EYLEA 40 mg/ml intravitreal enjeksiyon için çöz. içeren 1 flakon Kısa Ürün Bilgisi
{ Aflibersept }
1. BEŞERİ TIBBİ ÜRÜNÜN ADI
EYLEA® 40 mg/mL intravitreal enjeksiyon için çözelti içeren kullanıma hazır enjektör
Steril
2. KALİTATİF VE KANTİTATİF BİLEŞİM
Etkin madde
1 mL intravitreal enjeksiyon için çözeltide, Aflibersept* 40 mg
Bir kullanıma hazır enjektör, en az 3,6 mg aflibersepte eşdeğer en az 0,09 mL çekilebilir hacim içerir. Bu 2 mg aflibersept içeren 0,05 mL tek bir dozun uygulanabilmesi için yeterli miktar ihtiva eder.
*İnsan VEGF (Vasküler Endotelyal Büyüme Faktörü) reseptörleri 1 ve 2'nin ekstraselüler bölgelerinin parçalarından meydana gelen, insan IgG1'in Fc parçası ile birleştirilmiş ve Çin hamsteri yumurtalık (CHO) K1 hücrelerinde rekombinant DNA teknolojisi ile üretilmiş füzyon proteini.
Yardımcı maddeler
Sodyum fosfat, monobazik monohidrat (pH ayarlaması için) 1,104 mg/mL Sodyum fosfat, dibazik heptahidrat (pH ayarlaması için) 0,537 mg/mL Sodyum klorür 2,338 mg/mL
Yardımcı maddelerin tam listesi için 6.1'e bakınız.
3. FARMASÖTİK FORMU
İntravitreal enjeksiyon için çözelti
Berrak, renksiz ila soluk sarı arası renkte ve izo-ozmotik çözelti.
4. KLİNİK ÖZELLİKLER
4.1. Terapötik endikasyonlar
EYLEA, yetişkinlerde aşağıdakilerin tedavisi için endikedir:
Neovasküler (yaş tip) yaşa bağlı makula dejenerasyonu (YBMD) (bkz. Bölüm 5.1),
4.2. Pozoloji ve uygulama şekli
EYLEA yalnızca intravitreal enjeksiyon içindir.
EYLEA, intravitreal enjeksiyon uygulamasında deneyimli, göz hastalıkları uzmanı olan bir
hekim tarafından uygulanmalıdır.
Pozoloji/uygulama sıklığı ve süresi:
Yaş tip YBMD (yaşa bağlı makula dejenerasyonu)
Önerilen EYLEA dozu, 0,05 mL'ye eşdeğer 2 mg aflibersepttir.
EYLEA tedavisinin, birbirini takip eden üç doz şeklinde ayda bir kez tek enjeksiyon olarak
başlatılması ve daha sonra, tedavi aralığının iki ay olacak şekilde uzatılması önerilir.
Hekimin görmeyle ilgili ve/veya anatomik sonuçlara ilişkin değerlendirmesi doğrultusunda, tedavi aralığı iki ay olarak kalabilir ya da tedavi et ve uzat dozlama rejimi kullanılarak uzatılabilir, bu yaklaşımda görmeyle ilgili ve/veya anatomik sonuçların korunması için enjeksiyon aralıkları 2 veya 4 haftalık kademelerle artırılır. Görmeyle ilgili ve/veya anatomik sonuçlarda kötüleşme olması halinde, tedavi aralığı uygun şekilde kısaltılmalıdır.
Enjeksiyonlar arasında izlem yapılması gerekmez. Hekimin takdirine göre, izlem vizitlerinin
programı enjeksiyon vizitlerinden daha sık olabilir.
Enjeksiyonlar arasında dört aydan uzun veya 4 haftadan kısa tedavi aralıkları değerlendirilmemiştir (bkz. Bölüm 5.1).
RVO'ya sekonder makula ödemi (RVDO veya SRVO)
Önerilen EYLEA dozu, 0,05 mL'ye eşdeğer 2 mg aflibersepttir.
İlk enjeksiyondan sonra, tedavinin ayda bir kez uygulanması önerilir. İki doz arasında geçen
süre bir aydan kısa olmamalıdır.
Anatomi ve görmeyle ilgili sonuçların hastanın tedaviden fayda görmediğine işaret etmesi halinde EYLEA tedavisi kesilmelidir.
Aylık tedavinin maksimum görme keskinliği elde edilene ve/veya hastalık aktivitesine ilişkin belirti kalmayana kadar devam ettirilmesi önerilir. Ardışık üç veya daha fazla aylık enjeksiyon gerekebilir.
Tedavi daha sonra tedavi et ve uzat rejimiyle devam ettirilebilir, görmeyle ilgili ve/veya anatomik sonuçların stabil kalması sağlanarak tedavi aralıkları kademeli şekilde uzatılabilir, ancak bu aralıkların uzunluklarının belirlenmesi için gerekli veriler yetersizdir. Görmeyle ilgili ve/veya anatomik sonuçların kötüleşmesi halinde, tedavi aralığı buna uygun şekilde kısaltılmalıdır.
İzlem ve tedavi programı tedavi eden hekim tarafından hastaya özgü yanıt doğrultusunda belirlenmelidir.
Hastalık aktivitesinin izlemi klinik muayene, fonksiyon testi ya da görüntüleme tekniklerini (örn. optik koherens tomografisi ya da floresan anjiyografi) içerebilir.
Diyabetik makula ödemi
EYLEA için önerilen tedavi şekli, 50 mikrolitreye (0,05 mL) eşdeğer 2 mg afliberseptin ardışık beş doz olmak üzere ayda bir kez tek enjeksiyonudur ve takiben iki ayda bir tek enjeksiyon olarak devam ettirilmesidir.
Hekimin görmeyle ilgili ve/veya anatomik sonuçları değerlendirmesi doğrultusunda tedavi aralığı 2 ay olarak tutulabilir veya bireyselleştirilebilir; örneğin görmeyle ilgili ve/veya anatomik sonuçların korunması için tedavi aralıklarının genellikle 2 haftalık kademelerle artırıldığı tedavi et ve uzat dozlama rejimi kullanılabilir. Dört aydan daha uzun tedavi aralıkları ile ilgili olarak sınırlı veri bulunmaktadır. Görmeyle ilgili ve/veya anatomik sonuçların kötüleşmesi halinde, tedavi aralığı buna uygun şekilde kısaltılmalıdır. 4 haftadan daha kısa tedavi aralıkları değerlendirilmemiştir (bkz. Bölüm 5.1).
İzlem programı tedavi eden hekim tarafından belirlenmelidir.
Anatomi ve görmeyle ilgili sonuçların hastanın tedaviden fayda görmediğine işaret etmesi halinde EYLEA tedavisi kesilmelidir.
