GLIVEC 400 mg 30 film kapl� tablet K�sa �r�n Bilgisi
{ Imatinib }
1. BE�ER� TIBB� �R�N�N ADI
GL�VEC 400 mg film kapl� tablet
2. KAL�TAT�F VE KANT�TAT�F B�LE��M
Bir film kapl� tablet, 400 mg imatinib (478 mg imatinib mesilat beta kristali olarak)
i�erir.
Boyar madde olarak k�rm�z� demir oksit (E172) ve sar� demir oksit (E172) i�erir.
3. FARMAS�T�K FORMU
Film Kapl� Tablet
Kenarlar� d��a do�ru e�imli, oval, bir y�z�nde “NVR”, di�er y�z�nde “SL” harfleri bas�l� olan, �ok koyu sar� ile kahverengimsi turuncu aras�nda film kapl� tabletler.
ANC < 1.0 x 10/l ve/veya trombosit say�s�n�n
ANC < 1.0 x10/l ve/veya trombosit < 50 x10/l'nin tekrarlamas� durumunda, 1. ad�m� tekrarlay�n ve 260 mg/m'lik azalt�lm�� dozda GL�VEC'e devam edin.
Sitopeni 2 hafta devam ederse, 300 mg'a kadar azalt�n.
Sitopeni 2 hafta devam ederse, 200 mg/m'ye azalt�n.
ANC < 1.0 x 10/l ve/veya trombositlerin < 50 x 10/l olmas� durumunda, 1. ad�m� tekrarlay�n ve 400 mg'l�k azalt�lm�� dozda GL�VEC'e devam edin.
4. KL�N�K �ZELL�KLER
4.1. Terap�tik endikasyonlar
GL�VEC, a�a��dakilerin tedavisinde endikedir:
Birinci basamak tedavi olarak kemik ili�i transplantasyonunun d���n�lmedi�i, yeni tan� konmu� Philadelphia kromozomu (bcr-abl) pozitif (Ph+) kronik miyeloid l�semi (KML) olan eri�kin ve pediyatrik hastalar.
4.2. Pozoloji ve uygulama �ekli
Tedavi, uygun oldu�u �ekilde, hematolojik maligniteleri ve malign sarkomlar� olan hastalar�n tedavisinde deneyimli bir doktor taraf�ndan ba�lat�lmal�d�r.
400 mg ve 800 mg d���ndaki dozlar i�in (a�a��daki dozaj �nerisine bak�n) 100 mg b�l�nebilir tablet mevcuttur.
400 mg ve �zerindeki dozlar i�in (a�a��daki dozaj �nerisine bak�n�z) 400 mg'l�k bir tablet (b�l�nemez) mevcuttur.
Re�ete edilen doz, gastrointestinal iritasyon riskini en aza indirmek i�in bir yemek ve b�y�k bir bardak su ile oral olarak uygulanmal�d�r. 400 mg veya 600 mg'l�k dozlar g�nde bir kez, 800 mg'l�k doz ise sabah ve ak�am olmak �zere g�nde iki kez 400 mg olarak uygulanmal�d�r.
Film kapl� tabletleri yutamayan hastalar i�in, tabletler bir bardak su veya elma suyu i�inde da��t�labilir. Gerekli say�da tablet, uygun hacimdeki i�ece�in (100 mg tablet i�in yakla��k 50 ml, 400 mg tablet i�in yakla��k 200 ml) i�erisine konulmal� ve ka��kla kar��t�r�lmal�d�r. Tabletin/tabletlerin tamamen par�alanmas�ndan hemen sonra s�spansiyon uygulanmal�d�r.
Eri�kin hastalarda KML i�in pozoloji
Kronik faz KML'de eri�kin hastalar i�in �nerilen GL�VEC dozu 400 mg/g�n'd�r. Kronik faz KML, a�a��daki kriterlerin t�m�n�n kar��lanmas� olarak tan�mlan�r: kan ve kemik ili�inde < %15 blastlar, < %20 periferik kan bazofil, > 100 x 109/l trombosit say�s�.
Akselere fazdaki eri�kin hastalar i�in �nerilen GL�VEC dozu 600 mg/g�n'd�r. Akselere faz, a�a��dakilerden herhangi birinin varl��� olarak tan�mlan�r: kan veya kemik ili�inde ≥
%15 fakat < %30 blast, kan veya kemik ili�inde ≥ %30 blast art� promiyelosit say�s� (<
%30 blast sa�layan), ≥ %20 periferik kan bazofil say�s�, tedaviyle ili�kisiz olarak < 100 x 109/l trombosit say�s�.
Blast krizindeki eri�kin hastalar i�in �nerilen GL�VEC dozu 600 mg/g�n'd�r. Blast krizi, kan veya kemik ili�inde ≥ %30 blast veya hepatosplenomegali d���ndaki ekstramed�ller hastal�k olarak tan�mlan�r.
Tedavi s�resi: Klinik �al��malarda, hastal�k progresyonuna kadar GL�VEC tedavisine devam edilmi�tir. Tam bir sitogenetik yan�t elde edildikten sonra tedaviyi durdurman�n etkisi ara�t�r�lmam��t�r.
Ciddi advers ila� reaksiyonu ve l�semi ile ili�kili olmayan ciddi n�tropeni veya trombositopeni yoklu�unda, a�a��daki durumlarda, kronik faz hastal��� olan hastalarda 400 mg'dan 600 mg'a veya 800 mg'a veya akselere faz veya blast krizi olan hastalarda 600 mg'dan maksimum 800 mg'a (g�nde iki kez 400 mg olarak verilir) kadar doz art��lar� d���n�lebilir: hastal�k progresyonu (herhangi bir zamanda); en az 3 ayl�k tedaviden sonra tatmin edici bir hematolojik yan�t elde edilememesi; 12 ayl�k tedaviden sonra sitogenetik yan�t elde edilememesi; veya �nceden elde edilmi� bir hematolojik ve/veya sitogenetik yan�t�n kayb�. Daha y�ksek dozlarda advers reaksiyon insidans�nda art�� potansiyeli g�z �n�ne al�nd���nda, hastalar doz art�r�m�n�n ard�ndan yak�ndan izlenmelidir.
�ocuklarda KML i�in pozoloji
�ocuklar i�in dozaj ayarlamas�, v�cut y�zey alan�na (mg/m2) g�re yap�lmal�d�r. Kronik faz KML ve ileri faz KML'si olan �ocuklar i�in g�nl�k 340 mg/m2 doz �nerilir (toplam doz 800 mg'� ge�memelidir). Tedavi g�nde bir doz olarak verilebilir veya alternatif olarak g�nl�k doz, biri sabah ve biri ak�am olmak �zere iki uygulamaya b�l�nebilir. Doz tavsiyesi �u anda az say�da pediyatrik hastaya dayanmaktad�r (bkz. B�l�m 5.1 ve 5.2). 2 ya��n alt�ndaki �ocuklar�n tedavisine ili�kin deneyim bulunmamaktad�r.
Ciddi advers ila� reaksiyonu ve l�semi ile ili�kili olmayan ciddi n�tropeni veya trombositopeni yoklu�unda, a�a��daki durumlarda, �ocuklarda g�nl�k 340 mg/m2'den g�nde 570 mg/m2'ye (toplam 800 mg dozu a�mamak �zere) doz art��lar� d���n�lebilir: hastal�k progresyonu (herhangi bir zamanda); en az 3 ayl�k tedaviden sonra tatmin edici bir hematolojik yan�t elde edilememesi; 12 ayl�k tedaviden sonra sitogenetik yan�t elde edilememesi; veya �nceden elde edilmi� bir hematolojik ve/veya sitogenetik yan�t�n kayb�. Daha y�ksek dozlarda advers reaksiyon insidans�nda art�� potansiyeli g�z �n�ne al�nd���nda, hastalar doz art�r�m�n�n ard�ndan yak�ndan izlenmelidir.
Eri�kin hastalarda Ph+ ALL i�in pozoloji
Ph+ ALL'li eri�kin hastalar i�in �nerilen GL�VEC dozu 600 mg/g�n'd�r. Bu hastal���n y�netiminde uzman hematologlar, bak�m�n t�m a�amalar�nda tedaviyi denetlemelidir.
Tedavi �emas�: Mevcut verilere dayanarak, yeni tan� konulan Ph+ ALL'li eri�kin hastalar i�in kemoterapinin ind�ksiyon faz�nda, konsolidasyon ve idame fazlar�nda kemoterapi ile kombinasyon halinde 600 mg/g�n olarak uyguland���nda (bkz. B�l�m 5.1) GL�VEC'in etkili ve g�venli oldu�u kan�tlanm��t�r. GL�VEC tedavisinin s�resi, se�ilen tedavi program�na g�re de�i�ebilir, ancak genellikle GL�VEC'e daha uzun s�re maruziyet daha iyi sonu�lar vermi�tir.
N�kseden veya diren�li Ph+ALL'li eri�kin hastalar i�in 600 mg/g�n GL�VEC monoterapisi g�venlidir, etkilidir ve hastal�kta progresyon olu�ana kadar verilebilir.
�ocuklarda Ph+ ALL i�in pozoloji
�ocuklar i�in dozaj ayarlamas�, v�cut y�zey alan�na (mg/m2) g�re yap�lmal�d�r. Ph+ ALL'li �ocuklar i�in g�nl�k 340 mg/m2 doz �nerilir (toplam doz 600 mg'� ge�memelidir).
MDS/MPD i�in Pozoloji
MDS/MPD'li eri�kin hastalar i�in �nerilen GL�VEC dozu 400 mg/g�n'd�r.
Tedavi s�resi: �imdiye kadar ger�ekle�tirilen tek klinik �al��mada, hastal�k progresyonuna kadar GL�VEC tedavisine devam edilmi�tir (bkz. B�l�m 5.1). Analiz s�ras�nda, tedavi s�resi ortanca 47 ayd�r (24 g�n - 60 ay).
HES/CEL i�in pozoloji
HES/CEL'li eri�kin hastalar i�in �nerilen GL�VEC dozu 100 mg/g�n'd�r. De�erlendirmeler tedaviye yetersiz yan�t oldu�unu g�steriyorsa, advers ila�
reaksiyonlar�n�n yoklu�unda dozun 100 mg'dan 400 mg'a ��kar�lmas� d���n�lebilir. Hasta fayda g�rd��� s�rece tedaviye devam edilmelidir.
