
12 g�n boyunca ambrisentan (g�nde bir kez 10 mg) uygulamas�n�n etinil estradiol (35 mikrogram) ve noretindron (1 mg) i�eren oral kontraseptiflerin tek dozunun farmakokineti�i �zerine etkisi sa�l�kl� kad�n g�n�ll�lerde ara�t�r�lm��t�r. Cmaks ve EAA0-∞, etinil estradiol i�in hafif azalm�� (s�ras�yla %8 ve %4), noretindron i�in hafif artm��t�r (s�ras�yla %13 ve %14). Etinil estradiol ve noretindron maruziyet de�i�iklikleri k���k olmu�tur ve klinik olarak anlaml� olmalar� beklenmemektedir (bkz. B�l�m 4.5).
Eliminasyon:
Ambrisentan ve metabolitleri, hepatik ve/veya ekstrahepatik metabolizmay� takiben primer olarak safrada eliminde edilmektedir.Uygulanandozunyakla��k %22'si oral uygulamadan
�nsanlarda plazma eliminasyon yar�lanma �mr� yakla��k 13,6 ila 16,5 saattir.
�zel pop�lasyonlar:
Sa�l�kl� g�n�ll�ler ve PAH hastalar�nda yap�lan bir pop�lasyon farmakokineti�i analizinden elde edilen bulgulara g�re, ambrisentan�n farmakokineti�i cinsiyet veya ya�tan etkilenmemi�tir (bkz. B�l�m 4.2).
B�brek yetmezli�i
Ambrisentan, anlaml� renal metabolizma veya renal klirense (at�l�m) maruz kalmamaktad�r. Bir pop�lasyon farmakokineti�i analizinde, kreatinin klirensinin oral ambrisentan klirensini etkileyen istatistiksel a��dan anlaml� bir de�i�ken oldu�u belirlenmi�tir. Oral klirensteki azalman�n boyutu, orta �iddette b�brek yetmezli�i olan hastalarda orta d�zeyde (%20-40) oldu�undan bu d�����n klinik a��dan anlaml� olmas� beklenmemektedir. Bununla birlikte, �iddetli b�brek yetmezli�i olan hastalarda dikkatli olunmas� gereklidir (bkz. B�l�m 4.2).
Karaci�er yetmezli�i
Ambrisentan�n temel metabolizma yolu glukuronidasyon ve oksidasyon ile daha sonra safrada meydana gelen eliminasyon oldu�undan karaci�er yetmezli�inin ambrisentan maruziyetinde (C ve EAA) bir art��a neden olmas� beklenebilir. Bir pop�lasyon farmakokineti�i analizinde, oral klirensin artan bilirubin d�zeylerinin bir fonksiyonu olarak azald��� g�sterilmi�tir. Bununla birlikte, bilirubinin etkisi orta d�zeydedir (bilirubin d�zeyi 0,6 mg/dl olan tipik bir hastaya k�yasla, bilirubin d�zeyi artarak 4,5 mg/dl olan bir hastada oral ambrisentan klirensi yakla��k %30 daha d���k olacakt�r). �iddetli karaci�er yetmezli�i (siroz ile veya siroz olmadan) olan hastalarda ambrisentan�n farmakokineti�i ara�t�r�lmam��t�r. Bu nedenle, ambrisentan �iddetli karaci�er yetmezli�i veya klinik a��dan anlaml� �ekilde y�ksek hepatik aminotransferaz de�erleri (>3xULN) olan hastalarda kullan�lmamal�d�r (bkz. B�l�m 4.3 ve 4.4).
5.3. Klinik �ncesi g�venlilik verileri
S�n�fa �zg� primer farmakolojik etki nedeniyle, y�ksek tek doz ambrisentan (doz a��m�) uygulanmas� arteriyel bas�nc� d���rebilmi�tir ve hipotansiyona ve vazodilatasyon ile ili�kili semptomlara neden olma potansiyeline sahiptir.
Ambrisentan�n safra asidi ta��y�c�s�n�n bir inhibit�r� oldu�u veya a��r� hepatotoksisiteye neden
oldu�u g�sterilmemi�tir.
�nsanlardaki terap�tik d�zeylerin alt�ndaki maruziyetlerde kronik uygulamadan sonra kemirgenlerde nazal kavite epitelyumunda enflamasyon ve de�i�iklikler g�r�lm��t�r. K�peklerde, hastalarda g�zlenenin 20 kat�ndan daha y�ksek maruziyetlerde kronik y�ksek doz ambrisentan uygulanmas�n� takiben hafif enflamatuvar yan�tlar g�zlenmi�tir.