Protokol T çalışma tasarımına göre, EYLEA tedavisi aşağıda belirtilen şekilde uygulanabilir: Aflibersept başlangıçta ve ardından, uygunluk eşiğinin altında bir santral alt alan kalınlığı (SAAK) ile, görme keskinliği (GK) 20/20 ya da daha iyi değilse (Yaklaşık 85 harf değerinde GK skoruna eşdeğer Snellen) ve önceki son iki enjeksiyona cevaben iyileşme ya da kötüleşme olmamışsa 4 haftada bir enjekte edilmiştir (bkz. Bölüm 5.1). İlk yıl boyunca, takip vizitleri her dört haftada bir (±1 hafta) gerçekleşmiştir.
24. haftadaki vizitten itibaren, görme keskinliği ve SAAK ne olursa olsun, iki ardışık enjeksiyondan sonra iyileşme veya kötüleşme olmamışsa enjeksiyon durdurulmuş, ancak görme keskinliği harf skoru veya SAAK kötüleşmişse tedavi yeniden başlatılmıştır (bkz. Bölüm 5.1)
Miyopik koroidal neovaskülarizasyon
EYLEA için önerilen doz 0,05 mL'ye eşdeğer 2 mg aflibersept ile tek bir intravitreal enjeksiyondur.
Görmeyle ilgili ve/veya anatomik sonuçlar hastalığın persistan olduğunu düşündürdüğü takdirde ilave dozlar uygulanabilir. Tekrarlama durumunda, hastalık yeni ortaya çıkmış gibi tedavi edilmesi önerilir.
İzlem programı tedavi eden hekim tarafından belirlenmelidir.
İki doz arasında geçen süre bir aydan kısa olmamalıdır.
Uygulama şekli:
İntravitreal enjeksiyonlar tıbbi standartlar ile geçerli kılavuzlara uygun şekilde ve intravitreal enjeksiyon konusunda deneyimli bir uzman hekim tarafından yapılmalıdır. Genel olarak, yeterli anestezi ve topikal geniş spektrumlu mikrobisit dahil aseptik koşullar (örn. perioküler deri, göz kapağı ve oküler yüzeye povidon iyodür uygulama) sağlanmalıdır. Cerrahi el dezenfenksiyonu, steril eldiven, steril örtü ve steril göz kapağı spekulumu (veya eşdeğeri) önerilmektedir.
Enjeksiyon iğnesi limbusa 3,5-4,0 mm posterior konumdan vitreus boşluğuna batırılmalı, yatay meridyenden kaçınılarak kürenin merkezi hedeflenmelidir. Enjeksiyon hacmi 0,05 mL olmalı; sonraki enjeksiyonlar için diğer bir sklera bölgesi kullanılmalıdır.
İntravitreal enjeksiyondan hemen sonra hastalar göz içi basınçta artış için kontrol edilmelidir. Uygun takip yöntemi optik sinir başı için perfüzyon kontrolü veya tonometri şeklinde olabilir. Gerektiğinde parasentez için uygun steril ekipman bulundurulmalıdır.
İntravitreal enjeksiyondan sonra hastalar endoftalmi düşündüren her türlü semptomu gecikmeden bildirmeleri gerektiği konusunda bilgilendirilmelidir (örn. göz ağrısı, gözde kızarıklık, fotofobi, bulanık görme).
Her bir kullanıma hazır enjektör yalnızca tek bir gözün tedavisinde kullanılmalıdır. Tek bir kullanıma hazır enjektörden birden fazla dozun verilmesi kontaminasyon ile bunu takiben enfeksiyon riskini artırabilir.
Kullanıma hazır enjektör önerilen 2 mg'lık aflibersept (0,05 mL enjeksiyonluk çözeltiye eşdeğer) dozundan fazlasını içerir. Enjektörden çekilebilen hacim, enjektörden çıkabilen miktardır ve tamamı kullanılmayacaktır. EYLEA kullanıma hazır enjektörden çekilebilecek hacim en az 0,09 mL'dir. Önerilen dozun enjeksiyonundan önce fazla hacim mutlaka atılmalıdır (bkz. Bölüm 6.6).
Kullanıma hazır enjektör içindeki hacmin tamamının enjekte edilmesi doz aşımına neden olabilir. Fazla tıbbi ürünle birlikte hava kabarcığının uzaklaştırılması için piston yavaşça bastırılmalı ve piston kubbesinin tabanı (kubbenin ucu değil) ile şırınga üzerindeki siyah dozlama çizgisinin (0,05 mL, yani 2 mg aflibersepte eşdeğer) aynı hizaya gelmesi sağlanmalıdır (bkz. Bölüm 4.9 ve 6.6).
Enjeksiyondan sonra kullanılmamış olan ürün atılmalıdır.
Uygulama öncesinde tıbbi ürünün hazırlanmasıyla ilgili talimatlar için, bkz. Bölüm 6.6.
Özel popülasyonlara ilişkin ek bilgiler:
Böbrek/Karaciğer yetmezliği:
Karaciğer ve/veya böbrek yetmezliği olan hastalarda EYLEA ile ilgili özel bir çalışma yapılmamıştır.
Mevcut veriler bu hastalarda EYLEA için doz ayarlaması gerektiğine işaret etmemektedir (bkz. Bölüm 5.2).
Pediyatrik popülasyon:
EYLEA'nın çocuklarda ve adolesanlarda güvenlilik ve etkililiği belirlenmemiştir. EYLEA'nın yaş tip YBMD, SRVO, RVDO, DMÖ ve miyopik KNV endikasyonları için pediyatrik popülasyonda kullanımı söz konusu değildir.
Geriyatrik popülasyon:
Özel bir uygulama yapılması gerekmez. 75 yaşın üzerindeki DMÖ'lü hastalara ilişkin deneyim sınırlıdır.
4.3. Kontrendikasyonlar
Etkin madde
herhangi birine aşırı duyarlılık,
4.4. Özel kullanım uyarıları ve önlemleri
İntravitreal enjeksiyon ile ilişkili reaksiyonlar
EYLEA ile uygulananlar dahil intravitreal enjeksiyonlar, endoftalmi, intraoküler enflamasyon, regmatojen retina dekolmanı, retina yırtılması ve iatrojenik travmatik katarakt ile ilişkilendirilmiştir (bkz. Bölüm 4.8). EYLEA uygulanırken daima uygun aseptik enjeksiyon teknikleri kullanılmalıdır. Hastalar ayrıca, enfeksiyon oluşursa erken tedaviye olanak sağlamak için enfeksiyonu takiben hafta boyunca gözlenmelidir. Hastalardan, endoftalmi ya da yukarıda bahsi geçen diğer olayları düşündüren bir semptomla karşılaşmaları halinde gecikmeden durumu bildirmesi istenmelidir.