G�ST i�in pozoloji
Rezeke edilemeyen ve/veya metastatik malign GIST'li eri�kin hastalar i�in �nerilen GL�VEC dozu 400 mg/g�n'd�r.
Daha d���k dozda progrese olan hastalarda 400 mg'dan 600 mg'a veya 800 mg'a doz art��lar�n�n etkisine ili�kin s�n�rl� veriler mevcuttur (bkz. B�l�m 5.1).
Tedavi s�resi: GIST hastalar�nda yap�lan klinik �al��malarda, GL�VEC tedavisine hastal�k progresyonuna kadar devam edilmi�tir. Analiz s�ras�nda, tedavi s�resi medyan 7 ayd�r (7 g�n ila 13 ay). Bir yan�t elde edildikten sonra tedaviyi durdurman�n etkisi ara�t�r�lmam��t�r.
G�ST rezeksiyonu sonras� eri�kin hastalar�n adjuvan tedavisi i�in �nerilen GL�VEC dozu
400 mg/g�n'd�r. Optimal tedavi s�resi hen�z belirlenmemi�tir. Bu endikasyonu
destekleyen klinik �al��mada tedavi s�resi 36 ay olmu�tur (bkz. B�l�m 5.1).
DFSP i�in pozoloji
DFSP'li eri�kin hastalar i�in �nerilen GL�VEC dozu 800 mg/g�n'd�r.
Advers reaksiyonlar i�in doz ayarlamas� Hematolojik olmayan advers reaksiyonlar
GL�VEC kullan�m�yla ciddi hematolojik olmayan bir advers reaksiyon geli�irse, olay d�zelene kadar tedavi durdurulmal�d�r. Daha sonra, olay�n ba�lang��taki ciddiyetine ba�l� olarak uygun �ekilde tedaviye devam edilebilir.
Bilirubinde > 3 x normal s�n�r�n �st limitini (NK�S) veya karaci�er transaminazlar�nda
> 5 x NK�S y�kselmeler olu�ursa, bilirubin seviyeleri < 1.5 x NK�S'e ve transaminaz seviyeleri < 2.5 x NK�S'e d�nene kadar GL�VEC kesilmelidir. Daha sonra GL�VEC ile tedaviye azalt�lm�� g�nl�k dozda devam edilebilir. Eri�kinlerde doz 400'den 300 mg'a veya 600'den 400 mg'a veya 800 mg'dan 600 mg'a ve �ocuklarda 340'tan 260 mg/m2/g�n'e d���r�lmelidir.
Hematolojik advers reaksiyonlar
Ciddi n�tropeni ve trombositopeni i�in a�a��daki tabloda belirtildi�i gibi doz azalt�lmas� veya tedaviye ara verilmesi �nerilir.
N�tropeni ve trombositopeni i�in doz ayarlamalar�:
HES/CEL (ba�lang�� dozu 100 mg) | ANC < 1.0 x 10/l ve/veya trombosit < 50 x 10/l | ANC ≥ 1.5 x 10/l ve trombosit ≥ 75 x 10/l olana kadar GL�VEC'i durdurun. Sitopeni 4 hafta devam ederse ve hala l�semi ile ili�kili de�ilse, ANC ≥ 1 x 10/l ve trombosit Sitopeni 4 hafta devam ederse ve hala l�semi ile ili�kili de�ilse, ANC ≥ 1 x 10/l ve trombosit 4.3. KontrendikasyonlarAktif maddeye veya eksipiyanlardan herhangi birine kar�� a��r� duyarl�l�k (bkz. B�l�m 6.1). 4.4. �zel kullan�m uyar�lar� ve �nlemleriGL�VEC, ba�ka ila�larla birlikte uyguland���nda, ila� etkile�imleri g�r�lme potansiyeli bulunmaktad�r. GL�VEC, proteaz inhibit�rleri, azol antifungaller, belirli makrolitler (bkz. B�l�m 4.5), dar terap�tik pencereye sahip CYP3A4 substratlar� (�rn. siklosporin, pimozid, takrolimus, sirolimus, ergotamin, diergotamin, fentanil, alfentanil, terfenadin, bortezomib, dosetaksel, kinidin) veya varfarin ve di�er kumarin t�revleri ile birlikte verildi�inde dikkatli olunmal�d�r (bkz. B�l�m 4.5). �matinib ve CYP3A4 enzimini ind�kleyen t�bbi �r�nlerin (�rn. deksametazon, fenitoin, karbamazepin, rifampisin, fenobarbital veya Hypericum perforatum [Sar� kantaron]) birlikte e�zamanl� kullan�m�, GL�VEC maruziyetini �nemli �l��de azaltarak terap�tik ba�ar�s�zl�k riskini art�rabilir. Bu nedenle kuvvetli CYP3A4 ind�kleyicilerinin ve imatinibin birlikte e�zamanl� uygulamas�ndan ka��n�lmal�d�r (bkz. B�l�m 4.5). Hipotiroidizm: GL�VEC tedavisi s�ras�nda levotiroksin replasman� yap�lan tiroidektomi hastalar�nda klinik hipotiroidizm olgular� bildirilmi�tir (bkz. B�l�m 4.5). Bu t�r hastalarda tiroid stimule edici hormon (TSH) d�zeyleri yak�ndan izlenmelidir. Hepatotoksisite: GL�VEC temel olarak karaci�erde metabolize olur ve at�l�m�n yaln�zca %13'� b�brekler arac�l���ylad�r. Karaci�er disfonksiyonu (hafif, orta ve �iddetli) olan hastalarda, periferik kan say�mlar� ve karaci�er enzimleri dikkatli bir �ekilde izlenmelidir (bkz. B�l�m 4.2, 4.8, 5.1, 5.2). GIST hastalar�nda karaci�er yetmezli�ine sebebiyet verebilecek karaci�er metaztazlar� g�r�lmesi olas�d�r. �matinib ile karaci�er yetmezli�i ve hepatik nekroz dahil karaci�er hasar� vakalar� g�zlenmi�tir. �matinib, y�ksek doz kemoterapi rejimleri ile kombine edildi�inde, ciddi hepatik reaksiyonlarda bir art�� bildirilmi�tir. �matinibin karaci�er fonksiyon bozuklu�u ile ili�kili oldu�u bilinen kemoterapi rejimleriyle kombine edildi�i durumlarda karaci�er fonksiyonu dikkatle izlenmelidir (bkz. B�l�m 4.5 ve 4.8). S�v� retansiyonu: GL�VEC alan yeni tan� konulmu� KML hastalar�n�n yakla��k % 2,5'inde ciddi s�v� retansiyonu (plevral ef�zyon, �dem, pulmoner �dem, assit, y�zeysel �dem) ortaya ��kt��� bildirilmi�tir. Bu nedenle, hastalarda d�zenli aral�klarla kilo kontrol� �nerilir. Beklenmedik, ani bir kilo art��� dikkatli ara�t�r�lmal� ve gerekti�inde uygun destek tedavisi uygulanmal� ve terap�tik �nlemler al�nmal�d�r. Klinik �al��malarda, ya�l� hastalarda ve daha �nceden kardiyak hastal�k hikayesi bulunanlarda bu olaylar�n insidanslar�n�n artt��� saptanm��t�r. Bu nedenle kardiyak disfonksiyonu olan hastalarda dikkatli olunmal�d�r. Kalp hastal��� olan hastalar: Kalp hastal���, kalp yetmezli�i a��s�ndan risk fakt�rleri bulunan veya b�brek yetmezli�i hikayesi olan hastalar dikkatlice takip edilmeli, kalp veya b�brek yetmezli�ini d���nd�ren belirti ve semptomlar� olan her hasta de�erlendirilmeli ve tedavi edilmelidir. Miyokardiyum i�inde hipereozinofili sendromu (HES) h�crelerinin ok�lt infiltrasyon g�r�ld��� hastalarda izole kardiyojenik �ok/sol ventrik�l disfonksiyonu olgular�, imatinib tedavisine ba�lanmas�yla beraber olu�an HES h�cre degran�lasyonu ile ili�kilendirilmi�tir. Bu durumun sistemik steroidler kullan�larak, dola��m� destekleyen �nlemler alarak ve imatinib tedavisini ge�ici olarak durdurarak geri d�n��ebilece�i bildirilmi�tir. �matinib ile yayg�n olmayan kardiyak yan etkiler bildirildi�i i�in, HES/CEL (kronik eozinofilik l�semi) pop�lasyonunda GL�VEC tedavisine ba�lamadan �nce dikkatli bir yarar/zarar (risk) de�erlendirmesi yap�lmal�d�r. PDFGR gen yeniden d�zenlemeleri ile miyelodisplastik/miyeloproliferatif hastal�klar (MDS/MPD) ve sistemik mastositoz y�ksek eozinofil d�zeyleri ile ili�kili olabilir. Bu nedenle, eozinofil d�zeylerinin y�ksek oldu�u MDS/MPD hastalar�nda, sistemik mastositoz (SM) hastalar�nda ve HES/CEL hastalar�nda imatinib uygulanmadan �nce kardiyoloji uzman� taraf�ndan de�erlendirme yap�lmal�, ekokardiyografik inceleme yap�lmal� ve serum troponin d�zeyleri �l��lmelidir. Bunlardan birinde anormallik tespit edilirse, kardiyoloji uzman� ile beraber takip edilmeli ve tedavi ba�lang�c�nda imatinible birlikte 1-2 hafta boyunca 1-2 mg/kg dozunda sistemik steroid kullan�lmas� d���n�lmelidir. Gastrointestinal kanama: Rezeke edilemeyen ve/veya metastatik GIST'li hastalarda y�r�t�len bir �al��mada gerek gastrointestinal gerekse t�m�r i�i hemorajiler bildirilmi�tir (bkz. B�l�m 4.8). Eldeki verilere dayan�larak, GIST'li hastalar� her iki hemoraji tipi a��s�ndan daha y�ksek risk alt�na sokan herhangi bir predispozan fakt�r tan�mlanmam��t�r (�rn. t�m�r b�y�kl���, t�m�r yeri, p�ht�la�ma bozukluklar�). Vask�larite art��� ve kanamaya yatk�nl�kta art��, GIST'in do�as�nda yer ald���ndan ve hastal���n klinik seyrinin par�as� oldu�undan, t�m hastalarda hemoraji izlemi ve kontrol�ne y�nelik standart uygulamalar ve prosed�rler