Klinik EAA'n�n 3 kat� maruziyet d�zeylerinde ambrisentan uygulanan s��anlar�n nazal kavitesinde etmoid konkada nazal kemik hiperplazisi g�zlenmi�tir. Nazal kemik hiperplazisi fare veya k�peklerde ambrisentan ile g�zlenmemi�tir. S��anlarda, di�er bile�iklerle edinilen deneyime g�re, nazal konkalarda kemikteki hiperplazi nazal enflamasyona kar�� bilinen bir yan�tt�r.
Ambrisentan memeli h�crelerinde in vitro y�ksek konsantrasyonlarda test edildi�inde
klastojenik �zellik g�stermi�tir. Bakterilerdeveyaiki invivo kemirgen �al��mas�nda
ambrisentan i�in mutajenik veya genotoksik etki kan�t� saptanmam��t�r.
S��an ve farelerde 2 y�ll�k oral dozaj �al��malar�nda karsinojenik potansiyele dair bir kan�t saptanmam��t�r. Yaln�zca en y�ksek dozda erkek s��anlarda selim meme fibroadenomunda (bir benign t�m�r) hafif art�� g�zlenmi�tir. Bu dozda erkek s��anlar�n sistemik ambrisentan maruziyeti (kararl� durum EAA'ya g�re) 10 mg/g�n klinik dozuyla eri�ilenin 6 kat� olmu�tur.
Erkek s��anlar ve farelerde g�venlilik marj� olmaks�z�n yap�lan oral tekrarl� doz toksisite ve fertilite �al��malar�nda bazen aspermi ile ili�kilendirilen testik�ler t�b�ler atrofi g�zlenmi�tir. Testik�ler de�i�iklikler, doz uygulanmayan periyotta de�erlendirildi�inde tam olarak d�zeltilebilir olmam��t�r. Bununla birlikte 39 haftaya kadarki �al��malarda EAA'ya g�re, insanlarda g�r�lenden 35 kat y�ksek bir maruziyette k�peklerde testik�ler de�i�iklik g�zlenmemi�tir. Erkek s��anlarda, (300mg/kg/g�n'e kadar olan) test edilen hi�bir dozda ambrisentan�n sperm motilitesine etkisi g�r�lmemi�tir. 300mg/kg/g�n dozunda morfolojik olarak normal spermlerin y�zdesinde k���k bir azalma (%10'dan daha az) g�zlemlenmi�tir, ancak bu 100 mg/kg/g�n dozunda (10 mg/g�n klinik dozunun 9 kat�ndan fazla olan klinik maruziyet) g�zlemlenmemi�tir. Ambrisentan�n erkeklerde fertilite �zerindeki etkisi bilinmemektedir.
Ambrisentan�n tav�an ve s��anlarda teratojenik oldu�u g�sterilmi�tir. Test edilen t�m dozlarda alt �ene, dil ve/veya damak anomalileri g�r�lm��t�r. Buna ek olarak s��anlarda yap�lan �al��mada interventrik�ler septal defektler, g�vde damarlar�nda damar defektleri, tiroid ve timus anomalileri, sfenoid kemik taban�nda osifikasyon ve umblikal arterin mesanenin sa� taraf�nda de�il sol taraf�nda yer almas� insidans�nda art�� g�r�lm��t�r. Teratojenisitenin ERA maddelerinin bir s�n�f etkisi oldu�undan ��phelenilmektedir.
Ge� gebelik ile laktasyon d�neminde di�i s��anlara insanlarda �nerilen maksimum dozda EAA'n�n 3 kat� maruziyette ambrisentan uygulanmas� maternal davran��larda yan etkilere neden olmu�, yavru hayatta kal�m�n� azaltm�� ve yavrular�n �reme yetene�inde bozulmaya (nekropside k���k testis bulgusu ile) neden olmu�tur.
Postnatal 7. g�nden 26., 36. veya 62. g�ne kadar g�nde bir defa oral olarak ambrisentan uygulanan j�venil s��anlarda; nefes alma sesleri, apne ve hipoksi g�r�lmesinden sonra morfolojik veya n�rodavran��sal de�i�iklik olmaks�z�n beyin a��rl���nda azalma (-%3 ila -%8) g�r�lm��t�r. Bunlar, EAA'ya g�re 10 mg'daki insan pediyatrik maruziyetlerinin (9 ila 15 ya�) yakla��k 1,8 ila 7 kat� maruziyetlerde ger�ekle�mi�tir. Bu bulgunun pediyatrik pop�lasyon i�in klinik a��dan �nemi hen�z bilinmemektedir.