Kullanıma hazır enjektör, önerilen 2 mg aflibersept dozundan (0,05 mL'ye eşdeğer) fazlasını
içerir. Fazla hacim uygulamadan önce dışarı atılmalıdır (bkz. bölüm 4.2 ve 6.6).
EYLEA enjeksiyonu dahil intravitreal enjeksiyonlar sonrasında 60 dakika içinde göz içi basıncın arttığı görülmüştür (bkz. Bölüm 4.8). Glokomun yeterli kontrol altında olmadığı hastalarda özellikle dikkatli olunmalıdır (göz içi basıncı ≥30 mmHg olanlara EYLEA enjeksiyonu yapılmamalıdır). Bu nedenle göz içi basınç ve optik sinir başının perfüzyonu tüm olgularda izlenmeli ve uygun şekilde kontrol altına alınmalıdır.
İmmünojenisite
EYLEA terapötik bir protein olduğundan, immünojenisite potansiyeli bulunmaktadır (bkz. Bölüm 4.8). Hastalardan, aşırı duyarlılığa atfedilebilecek klinik bulgular olabileceği için ağrı, fotofobi ya da kızarıklık gibi intraoküler enflamasyon belirti veya semptomlarını bildirmeleri istenmelidir.
Sistemik etkiler
Oküler olmayan hemorajiler ve arteriyel tromboembolik olaylar dahil sistemik advers olaylar, VEGF inhibitörlerinin intravitreal enjeksiyonunu takiben bildirilmiştir ve bunların VEGF inhibisyonu ile ilişkili olabileceğine yönelik teorik bir risk vardır. Son 6 ay içinde inme, geçici iskemik atak ya da miyokard enfarktüsü öyküsü olan SRVO, RVDO, DMÖ ya da miyopik KNV'li hastaların tedavisinde güvenlilik ile ilgili veriler sınırlıdır. Bu tip hastaların tedavisinde dikkatli olunmalıdır.
Diğer
YBMD, SRVO, RVDO, DMÖ ve miyopik KNV'nin diğer intravitreal anti-VEGF tedavilerinde de olduğu gibi aşağıdaki durumlar geçerlidir:
Her iki göze eşzamanlı olarak uygulanan EYLEA'nın güvenlilik ve etkililiği sistematik olarak çalışılmamıştır (bkz. Bölüm 5.1). Aynı zamanda bilateral tedavi uygulanıyorsa, bu durum sistemik maruziyette artışa neden olabilir ve sistemik advers olay riskini artırabilir.
4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri
Herhangi bir etkileşim çalışması yapılmamıştır.
Verteporfin ile fotodinamik tedavi (PDT) ve EYLEA'nın birlikte kullanımı çalışılmamıştır; bu nedenle, güvenlilik profili belirlenmemiştir.
Özel popülasyonlara ilişkin ek bilgiler
Özel popülasyonlara ilişkin herhangi bir etkileşim çalışması yapılmamıştır.
Pediyatrik popülasyon:
Pediyatrik popülasyona ilişkin herhangi bir etkileşim çalışması yapılmamıştır.
4.6. Gebelik ve laktasyon
Gebelik kategorisi: C
Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon) Doğurganlık çağındaki kadınlar aflibersept tedavisi sırasında ve son intravitreal enjeksiyondan sonra en az 3 ay süreyle etkili bir kontrasepsiyon yöntemi kullanmalıdır (bkz. Bölüm 4.4).
Gebelik dönemi
Gebe kadınlarda aflibersept kullanımıyla ilgili veri bulunmamaktadır.
Hayvanlardaki çalışmalarında embriyo-fetal toksisite gösterilmiştir (bkz. Bölüm 5.3).
Oküler uygulama sonrasında sistemik maruziyet son derece düşük olmakla birlikte, olası fayda fetüs ile ilgili olası riske üstün gelmediği sürece gebelik sırasında EYLEA kullanılmamalıdır.
Laktasyon dönemi
Afliberseptin insanlarda anne sütüne geçip geçmediği bilinmemektedir. Anne sütü almakta olan çocuğa yönelik risk dışlanamaz.
Laktasyon döneminde EYLEA kullanımı önerilmez. Emzirmeye ara verilmesi veya EYLEA kullanımından kaçınılması konusu, emzirmenin çocuğa getirdiği yarar ile tedavinin kadına getirdiği yarar dikkate alınarak kararlaştırılmalıdır.
Üreme yeteneği/Fertilite
Yüksek sistemik maruziyet ile gerçekleştirilen hayvan çalışmalarında elde edilen bulgular afliberseptin erkek ve kadın fertilitesine zarar verebileceğini göstermektedir (bkz. Bölüm 5.3). Sistemik maruziyetin son derece düşük olduğu oküler uygulamalardan sonra bu tip etkiler beklenmez.
4.7. Araç ve makine kullanımı üzerindeki etkiler
EYLEA enjeksiyonu, enjeksiyon veya göz muayenesiyle ilişkili olası geçici görme bozukluğu nedeniyle araç ve makine kullanma üzerinde minör bir etkiye sahiptir. Hastalar görme fonksiyonu tamamen normale dönmedikçe araç veya makine kullanmamalıdır.
4.8. İstenmeyen etkiler
Güvenlilik profilinin özeti:
Sekiz faz III çalışmanın güvenlilik popülasyonu toplam 3102 hastadan oluşmaktadır. Bu hastaların 2501'i önerilen doz olan 2 mg dozuyla tedavi edilmiştir.
Enjeksiyon prosedürüne bağlı olarak çalışılan gözde meydana gelen ciddi oküler advers reaksiyonlar, EYLEA ile 1900 intravitreal enjeksiyonun 1'inden azında meydana gelmiş ve körlük, endoftalmi, retina dekolmanı, travmatik katarakt, katarakt, vitreus kanaması, vitreus dekolmanı ve göz içi basınçta artışı içermiştir (bkz. Bölüm 4.4).
En sık görülen advers reaksiyonlar (EYLEA ile tedavi edilen hastaların en az %5'i); konjonktival hemoraji (%25), retinal hemoraji (%11), görme keskinliğinde azalma (%11), göz ağrısı (%10), katarakt (%8), göz içi basınçta artış (%8), vitreus dekolmanı (%7) ve vitreusta uçuşan noktalardır (%7).
Advers reaksiyonların listesi:
Aşağıda açıklanan güvenlilik verileri, yaş tip YBMD, SRVO, RVDO, DMÖ ve miyopik KNV ile ilgili sekiz faz III çalışmada ortaya çıkan ve enjeksiyon prosedürü veya tıbbi ürünler makul nedensellik olasılığı bulunan tüm advers reaksiyonları içermektedir.