uygulanmal�d�r. Ayr�ca, KML, ALL ve di�er hastal�klar� olan hastalarda pazarlama sonras� deneyimde nadir bir gastrointestinal hemoraji nedeni olarak gastrik antral vask�ler ektazi (GAVE) bildirilmi�tir (bkz. B�l�m 4.8). Gerekti�inde, GL�VEC tedavisinin b�rak�lmas� d���n�lmelidir. T�m�r lizis sendromu: T�m�r lizis sendromu (TLS) meydana gelme olas�l��� nedeniyle, GL�VEC ba�lat�lmadan �nce klinik a��dan anlaml� dehidrasyonun d�zeltilmesi ve y�ksek �rik asit d�zeylerinin tedavisi �nerilmektedir (bkz. B�l�m 4.8). Hepatit B reaktivasyonu: Hepatit B vir�s� (HBV) kronik ta��y�c�s� olan hastalarda, BCR-ABL tirozin kinaz inhibit�rleri ile tedavi sonras�, Hepatit B reaktivasyonu ortaya ��km��t�r. Baz� vakalar, karaci�er nakli veya �l�me sebep olan akut karaci�er yetmezli�i veya fulminan hepatit ile sonu�lan�r. GL�VEC tedavisine ba�lanmadan �nce, hastalar HBV enfeksiyonu a��s�ndan test edilmelidir. Pozitif HBV serolojisine sahip (aktif hastal��� olanlar dahil) ve tedavi s�ras�nda HBV enfeksiyonu i�in pozitif test sonucu veren hastalarda, tedavi ba�lat�lmadan �nce karaci�er hastal��� ve HBV tedavisi konusunda uzman hekimlere dan���lmal�d�r. GL�VEC ile tedaviye ihtiya� duyan HBV ta��y�c�lar�, tedavi boyunca ve tedavi sonland�r�ld�ktan sonra birka� ay boyunca aktif HBV enfeksiyonu bulgu ve belirtileri i�in yak�ndan izlenmelidir (bkz. B�l�m 4.8). Fototoksisite: GL�VEC tedavisi ile ili�kili fototoksisite riski nedeniyle do�rudan g�ne� �����na maruziyetten ka��n�lmal� ya da maruziyet en aza indirilmelidir. Hastalara, koruyucu k�yafetler ya da y�ksek g�ne� koruma fakt�r�ne (SPF) sahip g�ne� kremlerinin kullan�m� gibi �nlemler almalar� s�ylenmelidir. Trombotik mikroanjiyopati: BCR-ABL tirozin kinaz inhibit�rleri (TK�'ler), GL�VEC i�in bireysel vaka raporlar� dahil olmak �zere trombotik mikroanjiyopati (TMA) ile ili�kilendirilmi�tir (bkz. B�l�m 4.8). E�er GL�VEC alan bir hastada TMA ile ili�kili laboratuar ya da klinik bulgular meydana gelirse, tedavi b�rak�lmal� ve ADAMTS13 aktivitesi ve anti-ADAMTS13- antikorunun belirlenmesi dahil olmak �zere TMA i�in kapsaml� bir de�erlendirme yap�lmal�d�r. E�er d���k ADAMTS13 aktivitesi ile birlikte anti-ADAMTS13-antikoru y�kselmi�se, GL�VEC tedavisi yeniden ba�lat�lmamal�d�r. Laboratuvar testleri: GL�VEC ile tedavi s�ras�nda d�zenli olarak tam kan say�mlar� yap�lmal�d�r. KML hastalar�nda GL�VEC ile tedavi, n�tropeni ya da trombositopeni ile ili�kilendirilmi�tir. Bununla birlikte, bu sitopenilerin ortaya ��k���, hastal���n tedavi edildi�i evreye ba�l�d�r ve kronik fazda KML bulunan hastalarla kar��la�t�r�ld���nda, h�zlanm�� fazda KML ya da blast krizinde bulunan hastalarda daha s�k olmaktad�r. Bu durumda, B�l�m 4.2'de �nerildi�i gibi GL�VEC tedavisi kesilebilir ya da dozu azalt�labilir. GL�VEC alan hastalarda karaci�er fonksiyonu (transaminazlar, bilirubin, alkalen fosfataz) d�zenli olarak takip edilmelidir. B�brek fonksiyonu bozuk olan hastalarda, imatinib plazma maruziyetinin, b�brek fonksiyonu normal olan hastalara k�yasla daha y�ksek oldu�u g�r�lmektedir; bunun olas� nedeni imatinibe ba�lanan bir protein olan alfa-asit glikoproteinin (AGP) plazma d�zeylerinin bu hastalarda daha y�ksek olmas�d�r. B�brek bozuklu�u olan hastalarda en d���k ba�lang�� dozu verilmelidir. �iddetli b�brek bozuklu�u olan hastalar dikkatle tedavi edilmelidir. Doz, tolere edilmiyorsa azalt�labilir (bkz. B�l�m 4.2 ve 5.2). Uzun s�reli imatinib ile tedavi, b�brek fonksiyonunda klinik olarak anlaml� azalma ile ili�kili olabilir. Bu nedenle imatinib tedavisine ba�lanmadan �nce b�brek fonksiyonu de�erlendirilmeli ve tedavi s�ras�nda yak�ndan izlenmeli, b�brek fonksiyon bozuklu�u a��s�ndan risk fakt�rleri g�steren hastalara �zellikle dikkat edilmelidir. B�brek fonksiyon bozuklu�u g�zlenirse, standart tedavi k�lavuzlar� uyar�nca uygun y�netim ve tedavi re�ete edilmelidir. Pediyatrik pop�lasyon �matinib alan �ocuklarda ve ergenlik �ncesi �ocuklarda g�r�len b�y�me gerili�ine ili�kin vaka raporlar� al�nm��t�r. KML pediyatrik pop�lasyonundaki g�zlemsel bir �al��mada iki k���k alt k�mede pubertal durum veya cinsiyet fark etmeksizin medyan boy standart sapma skorlar�nda 12 ve 24 ay sonra istatistiksel olarak anlaml� (fakat klinik anlaml�l��� belirsiz) bir azalma bildirilmi�tir. �matinib tedavisi g�rmekte olan �ocuklarda b�y�menin yak�ndan izlenmesi �nerilir (bkz. B�l�m 4.8). 4.5. Di�er t�bbi �r�nler ile etkile�imler ve di�er etkile�im �ekilleri�matinibin plazma konsantrasyonlar�n� art�rabilen ila�lar: Sitokrom P450 izoenzimlerinden CYP3A4 aktivitesini inhibe eden maddeler (�rn. indinavir, lopinavir/ritonavir, ritonavir, sakinavir, telaprevir, nelfinavir ve boseprevir gibi proteaz inhibit�rleri; ketokonazol, itrakonazol, posakonazol ve varikonazol gibi azol antifungal ajanlar; eritromisin, klaritromisin ve telitromisin gibi belirli makrolidler) metabolizmay� azaltabilir ve imatinib konsantrasyonlar�n� art�rabilirler. Sa�l�kl� deneklere tek doz ketokonazol (bir CYP3A4 inhibit�r�) ile birlikte uyguland���nda, imatinibe maruz kalma durumunda anlaml� bir art�� ortaya ��km��t�r (imatinibin ortalama Cve EAA de�erleri s�ras�yla % 26 ve % 40 artm��t�r). GL�VEC, CYP3A4 ailesinin inhibit�rleri ile birlikte verilirken dikkatli olunmal�d�r. �matinibin plazma konsantrasyonlar�n� azaltabilen ila�lar: CYP3A4 aktivitesini ind�kleyen maddeler (�rn. deksametazon, fenitoin, karbamazepin, rifampisin, fenobarbital, fosfenitoin, pirimidon ya da St. John's Wort olarak da bilinen Hypericum perforatum) GL�VEC'e maruz kalmay� anlaml� �ekilde azaltabilir ve potansiyel olarak tedavinin ba�ar�s�zl�k riskini artt�rabilir. Tedavi �ncesi verilen birden fazla 600 mg rifampisin dozunun ard�ndan tek bir 400 mg GL�VEC dozunun uygulanmas�, Cve EAAde�erlerinde, rifampisin tedavisinin olmad��� durumdaki ilgili de�erlerin en az %54 ve %74'� oran�nda d����e neden olmu�tur. GL�VEC ile tedavi edilen malign gliomal� hastalarda karbamazepin, okskarbazepin ve fenitoin gibi enzim ind�kleyici antiepileptik ila�lar (EIAED'ler) al�rken benzer sonu�lar g�zlenmi�tir. �matinib i�in plazma EAA, EIAED kullanmayan hastalara k�yasla %73 azalm��t�r. Rifampisin veya di�er g��l� CYP3A4 ind�kleyicileri ile imatinibin birlikte e�zamanl� kullan�m�ndan ka��n�lmal�d�r. GL�VEC ile plazma konsantrasyonu de�i�ebilen ila�lar: �matinib, simvastatinin (CYP3A4 substrat�) ortalama Cve EAA de�erlerini s�ras�yla 2- ve 3,5 kat art�rmaktad�r ve bu durum CYP3A4'�n imatinib taraf�ndan inhibe edildi�ini g�stermektedir. Bu nedenle GL�VEC, dar bir terap�tik pencereye sahip CYP3A4 substratlar�yla (�rn. siklosporin, pimozid, takrolimus, sirolimus, ergotamin, diergotamin, fentanil, alfentanil, terfenadin, bortezomib, dosetaksel, kinidin) birlikte uyguland���nda dikkatli olunmal�d�r. GL�VEC, CYP3A4 taraf�ndan metabolize edilen di�er ila�lar�n plazma konsantrasyonunu art�rabilir (�rn. triazolo-benzodiazepinler, dihidropiridin kalsiyum kanal blok�rleri, baz� HMG-KoA red�ktaz inhibit�rleri, �rn. statinler, vs.). �matinib kullan�m� ile birlikte bilinen artm�� kanama riski nedeniyle (�rn. hemoraji), anti-koag�lasyon gerektiren hastalar, varfarin gibi kumarin t�revleri yerine d���k molek�l a��rl�kl� ya da standart heparin ile tedavi edilmelidir. GL�VEC, in