6. FARMAS�T�K �ZELL�KLER
6.1. Yard�mc� maddelerin listesi
Tablet �ekirde�i:
Mikrokristalin sell�loz
Laktoz monohidrat (inek s�t�nden elde edilir) Kroskarmelloz sodyum
Magnezyum stearat
Film kaplama: Hipromelloz
Titanyum dioksit (E 171)
Talk
K�rm�z� demir oksit (E 172)
6.2. Ge�imsizlikler
Ge�erli de�ildir.
6.3. Raf �mr�
36 ay
6.4. Saklamaya y�nelik �zel tedbirler
25ºC'nin alt�ndaki oda s�cakl���nda saklay�n�z.
6.5. Ambalaj�n niteli�i ve i�eri�i
30 tablet i�eren PVC/PVDC/al�minyum folyo blister ambalajda
6.6. Be�eri t�bbi �r�nden arta kalan maddelerin imhas� ve di�er �zel �nlemler
Kullan�lmam�� �r�nler ya da at�k materyaller “T�bbi At�klar�n Kontrol� Y�netmeli�i” ve “Ambalaj ve Ambalaj At�klar� Kontrol� Y�netmeli�i”'ne uygun olarak imha edilmelidir.
 Tiroid Kanseri
En s�k g�r�len tiroid kanseri t�r� olan papiller tiroid kanseri, t�m tiroid kanserlerinin yakla��k %70'ini olu�turur.
Tiroid Kanseri
En s�k g�r�len tiroid kanseri t�r� olan papiller tiroid kanseri, t�m tiroid kanserlerinin yakla��k %70'ini olu�turur. |
 Omurilik zedelenmeleri
Omurilik zedelenmesini takip eden birka� g�n i�inde, hi�kimse hasarin ne kadar olacagini tahmin edemez. Buradaki sorun, omuriligin herhangi bir zedelenmesinden hemen sonra, bir omurilik sokunun olusmasidir.
Omurilik zedelenmeleri
Omurilik zedelenmesini takip eden birka� g�n i�inde, hi�kimse hasarin ne kadar olacagini tahmin edemez. Buradaki sorun, omuriligin herhangi bir zedelenmesinden hemen sonra, bir omurilik sokunun olusmasidir. |
�LA� E�DE�ERLER�
| E�de�er �la� Ad� | Barkodu | �la� Fiyat� |
|---|---|---|
| VOLIBRIS | 8699522091263 | 33,087.45TL |
| VOLIMRA | 8699525090799 | 31,510.44TL |
| Di�er E�de�er �la�lar |
 |
Asperger Sendromu Asperger sendromu, otistik gurubun bir b�l�m� olan bir �z�rd�r. Bu genelde, gurubun daha ”y�ksek” taraf�nda yer ald��� d���n�len ki�ilere uygun bir tan�d�r. |
 |
�izofrenlik �izofrenli�in psikiatrik te�hisi hakk�nda �ok fazla anla�mazl�k vard�r. Bu sayfadaki bilgiler, �izofrenli�in te�hisi, nedenleri ve tedavisi hakk�ndaki fakl� teoriler hakk�nda bilgi verecektir. |
 Kanseri") |
Rahim Boyu ( Serviks ) Kanseri Rahim boynu (serviks) kanseri 35 ya� alt� kad�nlarda g�r�len vakalarda meme kanserinden sonra ikinci s�ray� al�r.Serviks kanserinin geli�mesi y�llarca s�rebilir. |
�LA� GENEL B�LG�LER�
Sanofi Sa�l�k �r�nleri Ltd.�ti
| Geri �deme Kodu | A16665 |
| Sat�� Fiyat� | 33175.6 TL [ 1 Dec 2025 ] |
| �nceki Sat�� Fiyat� | 33175.6 TL [ 24 Nov 2025 ] |
| Original / Jenerik | Jenerik �la� |
| Re�ete Durumu | Normal Re�eteli bir ila�d�r. |
| Barkodu | 8699502094574 |
| Etkin Madde | Ambrisentan |
| Yerli ve Be�eri bir ila�d�r. |