Advers reaksiyonlar sistem organ sınıfına ve sıklığa göre aşağıdaki şekilde listelenmiştir:
Çok yaygın (≥1/10); yaygın (≥1/100 ila <1/10); yaygın olmayan (≥1/1000 ila <1/100); seyrek (≥1/10.000 ila <1/1000); çok seyrek (<1/10.000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor).
Her sıklık grubunda, advers ilaç reaksiyonları azalan ciddiyet sıralamasında sunulmuştur.
Faz III çalışmalarda (yaş tip YBMD, SRVO, RVDO, DMÖ ve miyopik KNV endikasyonları için faz III çalışmalardan havuzlanmış veriler) veya pazarlama sonrası araştırmalarda hastalarda bildirilen tüm tedavi sonucu ortaya çıkan advers ilaç reaksiyonları
Bağışıklık sistemi hastalıkları
Yaygın olmayan: Aşırı duyarlılık***
Göz hastalıkları
Çok yaygın: Görme keskinliğinde azalma, retinal hemoraji, konjonktival hemoraji, göz ağrısı
Yaygın: Retina pigment epitelinin yırtılması*, retina pigment epitel dekolmanı, retinal dejenerasyon, vitreus kanaması, katarakt, kortikal katarakt, nükleer katarakt, subkapsüler katarakt, kornea erozyonu, kornea abrazyonu, göz içi basıncının yükselmesi (intraoküler basınç artışı), bulanık görme, vitreusta uçuşan noktalar, vitreus dekolmanı, enjeksiyon yerinde ağrı, gözlerde yabancı cisim hissi, lakrimasyon artışı, göz kapağında ödem, enjeksiyon yerinde hemoraji, punktat keratit, konjunktival hiperemi, oküler hiperemi
Yaygın olmayan: Endoftalmit**, retina dekolmanı, retina yırtılması, iritis, üveit, iridosiklit, lentiküler opasiteler, kornea epitel defekti, enjeksiyon yerinde iritasyon, gözde anormal his, göz kapağı iritasyonu, ön kamarada flare, kornea ödemi
Seyrek: Körlük, travmatik katarakt, vitrit, hipopiyon
* Yaş tip YBMD ile ilişkili olduğu bilinen durumlardır; sadece yaş tip YBMD çalışmalarında
gözlenmiştir.
** Kültür pozitif ve kültür negatif endoftalmit
*** Pazarlama sonrası dönemde, aşırı duyarlılık raporları döküntü, pruritus, ürtiker ve ciddi anafilaktik/anaflaktoid reaksiyon olgularının izole vakalarını içermiştir.
Seçili advers reaksiyonların tanımı:
Yaş tip YBMD ile ilgili faz III çalışmalarında antitrombotik ilaç alan hastalarda konjonktival kanama insidansının daha yüksek olduğu görülmüştür. Bu insidans artışı, ranibizumab ve EYLEA tedavisi alan hastalar arasında karşılaştırılabilir düzeydedir.
Arteriyel tromboembolik olaylar (ATO'lar) sistemik VEGF inhibisyonuyla potansiyel ilişkisi olan advers olaylardır. İntravitreal VEGF inhibitörlerinin kullanıldıktan sonra, inme ve miyokard enfarktüsü dahil arteriyel tromboembolik olay gelişmesi bakımından teorik bir risk bulunmaktadır.
YBMD, DMÖ, RVO ve miyopik KNV'li hastalarla yürütülen EYLEA klinik çalışmalarında, arteriyel tromboembolik olaylar açısından düşük bir insidans oranı görülmüştür. Endikasyonlar genelinde, aflibersept tedavisi alan gruplar ile karşılık gelen komparatör grupları arasında kayda değer bir fark gözlenmemiştir.
Tüm terapötik proteinler gibi, EYLEA'nın da immünojenisite potansiyeli bulunmaktadır.
EYLEA'nın uygulanmasının ardından gözün vitreusunda silikon yağıyla ilişkili uçuşan nokta izole vakaları bildirilmiştir.
Şüpheli advers reaksiyonların raporlanması
Ruhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar / risk dengesinin sürekli olarak izlenmesine olanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu Türkiye Farmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e- posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks: 0 312 218 35 99).
4.9. Doz aşımı ve tedavisi
Klinik çalışmalarda aylık aralıklarla verilen 4 mg'a kadar olan dozlar kullanılmış ve 8 mg ile izole doz aşımı olgularına rastlanmıştır.
Enjeksiyon hacminin artırılmasıyla gelişen doz aşımı, göz içi basıncın artmasına neden olabilir. Bu nedenle, doz aşımı durumunda göz içi basınç izlenmeli ve tedavi eden hekimin takdirine göre gerekli bulunduğunda, uygun tedavi uygulanmalıdır (bkz. Bölüm 6.6).
5. FARMAKOLOJİK ÖZELLİKLER
5.1. Farmakodinamik özellikler
Farmakoterapötik grup: Oftalmolojikler/Antineovaskülarizasyon ilaçları ATC kodu: S01LA05
Aflibersept, insan VEGF reseptörünün 1. ve 2. ekstraselüler parçaları ile insan IgG1'inin Fc kısmının birleştirildiği rekombinant bir füzyon proteinidir.
Aflibersept, rekombinant DNA teknolojisi ile Çin hamster over (CHO) K1 hücrelerinde üretilmektedir.
Aflibersept VEGF-A ile PlGF'e doğal reseptörlerinden daha yüksek afiniteyle bağlanan çözünür bir tuzak reseptör görevi görür ve böylece aynı kökenden gelen bu VEGF reseptörlerinin bağlanmasını ve aktivasyonunu inhibe edebilir.
Etki mekanizması:
Vasküler endotelyal büyüme faktörü-A (VEGF-A) ve plasental büyüme faktörü (PlGF), anjiyogenik faktörlerin VEGF ailesinin endotel hücreleri için potent mitojenik, kemotaktik ve vasküler geçirgenlik sağlayan üyeleridir. VEGF; endotel hücrelerinin yüzeyinde bulunan iki tirozin kinaz reseptörü, VEGFR-1 ve VEGFR-2 üzerinden etki göstermektedir. PlGF yalnızca VEGFR-1'e bağlanmakta ve VEGFR-1, lökosit yüzeyinde de bulunmaktadır. Bu reseptörlerin VEGF-A tarafından aşırı derecede aktivasyonu, patolojik neovaskülarizasyon ve aşırı vasküler geçirgenlik ile sonuçlanabilir. PlGF bu süreçlerde VEGF-A ile sinerji gösterebilir ve ayrıca lökosit infiltrasyonu ile vasküler enflamasyonu da uyardığı bilinmektedir.