vitro ko�ullarda, CYP3A4 aktivitesini etkileyen benzer konsantrasyonlarda sitokrom P450 izoenzim CYP2D6 aktivitesini inhibe eder. G�nde iki kez 400 mg dozda uygulanan imatinibin, CYP2D6-arac�l� metoprolol metabolizmas� �zerinde bir inhibit�r etkisi vard�r; metoprolol Cve EAA de�erleri yakla��k %23 kadar artar (%90 GA [1,16-1,30]). �matinib, CYP2D6 substratlar� ile birlikte uyguland���nda doz ayarlamalar�n�n gerekli olmad��� g�r�lmektedir ancak metoprolol gibi dar terap�tik pencereye sahip CYP2D6 substratlar� ile dikkatli olunmas� tavsiye edilir. Metoprolol ile tedavi edilen hastalarda klinik izlem g�z �n�nde bulundurulmal�d�r. GL�VEC, in vitro ortamda parasetamol O- glukuronidasyon 58,5 mikromol/l Ki de�eri ile inhibe eder. Bu inhibisyon in vivo ko�ullarda, 400 mg GL�VEC ve 1000 mg parasetamol uygulamas�n�n ard�ndan g�r�lmemi�tir. Daha y�ksek GL�VEC ve parasetamol dozlar� �al���lmam��t�r. Bu nedenle y�ksek dozda GL�VEC ve parasetamol birlikte e�zamanl� kullan�l�rken dikkatli olunmal�d�r. Levotiroksin kullanan tiroidektomi hastalar�nda GL�VEC birlikte uyguland���nda levotiroksine plazma maruziyeti azalabilir (bkz. B�l�m 4.4). Bu nedenle dikkat �nerilir. Bununla birlikte g�zlenen etkile�imin mekanizmas� halen bilinmemektedir. Ph+ ALL hastalar�nda kemoterapiyle birlikte GL�VEC uygulanmas�yla ilgili klinik deneyim bulunmaktad�r (bkz B�l�m 5.1), ancak imatinib ve kemoterapi rejimleri aras�ndaki ila�-ila� etkile�imleri iyi tan�mlanmam��t�r. �matinibin advers etkileri, �rn. hepatotoksisite, miyelosupresyon ya da di�erleri art�� g�sterebilir ve L-asparaginaz ile e�zamanl� kullan�m�n hepatatoksite art���yla ili�kili olabilece�i bildirilmi�tir (bkz. B�l�m 4.8). Bu nedenle, GL�VEC'in kombinasyonda kullan�m� �zel dikkat gerektirmektedir. �zel pop�lasyonlara ili�kin ek bilgiler�zel pop�lasyonlara ili�kin klinik etkile�im �al��mas� y�r�t�lmemi�tir. Pediyatrik pop�lasyonPediyatrik pop�lasyona ili�kin klinik etkile�im �al��mas� y�r�t�lmemi�tir. 4.6. Gebelik ve laktasyonGebelik kategorisi: D �ocuk do�urma potansiyeli bulunan kad�nlar / Do�um kontrol� (Kontrasepsiyon)�ocuk do�urma potansiyeli bulunan kad�nlara tedavi s�ras�nda ve tedavi durdurulduktan sonra en az 15 g�n boyunca etkili bir kontrasepsiyon uygulamalar� �nerilmelidir. Gebelik d�nemi�matinibin gebe kad�nlarda kullan�m�na ili�kin s�n�rl� veri mevcuttur. GL�VEC alan kad�nlarda, spontan d���kler ve bebekte konjenital anomalilerle ilgili pazarlama sonras� raporlar mevcuttur. Ancak, hayvanlar �zerinde yap�lan ara�t�rmalar, �reme toksisitesinin bulundu�unu g�stermi�tir (bkz. B�l�m 5.3) ve fet�s i�in potansiyel risk bilinmemektedir. GL�VEC, kesinlikle gerekli olmad�k�a gebelik d�neminde kullan�lmamal�d�r. Gebelik s�ras�nda kullan�lmas� durumunda, hastaya fet�s �zerindeki potansiyel riskleri hakk�nda bilgi verilmelidir. Laktasyon d�nemi�matinibin insan s�t�ne ge�i�i hakk�nda s�n�rl� bilgi vard�r. Emziren iki kad�nda yap�lan �al��malar, hem imatinibin hem de aktif metabolitinin anne s�t�ne ge�ebilece�ini ortaya koymu�tur. Tek bir hastada incelenen s�t plazma oran�, imatinib i�in 0,5 ve metabolit i�in 0,9 olarak belirlenerek metabolitin s�te daha fazla ge�ti�ini d���nd�rm��t�r. �matinib ve metabolitinin toplam konsantrasyonu ve bebeklerin maksimum g�nl�k s�t al�m� d���n�ld���nde, toplam maruziyetin d���k olmas� beklenir (bir terap�tik dozun ~%10'u). Bununla birlikte, bebe�in imatinibe d���k dozlarda maruz kalmas�n�n etkileri bilinmedi�inden, anneler GL�VEC tedavisi s�ras�nda ve tedavi durdurulduktan sonra en az 15 g�n boyunca bebeklerini emzirmemelidir. �reme yetene�i/FertiliteYap�lan klinik d��� �al��malarda, �reme parametreleri �zerinde etkiler g�zlenmi� olsa da di�i ve erkek farelerin fertiliteleri etkilenmemi�tir. (bkz. B�l�m 5.3) GL�VEC alan hastalarda ilac�n fertilite ve gametogenez �zerindeki etkileri ile ilgili �al��malar yap�lmam��t�r. GL�VEC tedavisi g�ren ve fertilite konusunda endi�e duyan hastalar, hekimlerine dan��mal�d�r. 4.7. Ara� ve makine kullan�m� �zerindeki etkilerHastalara imatinib ile tedavi s�ras�nda ba� d�nmesi, bulan�k g�rme ya da somnolans gibi istenmeyen etkiler ya�ayabilecekleri bildirilmelidir. Bu nedenle, araba ya da ara� kullan�rken dikkatli olunmas� �nerilmelidir. 4.8. �stenmeyen etkiler�leri a�amalarda maligniteleri olan hastalarda, altta yatan hastal�k, progresyon ve say�s�z t�bbi �r�n�n birlikte uygulanmas� ile ba�lant�l� �e�itli semptomlar nedeniyle advers reaksiyonlar�n nedensellik ili�kisinin de�erlendirilmesini zorla�t�ran say�s�z karma��kla�t�r�c� t�bbi durum mevcut olabilir. KML klinik �al��malar�nda ila�la ili�kili advers reaksiyonlar nedeniyle ilac�n kesilmesi durumu, yeni tan� konan hastalar�n %2,4'�nde, interferon tedavisinin ba�ar�s�z olmas�ndan sonra ge� kronik fazdaki hastalar�n %4'�nde, interferon tedavisinin ba�ar�s�z olmas�ndan sonra h�zlanm�� fazdaki hastalar�n %4'�nde ve interferon tedavisinin ba�ar�s�z olmas�ndan sonra blast krizindeki hastalar�n %5'inde g�zlenmi�tir. GIST �al��mas�nda ila�, hastalar�n %4'�ne ila�la ili�kili advers reaksiyonlar nedeniyle kesilmi�tir. �ki istisna haricinde advers reaksiyonlar, t�m endikasyonlarda benzer olmu�tur. GIST ile kar��la�t�r�ld���nda KML hastalar�nda daha fazla miyelos�presyon g�r�lm��t�r, bu durum olas�l�kla altta yatan hastal�k ile ili�kilidir. Rezekte edilemeyen ve/veya metastatik GIST'li hastalarda y�r�t�len bir �al��mada 7 (%5) hasta CTC derece 3/4 GI kanamalar (3 hasta), t�m�r i�i kanamalar (3 hasta) ya da ikisini birden (1 hasta) ya�am��t�r. GI t�m�r b�lgeleri GI kanamalar�n kayna�� olmu� olabilir (bkz. B�l�m 4.4). GI ve t�m�r kanamalar�, ciddi ve bazen �l�mc�l olabilmektedir. Her iki endikasyonda en s�k bildirilen (≥%10) ila�la ili�kili advers reaksiyonlar, hafif bulant�, kusma, ishal, abdominal a�r�, yorgunluk, kas a�r�s�, kas kramplar� ve d�k�nt� olmu�tur. Y�zeysel �demler, t�m �al��malarda yayg�n bir bulgu olmu� ve temelde periorbital ya da alt uzuv �demleri �eklinde tarif edilmi�tir. Bununla birlikte, bu �demler nadiren �iddetli olmu� ve di�retiklerle, di�er destekleyici �nlemlerle veya GL�VEC dozu azalt�larak kontrol edilebilmi�tir. �matinib, Ph+ ALL hastalar�nda y�ksek doz kemoterapi ile kombine edildi�ine transaminaz y�kselmesi ve hiperbilirubinemi formunda ge�ici karaci�er toksisitesi g�zlenmi�tir. S�n�rl� g�venlilik veritaban� g�z �n�nde bulunduruldu�unda, �ocuklarda �u ana kadar bildirilen advers olaylar, eri�kin Ph+ ALL hastalar�nda bilinen g�venlilik profili ile uyumludur. Ph+ ALL hastas� �ocuklardaki g�venlilik veritaban� �ok s�n�rl� olmakla birlikte herhangi bir yeni g�venlilik endi�esi tan�mlanmam��t�r. Plevral ef�zyon, asit, pulmoner �dem ve y�zeysel �demin e�lik etti�i ya da etmedi�i h�zl� kilo art��� gibi �e�itli advers reaksiyonlar kolektif olarak “s�v� tutulumu” �eklinde tarif edilebilir. Bu reaksiyonlar genellikle GL�VEC tedavi ge�ici olarak durdurularak ve di�retiklerle ya da di�er uygun destekleyici bak�m �nlemleriyle kontrol edilebilmektedir. Di�er yandan, bu reaksiyonlar�n baz�lar� �iddetli ya da ya�am� tehdit edici olabilmektedir ve blast krizi olan �e�itli hastalar plevral ef�zyon, konjestif kalp yetmezli�i ve b�brek yetmezli�inden olu�an kompleks bir klinik �yk� ile ya�amlar�n� kaybetmi�tir. Pediyatrik klinik �al��malarda �zel bir g�venlilik bulgusu s�z konusu olmam��t�r. �zole bir vakadan daha fazlas� olarak bildirilen advers reaksiyonlar, sistem organ s�n�f�na ve s�kl��a g�re a�a��da listelenmi�tir. S�kl�k kategorileri �u standart kullan�larak tan�mlanm��t�r: �ok yayg�n (≥ 1/10); yayg�n (≥ 1/100, < 1/10); yayg�n olmayan(≥ 1/1.000, < 1/100); seyrek (≥ 1/10.000, < 1/1.000); �ok seyrek (< 1/10.000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). �stenmeyen etkiler, her s�kl�k grubu i�inde, en s�k g�r�len ba�ta olacak �ekilde s�kl�k s�ras�na g�re sunulmaktad�r. Advers reaksiyonlar ve s�kl�klar�, Tablo 3'te listelenmi�tir. Tablo 3: Advers reaksiyonlar�n tablo halinde �zeti