Farmakodinamik etkiler:
Yaş tip YBMD
Yaş tip YBMD patolojik koroidal neovaskülarizasyon (KNV) ile karakterizedir. KNV'den kan ve sıvı sızması retinanın kalınlaşmasına ya da ödem ve/veya sub/intra-retinal kanamaya neden olarak görme keskinliği kaybıyla sonuçlanabilmektedir.
EYLEA tedavisi uygulanan hastalarda (art arda üç ay süresince ayda bir kez tek enjeksiyon, ardından 2 ayda bir tek enjeksiyon) santral retina kalınlığı [SRK] tedavinin başlatılmasından kısa süre sonra azalmış ve ortalama KNV lezyonu boyutunun azaldığı görülmüştür; bu durum aylık 0,5 mg ranibizumab ile görülen bulgular ile uyumludur.
VIEW1 çalışmasında optik koherens tomografisinde (OKT) ortalama SRK azalmaları olduğu görülmüştür (iki ayda bir 2 mg EYLEA ve ayda bir 0,5 mg ranibizumab için 52. haftada sırasıyla -130 ve -129 mikron). Yine 52 haftalık zaman içerisinde, VIEW2 çalışmasında retina kalınlığı bakımından ortalama azalma olduğu OKT ile ortaya konmuştur (iki ayda bir 2 mg EYLEA ve her ay 0,5 mg ranibizumab için sırasıyla -149 ve -139 mikron). KNV boyutundaki azalma ile retina kalınlığındaki azalmanın çalışmaların ikinci yılında genel olarak korunduğu saptanmıştır.
ALTAIR çalışması önceden tedavi uygulanmamış yaş tip YBMD'li Japon hastalarla yürütülmüş, başlangıçta ayda bir kez 2 mg EYLEA olarak uygulanan 3 enjeksiyonun ardından 2 ay geçtikten sonra bir enjeksiyon daha yapılmış ve sonrasında maksimum aralık 16 hafta olmak üzere önceden belirlenmiş kriterlere göre değişen tedavi aralıklarıyla (2 haftalık veya 4 haftalık ayarlamalar) tedavi et ve uzat rejimi kullanılmış ve VIEW çalışmalarına benzer sonuçlar elde edilmiştir. Elli ikinci haftada, 2 haftalık ayarlama grubu ve 4 haftalık ayarlama grubu için OKT'de santral retina kalınlığı (SRK) değerlerinde sırasıyla ortalama -134,4 ve - 126,1 mikronluk ortalama azalmalar olduğu belirlenmiştir. Elli ikinci haftada 2 haftalık ve 4 haftalık ayarlama gruplarında OKT'de sıvı saptanmayan hastaların oranı sırasıyla %68,3 ve
%69,1 olarak kaydedilmiştir. SRK'deki azalma, ALTAIR çalışmasının ikinci yılında her iki tedavi kolunda da genel olarak korunmuştur.
ARIES çalışması, başlangıçtaki aylık 3 enjeksiyonun ve 2 aydan sonra bir ilave enjeksiyonun uygulanmasının hemen ardından başlatılan EYLEA 2 mg tedavi et ve uzat dozlama rejiminin,
bir yıllık tedaviden sonra başlatılan tedavi et ve uzat dozlama rejimine eşdeğerliğini araştırmak için tasarlanmıştır. Çalışmanın seyri sırasında en az bir kez Q8'den daha sık dozlama gerektiren hastalar için SRK yüksek kalmakla birlikte SRK'de başlangıçtan
104. haftaya kadar olan ortalama azalma, Q8 veya daha az sıklıktaki aralıklarla tedavi edilmiş hastalara benzer şekilde -160,4 mikrondur.
SRVO ve RVDO'ya sekonder makula ödemi
SRVO ve RVDO'da retinal iskemi gelişmekte ve VEGF salınımı için sinyal oluşmakta, buna karşılık olarak sıkı bağlantıların stabilizasyonu bozulmakta ve endotel hücre proliferasyonu uyarılmaktadır. VEGF'nin yukarı düzenlenmesi kan retina engelinin yıkımıyla, vasküler geçirgenliğin artmasıyla, retinal ödemle ve neovaskülarizasyon komplikasyonlarıyla ilişkilidir.
Ardışık 6 ayda aylık 2 mg EYLEA enjeksiyonlarıyla tedavi uygulanan hastalarda tutarlı, hızlı ve sağlam morfolojik yanıt elde edildiği (ortalama SRK iyileşmeleriyle ölçüldüğü üzere) gözlenmiştir. Yirmi dördüncü haftada, SRK azalmasının üç çalışmada da kontrole kıyasla istatistiksel açıdan üstün olduğu kaydedilmiştir (SRVO'da COPERNICUS: -457 ve -145 mikron; SRVO'da GALILEO: -449 ve -169 mikron; RVDO'da VIBRANT: -280 ve -128 mikron). Başlangıçtaki SRK değerinde kaydedilen azalma her bir çalışmanın sonuna kadar, yani COPERNICUS çalışmasında 100 hafta, GALILEO çalışmasında 76 hafta ve VIBRANT çalışmasında 52 hafta süreyle korunmuştur.
Diyabetik makula ödemi
Diyabetik makula ödemi, diyabetik retinopatinin bir sonucu olup, vasküler geçirgenlikte artış
ve retinal kapilerlerde görme keskinliğinin azalmasına yol açabilen hasarla karakterizedir.
Büyük bölümü Tip II diyabet olarak sınıflandırılan ve EYLEA tedavisi uygulanan hastalarda morfolojide (SRK, DRSS düzeyi) hızlı ve güçlü bir yanıt gözlenmiştir.
VIVIDDME ve VISTADME çalışmalarında EYLEA tedavisi alan hastalarda lazer kontrole kıyasla başlangıçtan 52. haftaya kadar istatistiksel olarak anlamlı şekilde daha fazla ortalama SRK azalması gözlenmiş; bu durum 2Q8 EYLEA grupları için -192,4 ve -183,1 mikron, kontrol grupları için sırasıyla -66,2 ve -73,3 mikron olarak kaydedilmiştir. VIVIDDME ve VISTADME çalışmalarında söz konusu azalma 100. haftada 2Q8 EYLEA grupları için sırasıyla
-195,8 ve -191,1 mikron olarak, kontrol gruplarında ise -85,7 ve -83,9 mikron olarak
korunmuştur.