Laboratuvar testi anormallikleriHematoloji KML'de ba�ta n�tropeni ve trombositopeni olmak �zere sitopeniler, t�m �al��malar�n devaml� bir bulgusu olmu�, 750 mg gibi daha y�ksek dozlarda daha s�k olduklar� d���n�lm��t�r (faz I �al��ma). Bununla birlikte, sitopenilerin ortaya ��k��� net bir �ekilde hastal���n evresine de ba�l� olmu�, 3. veya 4. derece n�tropenilerin (ANC < 1.0 x 109/l) ve trombositopenilerin (trombosit say�s� < 50 x 109/l) s�kl���, kronik faz KML'de yeni tan� alm�� hastalarla kar��la�t�r�ld���nda (%16,7 n�tropeni ve %8,9 trombositopeni) blast krizi ve akselere fazda 4 ila 6 kat daha y�ksek (n�tropeni ve trombositopeni i�in s�ras�yla % 59-64 ve % 44-63) bulunmu�tur. Yeni tan� konulmu� olan kronik faz KML vakalar�nda evre 4 n�tropeni (ANC < 0,5x109/l) ve trombositopeni (trombosit say�s� < 10x109/l), s�ras�yla yaln�zca % 3,6 ve < %1 oran�nda g�r�lm��t�r. N�tropenik ve trombositopenik epizodlar�n medyan s�resi, genellikle s�ras�yla 2 ve 3. haftalar aras�nda ve 3 ve 4. haftalar aras�nda yer alm��t�r. Bu olaylar, genellikle GL�VEC ile tedavinin dozu azalt�larak ya da tedavi kesilerek kontrol edilebilir, ancak baz� nadir vakalarda kal�c� olarak tedavinin b�rak�lmas�na neden olabilir. Pediyatrik KML hastalar�nda en s�k g�zlenen toksisiteler; n�tropeni, trombositopeni ve anemi dahil olmak �zere 3 ya da 4. derece sitopeniler olmu�tur. Bunlar genellikle tedavinin ilk birka� ay� i�erisinde ortaya ��kmaktad�r. Rezeke edilemeyen ve/veya metastatik GIST bulunan hastalarda yap�lan �al��mada, s�ras�yla hastalar�n % 5,4 ve % 0,7'sinde evre 3 ve 4 anemi bildirilmi�tir ve bu durum en az�ndan baz� hastalarda gastrointestinal ya da intra-t�m�ral kanamayla ili�kili olabilir. S�ras�yla hastalar�n % 7,5 ve %2,7'sinde evre 3 ve 4 n�tropeni ve hastalar�n % 0,7'sinde evre 3 trombositopeni g�r�lm��t�r. Hi�bir hastada evre 4 trombositopeni geli�memi�tir. �zellikle tedavinin ilk 6 haftas�nda beyaz kan h�cresi (WBC) ve n�trofil say�lar�nda azalmalar ortaya ��km��, bu de�erler daha sonra nispeten sabit kalm��t�r. Biyokimya KML hastalar�nda transaminazlarda (<%5) ya da bilirubinde (<%1) ciddi art��lar olmu�tur ve genellikle doz azalt�larak ya da kesilerek (Bu epizodlar�n medyan s�resi yakla��k 1 hafta olmu�tur.) kontrol alt�na al�nm��t�r. KML hastalar�n�n % 1'inden az�nda karaci�er laboratuar anormallikleri nedeniyle tedavi, s�rekli olarak kesilmi�tir. GIST hastalar�n�n (�al��ma B2222) %6,8'inde 3. veya 4. evre ALT (alanin aminotransferaz); %4,8'inde 3. veya 4. evre AST (aspartat aminotransferaz) y�kselmeleri g�zlemlenmi�tir. Bilirubin y�ksekli�i, %3'�n alt�nda olmu�tur. Sitolitik ve kolestatik hepatit ve karaci�er yetmezli�i olgular� s�z konusu olmu�tur; y�ksek doz parasetamol kullanan bir hasta dahil olmak �zere bunlar�n baz�lar� �l�mle sonu�lanm��t�r. Se�ili advers reaksiyonlar�n tan�mlanmas� Hepatit B reaktivasyonu BCR-ABL TK�'lerle ili�kili olarak hepatit B reaktivasyonu bildirilmi�tir. Baz� vakalar, karaci�er transplantasyonuna veya �l�mc�l sonu�lara yol a�an akut karaci�er yetmezli�i veya fulminan hepatit ile sonu�lanm��t�r (bkz. B�l�m 4.4). ��pheli advers reaksiyonlar�n raporlanmas� Ruhsatland�rma sonras� ��pheli ila� advers reaksiyonlar�n�n raporlanmas� b�y�k �nem ta��maktad�r. Raporlama yap�lmas�, ilac�n yarar / risk dengesinin s�rekli olarak izlenmesine olanak sa�lar. Sa�l�k mesle�i mensuplar�n�n herhangi bir ��pheli advers reaksiyonu T�rkiye Farmakovijilans Merkezi (T�FAM)'ne bildirmeleri gerekmektedir. (www.titck.gov.tr; e- posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks: 0 312 218 35 99) 4.9. Doz a��m� ve tedavisiTerap�tik dozlardan daha y�ksek dozlarla deneyim s�n�rl�d�r. GL�VEC doz a��m� ile ilgili bireysel vakalar, spontan olarak ve literat�rde bildirilmi�tir. Doz a��m� halinde, hasta g�zlem alt�nda tutulmal� ve uygun semptomatik tedavi uygulanmal�d�r. Genellikle, bu vakalarda bildirilen sonu�lar, d�zelme ya da iyile�me �eklinde olmu�tur. Farkl� doz aral�klar�nda bildirilen olaylar a�a��da verilmi�tir: Eri�kinlerde doz a��m�: 1200 ila 1600 mg (1 ila 10 g�n aras�nda de�i�en s�relerle): Bulant�, kusma, diyare, d�k�nt�, eritem, �dem, �i�me, yorgunluk, kas spazmlar�, trombositopeni, pansitopeni, kar�n a�r�s�, ba� a�r�s�, i�tahta azalma. 1800 ila 3200 mg (6 g�n boyunca g�nde 3200 mg'a kadar dozlar): G��s�zl�k, miyalji, CPK d�zeyinde y�kselme, bilirubin d�zeyinde y�kselme, gastrointestinal a�r�. 6400 mg (tek doz): Literat�rde yer alan bir vakada, bulant�, kusma, kar�n a�r�s�, pireksi, y�zde �i�me, n�trofil say�s�nda azalma, transaminaz d�zeylerinde y�kselme g�r�len bir hasta bildirilmi�tir. 8 ila 10 g (tek doz): Kusma ve gastrointestinal a�r� bildirilmi�tir. Pediyatrik doz a��m�: 400 mg'l�k tek doza maruz kalan 3 ya��ndaki bir erkek �ocukta kusma, diyare ve anoreksi; 980 mg'l�k tek doza maruz kalan 3 ya��ndaki di�er bir erkek �ocukta ise l�kosit say�s�nda azalma ve diyare g�r�lm��t�r. Doz a��m� durumunda hasta g�zlemlenmeli ve uygun destek tedavisi verilmelidir. 5. FARMAKOLOJ�K �ZELL�KLER5.1. Farmakodinamik �zelliklerFarmakoterap�tik grup: Antineoplastik ajanlar, BCR-ABL tirozin kinaz inhibit�rleri ATC kodu: L01EA01 Etki mekanizmas�: �matinib k���k bir molek�l yap�s�na sahip bir protein-tirozin kinaz inhibit�r�d�r; Bcr- Abl tirozin kinaz (TK) aktivitesini ve bir�ok resept�r TK'y� kuvvetli bir �ekilde inhibe etmektedir: KIT, c-KIT proto-onkogen taraf�ndan kodlanan k�k h�cre fakt�r� (Stem cell factor – SCF) resept�r�, diskoidin etki b�lgesine ait resept�rler (DDR1 ve DDR2), koloni uyar�c� fakt�r resept�r� (CSF-1R), trombosit k�kenli b�y�me fakt�r� (Platelet derived growth factor – PDGF) resept�rleri alfa ve beta (PDGFR-alfa ve PDGFR-beta). �matinib ayn� zamanda bu resept�r kinazlar�n aktivasyonunun arac�l�k etti�i h�cresel olaylar� da inhibe edebilmektedir. Farmakodinamik etkiler: �matinib, Bcr-Abl tirozin kinaz� in vitro, h�cresel ve in vivo seviyelerde g��l� bir �ekilde inhibe eden bir protein-tirozin kinaz inhibit�r�d�r. Bile�ik, Philadelphia kromozomu pozitif KML ve akut lenfoblastik l�semi (ALL) hastalar�ndan al�nan taze l�semik h�crelerin yan� s�ra Bcr-Abl pozitif h�cre dizilerinde proliferasyonu se�ici olarak inhibe eder ve apoptozu ind�kler. Bile�ik, in vivo olarak, Bcr-Abl pozitif t�m�r h�creleri kullan�lan hayvan modellerinde tek ajan olarak anti-t�m�r aktivite g�sterir. �matinib, ayn� zamanda trombosit t�revi b�y�me fakt�r� (Platelet derived growth factor – PDGF), PDGF-R ve k�k h�cre fakt�r� (Stem cell factor – SCF), c-KIT tirozin kinazlar�n resept�r� i�in bir inhibit�rd�r ve PDGF- ve SCF- arac�l� h�cresel olaylar� inhibe eder. In vitro olarak, imatinib, aktive edici bir KIT mutasyonunu ekspres eden GIST h�crelerinde proliferasyonu inhibe eder ve apopitozu uyar�r. PDGF resept�r�n�n veya Abl protein tirozin kinazlar�n �e�itli ortak proteinlere f�zyonunun veya yap�sal PDGF �retiminin bir sonucu olan yap�sal aktivasyonun, MDS/MPD, HES/CEL ve DFSP'nin patojenezinde rol oynad��� �ne s�r�lm��t�r. Ayr�ca, c-KIT ya da PDGFR'nin konstit�tif aktivasyonu SM'nin patojenezinde rol oynayan muhtemel nedendir. �matinib, d�zensiz PDGFR veya Abl kinaz aktivitesinin y�nlendirdi�i sinyalizasyonu ve h�cre proliferasyonunu inhibe eder. Kronik Miyeloid L�semide Klinik �al��malarGL�VEC'in etkinli�i, bir b�t�n olarak elde edilen hematolojik ve sitogenetik yan�t oranlar�n� ve hastal�ks�z sa�kal�m s�resini temel al�r. Yeni tan� alm�� kronik faz KML harici, hastal�k ili�kili semptomlar�n iyile�mesi veya sa�kal�m s�resinin artmas� gibi klinik faydalar�n oldu�unu g�steren kontroll� �al��ma yoktur. �leri evre, blast veya h�zland�r�lm�� faz hastal�kta Philadelphia kromozomu pozitif (Ph +) KML, di�er Ph + l�semiler veya kronik fazda KML'si olan fakat daha �nce interferon- alfa (IFN) tedavide ba�ar�s�z olunan hastalarda �� b�y�k, uluslararas�, a��k etiketli, kontroll� olmayan Faz II �al��ma yap�lm��t�r. Yeni tan� alm�� Ph + KML hastalar�nda b�y�k, a��k etiketli, �ok merkezli, uluslararas�, randomize bir Faz III �al��ma y�r�t�lm��t�r. Ek olarak, iki Faz I �al��mada ve bir Faz II �al��mada �ocuklar tedavi edilmi�tir. T�m klinik �al��malarda hastalar�n %38-40'� ≥ 60 ya��nda ve hastalar�n %10-12'si ≥ 70 ya��ndad�r. Kronik faz, yeni tan� konulmu�: Eri�kin hastalarda yap�lan bu faz III �al��mada, tek ajan GL�VEC veya interferon-alfa (IFN) art� sitarabin (Ara-C) kombinasyonu ile tedavi kar��la�t�r�lm��t�r. Yan�ts�zl�k (6 ayda tam hematolojik yan�t (THY) olmamas�, artan WBC, 24 ayda maj�r sitogenetik yan�t (MSY) olmamas�), yan�t kayb� (THY veya MSY kayb�) veya tedaviye �iddetli intolerans g�steren hastalar�n alternatif tedavi koluna ge�melerine izin verilmi�tir. GL�VEC kolunda hastalar, g�nl�k 400 mg ile tedavi edilmi�tir. IFN grubunda, hastalar 10 g�n / ay boyunca subkutan Ara-C 20 mg / m2 / g�n ile kombinasyon halinde subkutan olarak 5 MIU / m2 / g�n hedef IFN dozu ile tedavi edilmi�tir. Toplam 1106 (her grupta 553) hasta, randomize edilmi�tir. �ki kol aras�nda �al��ma ba�lang�c� �zellikleri iyi d�zeyde dengelenmi�tir. Medyan ya� 51 y�l (aral�k 18-70 y�l) olup, hastalar�n % 21,9'u 60 ya��nda veya �zerindedir. % 59'u erkek ve % 41'i kad�n; % 89,9'u beyaz ve % 4,7'si siyahi hastalardan olu�mu�tur. Son hastan�n �al��maya al�nmas�ndan yedi y�l sonra, GL�VEC ve IFN kollar�nda medyan birinci basamak tedavi s�resi s�ras�yla 82 ve 8 ay olmu�tur. GL�VEC ile ikinci basamak tedavinin medyan s�resi 64 ayd�r. Genel olarak, birinci basamak olarak GL�VEC alan hastalarda verilen ortalama g�nl�k doz, 406±76 mg'd�r. �al��man�n primer etkililik sonlan�m noktas�, progresyonsuz sa�kal�md�r. Progresyon, a�a��daki olaylardan herhangi biri olarak tan�mlanm��t�r: h�zlanm�� faz veya blast krizine progresyon, �l�m, THY veya MSY kayb� ya da uygun terap�tik tedaviye ra�men bir CHR'ye ula�amayan hastalarda WBC art���. Maj�r sitogenetik yan�t, hematolojik yan�t, molek�ler yan�t (minimal rezid�el hastal���n de�erlendirilmesi), h�zland�r�lm�� faza veya blast krizine kadar ge�en s�re ve hayatta kalma, ana sekonder sonlan�m noktalard�r. Yan�t verileri Tablo 4'te g�sterilmektedir.
Tablo 4: Yeni tan� konulan KML �al��mas�ndaki yan�tlar (84 ayl�k veri)
Birinci basamak tedavide tam hematolojik yan�t, maj�r sitogenetik yan�t ve tam sitogenetik yan�t oranlar�, son muayene tarihinde yan�ts�zl�klar�n sans�rlendi�i Kaplan- Meier yakla��m� kullan�larak hesaplanm��t�r. Bu yakla��m kullan�ld���nda, GL�VEC ile birinci basamak tedavi i�in hesaplanan k�m�latif yan�t oranlar� 12 ayl�k tedaviden 84 ayl�k tedaviye �u �ekilde d�zelme g�stermi�tir: THY %96,4'ten %98,4'e ve TSY %69,5'ten %87,2'ye. 7 y�ll�k takipte, GL�VEC grubunda 93 (%16,8) progresif olay olmu�tur: 37 (%6,7) h�zlanm�� faz/blastik kriz (AF/BK) ilerleme, 31 (%5,6) major sitogenetik yan�t (MSY) kayb�, 15 (%2,7) tam hematolojik yan�t (THY) kayb� ya da WBC (beyaz kan h�cresi) art��� ve 10 (%1,8) KML ile ili�kisiz �l�m. Buna kar��l�k IFN+Ara-C grubunda 165 (%29,8) olay olmu� ve bunlar�n 130'u birinci se�enek IFN+Ara-C tedavisi s�ras�nda meydana gelmi�tir. 84 ayda akselere faz veya blast krizine ilerlemeyen hastalar�n tahmini oran�, IFN koluna k�yasla GL�VEC kolunda �nemli �l��de daha y�ksekti (%92,5'e kar�� %85, p<0.001). Tedavide ge�en s�re ile birlikte h�zland�r�lm�� faza veya blast krizine y�ll�k progresyon oran� azalm�� ve d�rd�nc� ve be�inci y�llarda y�ll�k % 1'den az olmu�tur. 84 ayda progresyonsuz sa�kal�m tahmini oran� GL�VEC grubunda % 81,2 ve kontrol grubunda % 60,6 bulunmu�tur (p <0,001). GL�VEC i�in herhangi bir t�rdeki y�ll�k progresyon oranlar� da zamanla azalm��t�r. GL�VEC ve IFN+Ara-C gruplar�nda, s�ras�yla, toplam 71 (%12,8) ve 85 (%15,4) hasta �lm��t�r. 84 ayda randomize GL�VEC ve IFN+Ara-C gruplar�nda tahmin edilen genel sa�kal�m, s�ras�yla %86,4'e (83, 90) kar�� %83,3 (80, 87) d�zeyindedir (p=0,073, log- rank testi). Bu olaya kadar ge�en zaman sonlan�m noktas�, IFN + Ara-C'den GL�VEC'e y�ksek ge�i� oran�ndan b�y�k �l��de etkilenir. GL�VEC tedavisinin kronik fazdaki, yeni tan� konulmu� KML'deki sa�kal�m etkisi, ayn� rejimde IFN+Ara-C (n=325) kullan�lan ba�ka bir Faz III �al��madan elde edilen birincil verilerle birlikte yukar�da belirtilen GL�VEC verilerinin retrospektif analizinde ayr�nt�l� olarak incelenmi�tir. Bu retrospektif analizde, genel sa�kal�m bak�m�ndan GL�VEC'in IFN+Ara-C kar��s�ndaki �st�nl��� kan�tlanm��t�r (p<0,001); 42 ay i�inde 47 (%8,5) GL�VEC hastas� ve 63 (%19,4) IFN+Ara-C hastas� �lm��t�r. GL�VEC tedavisindeki hastalarda sitogenetik yan�t ve molek�ler yan�t derecesi, uzun d�nem sonu�lar �zerinde a��k bir etkiye sahip olmu�tur. 12 ayda TSY'si (KSY) olan hastalar�n tahmini %96's�nda (%93) 84 ayda akselere faza/blast krizine progresyon olmazken 12 ayda MSY'si olmayan hastalar�n sadece %81'inde 84 ayda ilerlemi� KML'ye progresyon olmad��� g�r�lm��t�r (genel p<0,001, TSY ile KSY aras�nda p=0,25). 12 ayda Bcr-Abl transkriptlerinde en az 3 logaritmal�k azalmas� olan hastalarda akselere faza/blast krizine progresyonsuz kalma olas�l��� 84 ayda %99 bulunmu�tur. 18 ayl�k d�n�m noktas� analizine dayan�larak benzer bulgular tespit edilmi�tir. Bu �al��mada g�nde 400 mg'dan 600 mg'a, ard�ndan g�nde 600 mg'dan 800 mg'a doz art�r�mlar�na izin verilmi�tir. 42 ayl�k izlem sonras�nda 11 hasta sitogenetik yan�tlar�nda do�rulanm�� bir kay�p (4 hafta i�inde) deneyimlemi�tir. Bu 11 hastan�n 4'�nde doz g�nde 800 mg'a art�r�lm�� olup hastalar�n 2'si sitogenetik yan�t� tekrar elde etmi� (1'inde k�smi, 1'inde tam; tam yan�t elde eden ayr�ca molek�ler yan�ta da ula�m��t�r), di�er yandan dozlar� art�r�lmayan 7 hastan�n sadece biri tam sitogenetik yan�t� tekrar elde etmi�tir. Doz art�r�m� �ncesindeki hasta pop�lasyonu (n=551) ile kar��la�t�r�ld���nda, dozun g�nde 800 mg'a y�kseltildi�i 40 hastada baz� advers reaksiyonlar�n y�zdesi daha y�ksek olmu�tur. Daha s�k g�r�len advers reaksiyonlar gastrointestinal hemorajileri, konjonktivit ve transaminazlar veya bilirubinde y�kselmeyi i�ermi�tir. Di�er advers olaylar daha d���k ya da e�it s�kl�kla bildirilmi�tir. Kronik faz, interferon ba�ar�s�zl���: 532 hasta, 400 miligraml�k ba�lang�� dozuyla tedavi edilmi�tir. Bu hastalar; hematolojik ba�ar�s�zl�k (%29), sitogenetik ba�ar�s�zl�k (%35) veya interferon intolerans� (%36) olmak �zere ba�l�ca 3 gruba ayr�lm��t�r. Hastalar daha �nce medyan 14 ay boyunca ≥25 x 106 IU/hafta dozlarda IFN tedavisi g�rm��t�r ve hepsi de ge� kronik fazdad�r; tan�dan itibaren ge�en medyan s�re 32 ayd�r. �al��man�n birincil etkililik de�i�keni maj�r sitogenetik yan�t oran�d�r (tam yan�t art� k�sm� yan�t, kemik ili�inde %0 ila %35 Ph+ metafaz). Bu �al��mada hastalar�n %65'i bir maj�r sitogenetik yan�ta ula�m��t�r; hastalar�n %53'�nde (do�rulanm�� %43) yan�t tamd�r (Tablo 3). Hastalar�n %95'inde tam hematolojik yan�ta ula��lm��t�r. H�zlanm�� faz: H�zlanm�� faz hastal��� olan 235 yeti�kin hasta kaydedilmi�tir. �lk 77'sinde tedaviye g�nde 400 mg ile ba�lanm��t�r; daha sonra �al��ma protokol�, daha y�ksek GL�VEC dozlar�n�n kullan�lmas�na olanak tan�yacak �ekilde d�zenlenmi� ve geriye kalan 158 hasta, ba�lang��ta 600 mg GL�VEC kullanm��t�r. Birincil etkililik de�i�keni, ya tam hematolojik yan�t, l�semi kan�t� olmamas� (yani ilik ve kandan blastlar�n temizlenmesi, fakat tam yan�tlar i�in t�m periferik kan parametrelerinde iyile�me olmamas�) ya da kronik faz KML'ye geri d�n�� olarak rapor edilen hematolojik yan�t oran� olmu�tur. Do�rulanm�� hematolojik yan�t, hastalar�n % 71,5'inde elde edilmi�tir (Tablo 3). �nemli olarak, hastalar�n %27,7'si ayn� zamanda maj�r bir sitogenetik yan�t elde etmi� olup bunlar�n %20,4'�nde (%16 do�rulanm��t�r) yan�t tam olmu�tur. 600 mg ile tedavi edilen hastalar i�in, medyan progresyonsuz sa�kal�m ve genel sa�kal�m i�in mevcut tahminler s�ras�yla 22,9 ve 42,5 ayd�r. Miyeloid blast krizi: Miyeloid blast krizi olan 260 hasta kaydedilmi�tir. Bu hastalar�n 95'i (%37'si), h�zlanm�� faz veya yine blast krizi nedeniyle daha �nce de kemoterapi g�rm��t�r (“�nceden tedavi edilmi� olan hastalar”), 165 (%63) hastada ise daha �nce kemoterapi uygulanmam��t�r (“�nceden tedavi edilmemi� olan hastalar”). �lk 37 hastaya 400 mg ile ba�lanm��, daha sonra protokol daha y�ksek doza izin verecek �ekilde de�i�tirilmi� ve geri kalan 223 hastaya 600 mg ba�lanm��t�r. Primer etkililik de�i�keni, h�zlanm�� faz �al��mas�nda oldu�u gibi ayn� kriterler kullan�larak tam hematolojik yan�t, l�semi kan�t�n�n mevcut olmamas� veya kronik faza d�n�� olarak tan�mlanan, hematolojik yan�t oran� olmu�tur. Bu �al��mada hastalar�n %31'inde hematolojik yan�t elde edilmi�tir (daha �nce tedavi g�rmemi� hastalarda %36, daha �nce tedavi g�rm�� hastalarda %22). 600 mg GL�VEC kullanan hastalardaki hematolojik yan�t oran�, 400 mg GL�VEC kullanm�� olanlara k�yasla daha y�ksektir (%16'ya kar��l�k %33, p=0,0220). Daha �nceden tedavi edilmemi� ve tedavi edilmi� hastalar�n mevcut medyan ortalama sa�kal�m� s�ras�yla 7,7 ve 4,7 ayd�r. Lenfoid blast krizi: Faz I �al��malara s�n�rl� say�da hasta kaydedilmi�tir (n=10). Hematolojik yan�t oran�, 2-3 ayl�k s�re ile %70 bulunmu�tur. Tablo 5: Yeti�kin KML �al��malar�nda elde edilen yan�tlar
Pediyatrik pop�lasyon: Kronik faz KML'si (n=11) veya blast krizi a�amas�nda KML'si ya da Ph+ akut l�semileri (n=15) olan, 18 ya� alt� toplam 26 pediyatrik hasta bir faz I doz y�kseltme �al��mas�na kaydedilmi�tir. Bu, yo�un �n tedavi g�rm�� hastalardan olu�an bir pop�lasyondur: hastalar�n %46's� �nceden BMT ve %73'� �nceden �oklu ajanl� kemoterapi g�rm��t�r. Hastalar 260 mg/m2/g�n (n=5), 340 mg/m2/g�n (n=9), 440 mg/m2/g�n (n=7) ve 570 mg/m2/g�n (n=5) GL�VEC dozlar� ile tedavi edilmi�tir. Kronik faz KML'si ve mevcut sitogenetik verileri olan 9 hastadan 4'� (%44) ve 3'� (%33) %77'lik bir MSY oran�yla s�ras�yla tam ve k�smi sitogenetik yan�t elde etmi�tir. Yeni tan� alm�� ve tedavi edilmemi�, kronik fazda KML'si olan toplam 51 pediyatrik hasta a��k-etiketli, �ok merkezli, tek kollu bir faz II �al��maya kaydedilmi�tir. Hastalar 340 mg/m2/g�n GL�VEC ile tedavi edilmi�, doz s�n�rlay�c� toksisitesi hari� ara verilmemi�tir. GL�VEC tedavisi yeni tan� konmu� pediyatrik KML hastalar�nda, 8 haftal�k tedavi sonras�nda %78 THY oran� ile h�zl� yan�t sa�lamaktad�r. Y�ksek THY oran�na, hastalar�n %65'inde tam sitojenik yan�t (TSY) geli�imi e�lik etmi� olup bu oran, eri�kinlerde g�zlenen sonu� ile kar��la�t�r�labilir niteliktedir. Ek olarak, hastalar�n %16's�nda k�sm� sitojenik yan�t (KSY) g�zlenmi�, bu da %81 MSY de�erini vermi�tir. TSY'ye ula�an hastalar�n b�y�k �o�unlu�u, Kaplan-Meier tahmine dayal� 5,6 ayl�k yan�ta kadar ge�en medyan s�re ile TSY'ye 3 ila 10'uncu aylar aras�nda ula�m��t�r. Avrupa �la� Ajans�, Philadelphia kromozomu (bcr-abl translokasyon) pozitif kronik faz kronik miyeloid l�semide pediyatrik pop�lasyonun t�m alt k�melerinde GL�VEC ile �al��malar�n sonu�lar� sunma zorunlulu�unu iptal etmi�tir (pediyatrik kullan�m ile ilgili bilgi i�in bkz. B�l�m 4.2). Ph+ ALL i�in klinik �al��malar Yeni te�his edilen Ph+ ALL: �matinibin, 55 ya� ve �zeri yeni tan� alm�� 55 hastada kemoterapi ind�ksiyonuyla kar��la�t�r�ld��� kontroll� bir �al��mada (ADE10), tek ajan olarak kullan�lan imatinib, kemoterapiye k�yasla anlaml� derecede daha y�ksek tam hematolojik yan�t oran� ile sonu�lanm��t�r (%96,3'e kar��l�k %50, p=0,0001). Kemoterapiye yan�t vermeyen veya zay�f yan�t veren hastalarda imatinib kurtarma tedavisi olarak kullan�ld���nda, 11 hastan�n 9'unda (%81,8) tam hematolojik yan�t elde edilmi�tir. Bu klinik etki, 2 haftal�k tedaviden sonra, kemoterapi kolu ile kar��la�t�r�ld���nda imatinib ile tedavi edilen hastalarda, bcr-abl transkriptlerinde daha b�y�k bir azalmayla ili�kilendirilmi�tir (p=0,02). T�m hastalar ind�ksiyon sonras�nda imatinib ve konsolidasyon kemoterapisi alm�� (bkz. Tablo 4) ve bcr-abl transkriptlerinin d�zeyleri sekizinci haftada iki kolda ayn� olmu�tur. �al��ma tasar�m� do�rultusunda beklendi�i �zere, iki grup aras�nda remisyon s�resi, hastal�ks�z sa�kal�m veya genel sa�kal�m a��s�ndan herhangi bir fark g�zlenmemi�, ancak tam molek�ler yan�t elde edilen ve minimal rezid�el hastal�k d�zeyinde kalan hastalarda gerek remisyon s�resi (p=0,01) gerekse hastal�ks�z sa�kal�m (p=0,02) bak�m�ndan sonu�lar daha iyi olmu�tur. Kontrol gruplar�na yer verilmeyen d�rt klinik �al��mada (AAU02, ADE04, AJP01 ve AUS01) yeni tan� alm�� 211 Ph+ ALL hastas�ndan olu�an bir pop�lasyonda g�zlenen sonu�lar, yukar�da tarif edilen sonu�lar ile uyumludur. Kemoterapi ind�ksiyonu ile kombinasyon halindeki imatinib (bkz. Tablo 4) %93'l�k bir tam hematolojik yan�t oran� (de�erlendirilebilir 158 hastan�n 147'si) ve %90'l�k bir maj�r sitogenetik yan�t oran� (de�erlendirilebilir 21 hastan�n 19'u) sonu�lar�n� vermi�tir. Tam molek�ler yan�t oran� %48 bulunmu�tur (de�erlendirilebilir 102 hastan�n 49'u). Hastal�ks�z sa�kal�m (DFS) ve genel sa�kal�m (OS) her durumda 1 y�l� ge�mi�tir ve iki �al��madaki (AJP01 ve AUS01) ge�mi� kontrolden �st�n olmu�tur (DFS p<0,001; OS p<0,0001). Tablo 6: �matinible kombinasyon halinde kullan�lan kemoterapi rejimi