VIVIDDME ve VISTADME çalışmalarında önceden belirlenmiş olmak üzere DRSS'de en az 2 basamak iyileşme olup olmadığı değerlendirilmiştir. VIVIDDME çalışmasındaki hastaların
%73,7'si ile VISTADME çalışmasındaki hastaların %98,3'ünde DRSS skorunun değerlendirilebilir olduğu kaydedilmiştir. Elli ikinci haftada, EYLEA 2Q8 gruplarının %27,7 ve %29,1'i ile kontrol gruplarının %7,5 ve %14,3'ünde DRSS'nin en az 2 basamak iyileştiği saptanmıştır. Söz konusu oranlar 100. haftada EYLEA 2Q8 grupları için %32,6 ve %37,1; kontrol grupları için %8,2 ve %15,6 olmuştur.
VIOLET çalışmasında, tedavinin 5 ardışık aylık dozla başlatıldığı ve ardından her 2 ayda bir doz verildiği sabit aralıklarla en az bir yıllık tedaviden sonra DMÖ tedavisi için EYLEA 2 mg'ın üç farklı doz rejimi karşılaştırılmıştır. Çalışmanın 52. haftasında ve 100. haftasında, yani tedavinin ikinci ve üçüncü yıllarında, SRK'deki ortalama değişiklikler tedavi et ve uzat
(2T&U), pro re nata (2PRN) ve 2Q8 için klinik olarak benzer olmuştur: 52. haftada -2,1, 2,2 ve -18,8 mikron ve 100. haftada 2,3, -13,9 ve -15,5 mikron.
Miyopik koroidal neovaskülarizasyon
Miyopik koroidal neovaskülarizasyon (miyopik KNV), patolojik miyopi bulunan yetişkinlerde görme kaybının sık rastlanan nedenlerinden biridir. Bruch membranındaki rüptürler sonucunda bir yara iyileşme mekanizması olarak gelişir ve patolojik miyopide görmeyi en fazla tehdit eden olayı temsil eder.
MYRROR çalışmasında EYLEA tedavisi uygulanan (tedavi başlangıcında yapılan bir enjeksiyon ile persistan hastalık veya rekürrens durumunda yapılan ilave enjeksiyonlar) hastalarda SRK, tedavi başladıktan kısa süre sonra EYLEA lehine olmak üzere 24 haftada azalmış (EYLEA 2 mg tedavi grubu ile kontrol grubu için sırasıyla -79 mikron ve -4 mikron) ve bu azalma 48 hafta süresince korunmuştur. Ayrıca, ortalama KNV lezyon boyutunun azaldığı saptanmıştır.
Klinik etkililik ve güvenlilik:
Yaş tip YBMD
EYLEA'nın güvenlilik ve etkililiği yaş tip YBMD'li hastaların yer aldığı iki randomize, çok merkezli, çift maskeli, aktif kontrollü çalışmada (VIEW1 ve VIEW2) değerlendirilmiş; toplam 2.412 hastanın etkililik açısından tedavi uygulanıp, değerlendirilebilir olduğu kaydedilmiştir (EYLEA alan 1.817 hasta). Hastalarda yaş aralığı 49 ila 99, ortalama yaşın 76'dır. Klinik çalışmalarda EYLEA tedavisine randomize edilen hastaların yaklaşık
%89'unun (1.616/1.817) 65 yaş ve üzeri grupta, yaklaşık %63'ünün de (1.139/1.817) 75 yaş ve üzeri grupta olduğu kaydedilmiştir.
Her bir çalışmada, hastalar aşağıdaki 4 dozlama rejiminden 1'i için 1:1:1:1 şeklinde rastgele
atanmıştır:
Ayda bir uygulanan ilk 3 dozdan sonra 8 haftada bir 2 mg EYLEA (EYLEA 2Q8);
5.2. Farmakokinetik özellikler
Genel ÖzelliklerEYLEA, gözde lokal etkiler oluşturmak üzere doğrudan vitröz içine uygulanır.
Emilim:
İntravitreal uygulamadan sonra afliberseptin gözden sistemik dolaşıma geçişi yavaştır ve sistemik dolaşımda ağırlıklı olarak VEGF ile inaktif, stabil bir kompleks halinde olduğu görülmektedir; bununla birlikte, yalnızca serbest afliberseptin endojen VEGF'ye bağlanabilir.
Dağılım:
Sık örnekleme yapılan 6 neovasküler yaş tip YBMD'li hastanın yer aldığı farmakokinetik bir alt çalışmada, plazmadaki maksimum serbest aflibersept konsantrasyonunun (sistemik C) düşük olup, 2 mg'lık intravitreal enjeksiyondan 1 ila 3 gün sonra ortalama yaklaşık 0,02 μg/mL (aralık: 0 ila 0,054) ve dozdan iki hafta sonra neredeyse hastaların tamamında saptanamayacak düzeyde olduğu saptanmıştır. İntravitreal yoldan 4 haftada bir uygulanan aflibersept plazmada birikmez.
Plazmadaki ortalama maksimum serbest aflibersept konsantrasyonu hayvan modellerinde sistemik VEGF'nin biyolojik aktivitesini %50 oranında inhibe etmek için gereken aflibersept konsantrasyonuna kıyasla 50 ila 500 kat düşüktür ve bu modellerde dolaşımdaki serbest aflibersept düzeyi yaklaşık 10 mikrogram/mL olduktan sonra kan basıncında değişiklik olduğu ve düzeyin 1 mikrogram/mL altına inmesiyle bunun normale döndüğü gözlenmiştir. Hastalara 2 mg intravitreal uygulama yapıldıktan sonra serbest afliberseptin ortalama maksimum plazma konsantrasyonu değerinin, sağlıklı gönüllülerle yürütülen bir çalışmada afliberseptin sistemik VEGF'ye (2,91 mikrogram/mL) yarı maksimal düzeyde bağlanması
için gereken konsantrasyondan en az 100 kat düşük olduğu tahmin edilmektedir. Bu nedenle
kan basıncı gibi sistemik farmakodinamik etkilerin ortaya çıkma olasılığı düşüktür.
SRVO, RVDO, DMÖ ya da miyopik KNV'li hastalarla yürütülen farmakokinetik alt çalışmalarda, plazmadaki ortalama serbest aflibersept Cdeğeri 0,03 ila 0,05 mikrogram/mL aralığındaki değerler ve 0,14 mikrogram/mL'yi aşmayan bireysel değerler ile benzerdir. Daha sonra, serbest aflibersept plazma konsantrasyonları, genellikle bir hafta içinde alt nicelik sınırı değerlerinin altına ya da bu sınıra yakın değerlere düşer. Tüm hastalarda 4 haftadan sonraki uygulamadan önce saptanamayan konsantrasyonlara ulaşılır.
Biyotransformasyon:
EYLEA, protein bazlı bir terapötik olduğu için, herhangi bir metabolizma çalışması yürütülmemiştir.
Eliminasyon:
Serbest aflibersept VEGF'ye bağlanarak stabil ve durağan bir kompleks oluşturmaktadır. Diğer büyük proteinler gibi, serbest ve bağlı afliberseptin proteolitik katabolizma ile uzaklaştırılması beklenir.
Doğrusallık/Doğrusal olmayan durum:
Doğrusallık/doğrusal olmayan durum geçerli değildir.
Hastalardaki karakteristik özellikler
Böbrek yetmezliği olan hastalar:
Böbrek yetmezliği olan hastalarda EYLEA ile ilgili çalışma yapılmamıştır.
VIEW2 çalışmasında yer alan ve %40'ında böbrek yetmezliği olan (%24 hafif, %15 orta dereceli, %1 şiddetli) hastalara ilişkin farmakokinetik analiz, 4 veya 8 haftada bir intravitreal uygulama sonrasında aktif ilacın plazma konsantrasyonu bakımından herhangi bir fark olmadığını göstermiştir.
Benzer sonuçlar, GALILEO çalışmasında SRVO'lu hastalarda, VIVIDDME çalışmasında DMÖ'lü hastalarda ve MYRROR çalışmasında miyopik KNV'li hastalarda görülmüştür.
5.3. Klinik öncesi güvenlilik verileri
Tekrarlanan doz toksisitesiyle ilgili klinik dışı çalışmalarda etkilerin yalnızca önerilen klinik dozun intravitreal yoldan uygulanmasıyla elde edilen maksimum insan maruziyetinden önemli oranda yüksek olan aşırı sistemik maruziyet ile ortaya çıktığının gözlenmesi, klinik kullanım bakımından anlamlı bir etki olmadığına işaret etmektedir.
İntravitreal aflibersept tedavisi uygulanan maymunlarda, maksimum insan maruziyetinden yüksek sistemik maruziyet ile nazal türbinatlardaki solunum yolu epitelinde erozyon ve ülserasyon geliştiği gözlenmiştir. Serbest afliberseptin Cve EAA değerleri doğrultusunda sistemik maruziyeti insanlarda intravitreal 2 mg'lık doz ile gözlenen değerlere kıyasla sırasıyla 200 ve 700 kat yüksek bulunmuştur. Maymunlarda 0,5 mg/göz olan Advers Etki Gözlenmeyen Düzeyde (NOAEL) Cve EAA değerlerine temel alındığında sistemik maruziyetin sırasıyla 42 ve 56 kat yüksek olduğu kaydedilmiştir.
Afliberseptin mutajenik veya karsinojenik potansiyeliyle ilgili çalışma yapılmamıştır.
İntravenöz (3 ila 60 mg/kg) ve subkutan (0,1 ila 1 mg/kg) uygulama yapılan gebe tavşanlarda yapılan embriyo-fetal gelişim çalışmalarında afliberseptin intrauterin gelişmeyi etkilediği gösterilmiştir. Maternal NOAEL, sırasıyla 3 mg/kg ve 1 mg/kg'dır. Gelişimle ilgili NOAEL belirlenmemiştir. Serbest afliberseptin 0,1 mg/kg doz düzeyindeki Cve kümülatif EAA değerleri doğrultusunda sistemik maruziyeti insanlarda intravitreal 2 mg'lık doz ile gözlenen değerlere kıyasla sırasıyla 17 ve 10 kat yüksek bulunmuştur.
![]()
Erkek ve dişi fertilitesi üzerine etkiler, maymunlarda 3 ila 30 mg/kg aralığındaki dozlarda intravenöz aflibersept uygulaması yapılan 6 aylık bir çalışma kapsamında değerlendirilmiştir. Tüm doz düzeylerinde, dişilerde üremeyle ilgili hormon seviyelerinde değişiklik olması nedeniyle menstrüasyon olmadığı veya menstrüasyonun düzensiz olduğu gözlenirken, erkeklerde sperm morfolojisi ve motilitesiyle ilgili değişiklikler olduğu saptanmıştır. Serbest aflibersept için 3 mg/kg intravenöz dozda gözlenen Cve EAA doğrultusunda, sistemik maruziyetlerin insanlarda intravitreal 2 mg'lık dozdan sonra gözlenen maruziyete kıyasla sırasıyla yaklaşık 4.,900 kat ve 1.500 kat yüksek olduğu belirlenmiştir. Tüm değişikliklerin geri dönüşümlü olduğu kaydedilmiştir.
6. FARMASÖTİK ÖZELLİKLER
6.1. Yardımcı maddelerin listesi
Polisorbat 20 (E 432)
Sodyum fosfat, monobazik monohidrat (pH ayarlaması için) Sodyum fosfat, dibazik heptahidrat (pH ayarlaması için) Sodyum klorür
Sukroz Enjeksiyonluk su
6.2. Geçimsizlikler
Geçimsizlik çalışması yapılmamış olduğu için, bu tıbbi ürün diğer tıbbi ürünlerle karıştırılmamalıdır.
6.3. Raf ömrü
24 ay
6.4. Saklamaya yönelik özel tedbirler
2°C - 8°C arasında (buzdolabında) saklayınız. Dondurmayınız. Işıktan korumak için orijinal ambalajında saklayınız.
Açılmamış blister buzdolabının dışında 25°C'nin altında 24 saate kadar saklanabilir. Blister
açıldıktan sonra aseptik koşullar altında gerekli işlemlere devam edilmelidir.
6.5. Ambalajın niteliği ve içeriği
Piston tıpası (elastomerik kauçuk) ve uç kapaklı (elastomerik kauçuk) bir Luer kilit adaptörü ile siyah dozlama çizgisi ile işaretlenmiş kullanıma hazır enjektörde (tip I cam) çözelti. Her bir kullanıma hazır enjektör en az 0,09 mL çekilebilir hacim içerir. Ambalaj boyutu 1 kullanıma hazır enjektör.
6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler
Kullanılmamış olan ürünler ya da atık materyaller “Tıbbi Atıkların Kontrolü Yönetmeliği†ve “Ambalaj Atıklarının Kontrolü Yönetmelikâ€lerine uygun olarak imha edilmelidir.
Kullanıma hazır enjektör sadece tek bir göz için tek kullanımlıktır.
Temiz uygulama odasının dışında steril kullanıma hazır enjektör blisterini açmayınız.
Kullanıma hazır enjektör, önerilen 2 mg aflibersept dozundan (0,05 mL'ye eşdeğer) fazlasını
içerir. Fazla hacim uygulamadan önce dışarı atılmalıdır.
Uygulama öncesinde çözelti görsel olarak yabancı partiküler maddeler ve/veya renk bozukluğu veya fiziksel görünüşte herhangi bir değişiklik yönünden incelenmelidir. Herhangi birinin tespit edilmesi durumunda, tıbbi ürün imha edilmelidir.
İntravitreal enjeksiyon için bir adet 30 G x ½ inç oftalmik amaçla kalifiye edilmiş enjeksiyon iğnesi kullanılması önerilmektedir. Vitreusta uçuşan noktalar riskini azaltmak için intravitreal enjeksiyonda silikon içermeyen enjeksiyon iğnesi kullanılması önerilmektedir.
Kullanıma hazır enjektörün kullanımı için talimatlar:
1. EYLEA'yı uygulamaya hazır olduğunuzda, kutuyu açınız ve steril blister paketini çıkartınız. Blister paketin dışını dikkatle, içindekilerin sterilitesini koruyacak şekilde soyarak açınız. Parçaları birleştirmeye hazır olana kadar enjektörü steril tepsi içinde bırakınız. | |
2. Aseptik teknik kullanarak enjektörü steril blister paketten çıkartınız. | |
3. Enjektör kapağını çıkartmak için bir elinizle enjektörü tutarken diğer elinizi kullanarak baş ve işaret parmaklarınızla enjektör kapağını sıkıca kavrayınız. Lütfen dikkat: Enjektör kapağını çevirip açınız (kopartmayınız). |
|
4. Ürünün sterilitesini bozmamak için pistonu geri çekmeyiniz. | |
5. Aseptik teknik kullanarak enjeksiyon iğnesini Luer kilitli enjektör ucuna, çevirerek sıkıca takınız. |
|
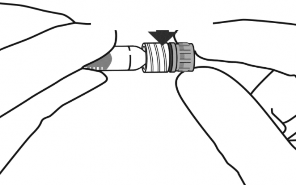
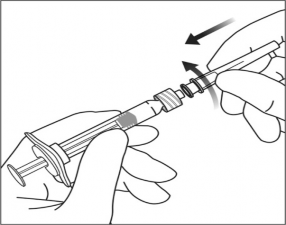
6. İğne yukarı bakacak şekilde enjektörü tutarak enjektörde hava kabarcığı olup olmadığını kontrol ediniz. Eğer hava kabarcığı varsa, kabarcıklar üst kısma çıkıncaya kadar parmağınızla enjektöre hafifçe vurunuz. |
|
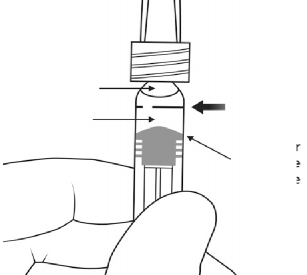
7. Tüm hava kabarcıklarını ve ilacın fazlasını uzaklaştırmak için, piston kubbesinin tabanı (kubbenin ucu değil) ile şırınga üzerindeki siyah dozlama çizgisi (0,05 mL, yani 2 mg aflibersepte eşdeğer) aynı hizaya gelecek şekilde pistona hafifçe bastırınız.
Not: Pistonun doğru konumlandırılması çok önemlidir, çünkü yanlış piston konumlandırması, etikette belirtilen dozdan daha fazlasının veya daha azının verilmesine yol açabilir. | |
Hava kabarcığı Dozlama Çözelti Çizgisi Piston Kubbe Tabanı |
Piston Kubbesi
Piston Kubbe Dozlama Tabanı Çizgisi |
8. Pistonu dikkatli bir şekilde ve sabit basınçla bastırarak enjekte ediniz. Piston enjektörün dibine ulaştığında daha fazla basınç uygulamayınız. Enjektörde gözlenen çözelti kalıntısı varsa uygulamayınız. | |
9. Kullanıma hazır enjektör sadece tek kullanımlıktır. Tek bir kullanıma hazır enjektörden çoklu dozların alınması kontaminasyon, sonrasında enfeksiyon riskini artırabilir. | |


 Pankreas Kanseri
Pankreas karnın alt kısmında yatay şekilde bulunan bir organdır. Sindirime yardımcı olan enzimleri ve kan şekerini yönetmeye yardımcı olan hormonları vücuda dağıtmakla görevlidir.
Pankreas Kanseri
Pankreas karnın alt kısmında yatay şekilde bulunan bir organdır. Sindirime yardımcı olan enzimleri ve kan şekerini yönetmeye yardımcı olan hormonları vücuda dağıtmakla görevlidir. |
 İnme
İnme, beynin hasar görmesinin sonucudur. Bu hasar, beynin bir kısmındaki ya bir kanama
ya da akut kan eksikliği nedeniyle o kısmın geçici ya da kalıcı olarak işlevini yapamamasına
yol açar.
İnme
İnme, beynin hasar görmesinin sonucudur. Bu hasar, beynin bir kısmındaki ya bir kanama
ya da akut kan eksikliği nedeniyle o kısmın geçici ya da kalıcı olarak işlevini yapamamasına
yol açar. |
İLAÇ EŞDEĞERLERİ
| Eşdeğer İlaç Adı | Barkodu | İlaç Fiyatı |
|---|---|---|
| YESAFILI | 8699514770008 | 15,786.98TL |
| ZALTRAP | 8699809980037 | 12,873.66TL |
| Diğer Eşdeğer İlaçlar |
 |
Doğum Sonrası Depresyonu Doğum sonrası depresyonu, doğumdan sonra her on kadından biri tarafından tecrübe edilen stresli bir durumdur. |
 |
Ruh ve Akıl Sağlığımızı Geliştirmek İyi akıl ve ruh sağlığı sahip olmaktan ziyade, yaptığınız şeylerdir. Akıl ve ruhsal olarak sağlıklı olmak için kendinize değer vermeli ve kendinizi kabul etmelisiniz. |
 |
Dış Gebelik Dış gebelik, her 100 gebelikten birini etkileyen, sık görülen ve ölüme sebep olabilecek bir durumdur. Bu, döllenen yumurta, rahimin dışına yerleşirse, oluşan bir durumdur. Gebelik ilerledikçe, ağrıya ve kanamalara sebep olur. |
İLAÇ GENEL BİLGİLERİ
Bayer Türk Kimya San. Tic. Ltd. Şti.
| Geri Ödeme Kodu | A14962 |
| Satış Fiyatı | 15786.98 TL [ 18 Apr 2025 ] |
| Önceki Satış Fiyatı | 15786.98 TL [ 14 Apr 2025 ] |
| Original / Jenerik | Original İlaç |
| Reçete Durumu | Normal Reçeteli bir ilaçdır. |
| Barkodu | 8699546770052 |
| Etkin Madde | Aflibersept |
| ATC Kodu | S01LA05 |
| Birim Miktar | 0.4 |
| Birim Cinsi | MG |
| Ambalaj Miktarı | 1 |
| Duyu Organları > Oküler Damar Bozukluğu Ajanları |
| İthal ( ref. ülke : Fransa ) ve Beşeri bir ilaçdır. |