Pediyatrik pop�lasyon: I2301 �al��mas�nda, Ph+ ALL'si olan toplam 93 pediyatrik, ergen ve gen� yeti�kin hasta (1 ila 22 ya�lar� aras�nda) a��k etiketli, �ok merkezli, s�ral� gruplu, randomize olmayan bir faz III �al��maya kaydedilmi� ve ind�ksiyon tedavisinden sonra yo�un kemoterapi ile kombinasyon halinde GL�VEC (340 mg/m2/g�n) ile tedavi edilmi�tir. GL�VEC, 1-5 aras� kohortlarda aral�kl� olarak uygulanm��, kohorttan kohorta s�re artm�� ve GL�VEC daha erken ba�lam��t�r: en d���k yo�unlukta GL�VEC'i grup 1 ve en y�ksek yo�unlukta GL�VEC'i grup 5 alm��t�r (ilk kemoterapi k�rleri s�ras�nda s�rekli g�nl�k GL�VEC dozlamas� ile g�n cinsinden en uzun s�re). Kohort 5 hastalar�nda (n=50) kemoterapi ile kombinasyon halinde tedavi k�r�n�n erken d�nemlerinde GL�VEC'e s�rekli g�nl�k maruziyet, GL�VEC'siz standart kemoterapinin uyguland��� tarihsel kontrollerle (n=120) kar��la�t�r�ld���nda 4 y�ll�k olays�z sa�kal�m� (EFS) art�rm��t�r (s�ras�yla %69,6'ya kar��l�k %31,6). Kohort 5 hastalar�nda tahmini 4 y�ll�k GS, tarihsel kontrollerdeki %44,8 de�eri ile kar��la�t�r�ld���nda %83,6 olmu�tur. Kohort 5'teki 50 hastadan 20'si (% 40) hematopoietik k�k h�cre nakli alm��t�r. Tablo 7: �al��ma I2301'de imatinib ile kombinasyon halinde kullan�lan kemoterapi rejimi
G-CSF = gran�losit koloni uyar�c� fakt�r, VP-16 = etoposid, MTX = metotreksat, IV = intraven�z, SC = subkutan, IT = intratekal, PO = oral, IM = intram�sk�ler, ARA-C = sitarabin, CPM = siklofosfamid, VCR = vinkristin, DEX = deksametazon, DAUN = daunorubisin, 6-MP = 6-merkaptopurin, E.Coli L-ASP = L- asparaginaz, PEG-ASP = PEG asparaginaz, MESNA = 2-merkaptoetan s�lfonat sodyum, iii = veya MTX d�zeyi <0,1 µM olana kadar, q6h= her 6 saatte bir, Gy = Gray �al��ma AIT07, kemoterapi ile kombinasyon halinde imatinib ile tedavi edilen 128 hastay� (1 ila <18 ya�) i�eren �ok merkezli, a��k etiketli, randomize, Faz II / III bir �al��mad�r. Bu �al��madan elde edilen g�venlilik verilerinin, imatinibin Ph + ALL hastalar�nda g�venlilik profili ile uyumlu oldu�u g�r�lmektedir. N�ksetmi�/tedaviye refrakter Ph+ ALL�matinib, yineleyen/refrakter Ph+ ALL hastalar�nda tek ajan olarak kullan�ld���nda, 411 hastan�n 53'�nde yan�t de�erlendirilebilmi�, hematolojik yan�t oran� %30 (%9'u tam) ve maj�r sitogenetik yan�t oran� ise %23 olarak bulunmu�tur (Not: 411 hastan�n 353'�, primer yan�t verileri toplanmaks�z�n geni�letilmi� eri�im �al��mas�nda tedavi edilmi�tir). 411 yineleyen/refrakter Ph+ ALL hastas�ndan olu�an toplam pop�lasyonda progresyona kadar ge�en medyan s�re 2,6 ile 3,1 ay aral���nda olurken, de�erlendirilebilir 401 hastada medyan genel sa�kal�m 4,9 ile 9 ay aral���nda bulunmu�tur. Bu veriler, sadece 55 ya� ve �zeri hastalar dahil edilecek �ekilde yeniden analiz yap�ld���nda da benzer olmu�tur. MDS/MPD'de klinik �al��malarBu endikasyonda GL�VEC ile deneyim �ok s�n�rl�d�r ve hematolojik ve sitogenetik yan�t oranlar�na dayanmaktad�r. Klinik bir fayda veya artan sa�kal�m� g�steren kontroll� �al��ma yoktur. Abl, Kit veya PDGFR protein tirozin kinazlarla ili�kili ya�am� tehdit eden hastal�klardan muzdarip �e�itli hasta pop�lasyonlar�nda GL�VEC'in test edildi�i bir a��k etiketli, �ok merkezli, faz II klinik �al��ma (�al��ma B2225) y�r�t�lm��t�r. Bu �al��ma, g�nde 400 mg GL�VEC ile tedavi edilen MDS/MPD'li 7 hastay� i�ermektedir. �� hasta tam hematolojik yan�t (THY) ve bir hasta k�smi hematolojik yan�t (KHY) vermi�tir. Orjinal analiz s�ras�nda, PDGFR gen yeniden d�zenlemeleri saptanan d�rt hastadan ���nde hematolojik yan�t (2 THY ve 1 KHY) geli�mi�tir. Bu hastalar�n ya�lar� 20 ile 72 aras�nda de�i�mektedir. PDGFR-β yeniden d�zenlemesi olan ve GL�VEC ile tedavi edilmi� miyeloproliferatif neoplazmalardan muzdarip hastalarda uzun vadeli g�venlilik ve etkililik verilerini toplamak i�in g�zlemsel bir kay�t �al��mas� (�al��ma L2401) yap�lm��t�r. Bu kay�t �al��mas�nda yer alan 23 hasta, medyan 7,2 y�l (0,1 ile 12,7 y�l) boyunca medyan g�nl�k 264 mg (aral�k: 100 ile 400 mg) dozda GL�VEC alm��t�r. Bu kay�t �al��mas�n�n g�zlemsel yap�s� nedeniyle, kay�tl� 23 hastan�n s�ras�yla 22, 9 ve 17'si i�in hematolojik, sitogenetik ve molek�ler de�erlendirme verileri mevcuttur. Konservatif olarak, eksik verileri olan hastalar�n yan�t vermeyenler oldu�u varsay�ld���nda, 20/23 (%87) hastada THY, 9/23 (%39,1) hastada TSY ve 11/23 (%47,8) hastada MY g�zlemlenmi�tir. En az bir ge�erli de�erlendirmesi olan hastalardan yan�t oran� hesapland���nda, THY, TSY ve MY i�in yan�t oran� s�ras�yla 20/22 (%90,9), 9/9 (%100) ve 11/17 (%64,7) olmu�tur. Ayr�ca 13 yay�nda MDS/MPD'li 24 hasta daha bildirilmi�tir. 21 hasta g�nl�k 400 mg GL�VEC ile tedavi edilirken, di�er 3 hasta daha d���k dozlar alm��t�r. On bir hastada PDGFR gen yeniden d�zenlemeleri tespit edilmi�tir, bunlardan 9'u bir THY ve 1 KHY elde etmi�tir. Bu hastalar�n ya�lar� 2 ile 79 aras�nda de�i�mi�tir. Yak�n tarihli bir yay�nda, bu 11 hastan�n 6's�ndan al�nan g�ncellenmi� bilgiler, t�m bu hastalar�n sitogenetik remisyonda (32-38 ay aral���nda) kald���n� ortaya koymu�tur. Ayn� yay�nda, PDGFR gen yeniden d�zenlemeleri olan 12 MDS/MPD hastas�ndan (B2225 �al��mas�ndan 5 hasta) uzun s�reli takip verileri bildirilmi�tir. Bu hastalar medyan 47 ay (24 g�n - 60 ay aral���nda) GL�VEC alm��t�r. Bu hastalar�n 6's�nda takip s�resi, art�k 4 y�l�n �zerindedir. On bir hasta h�zl� THY elde etmi�tir; RT-PCR ile �l��ld���nde 10'unda sitogenetik anormallikler tamamen d�zelmi� ve f�zyon transkriptleri azalm�� ya da kaybolmu�tur. Hematolojik ve sitogenetik yan�tlar, s�ras�yla medyan 49 ay (19-60 aral���) ve 47 ay (16-59 aral���) boyunca s�rd�r�lm��t�r. Genel sa�kal�m tan�dan itibaren 65 ayd�r (aral�k 25-234). Genetik translokasyonu olmayan hastalara GL�VEC uygulamas�, genellikle herhangi bir iyile�me sa�lamamaktad�r. MDS/MPD'li pediatrik hastalarda kontroll� �al��ma yoktur. D�rt yay�nda PDGFR gen yeniden d�zenlemeleriyle ili�kili MDS/MPD'li be� (5) hasta bildirilmi�tir. Bu hastalar�n ya�lar� 3 ay ile 4 y�l aras�nda de�i�mi�tir ve imatinib g�nde 50 mg dozda veya g�nde 92,5 ila 340 mg/m2 aras�nda de�i�en dozlarda verilmi�tir. T�m hastalarda tam hematolojik yan�t, sitogenetik yan�t ve/veya klinik yan�t elde edilmi�tir. HES/CEL ile �lgili Klinik �al��malarAbl, KIT ya da PDGFR protein tirozin kinazlarla ili�kili ya�am� tehdit edici hastal�klar� olan farkl� hasta pop�lasyonlar�nda GL�VEC'in test edildi�i a��k-etiketli, �ok merkezli bir faz II klinik �al��ma (�al��ma B2225) y�r�t�lm��t�r. Bu �al��mada HES/CEL'i olan 14 hasta, g�nde 100 mg ila 1000 mg dozda GL�VEC ile tedavi edilmi�tir. Yay�nlanm�� 35 vaka raporu ve vaka serisinde bildirilen HES/CEL'li 162 hasta daha g�nl�k 75 mg ila 800 mg dozlar�nda GL�VEC alm��t�r. 176 hastadan olu�an toplam pop�lasyonun 117'sinde sitogenetik anormallikler de�erlendirilmi�tir. Bu 117 hastan�n 61'inde FIP1L1- PDGFRα f�zyon kinaz tan�mlanm��t�r. Di�er 3 yay�nlanm�� raporda d�rt HES hastas�n�n daha FIP1L1-PDGFRα pozitif oldu�u bulunmu�tur. 65 FIP1L1-PDGFRα f�zyon kinaz pozitif hastan�n t�m�, aylarca s�rd�r�len bir THY elde etmi�tir (raporlama s�ras�nda sans�rlenen 1+ ila 44+ ay aras�nda). Yak�n tarihli bir yay�nda bildirildi�i gibi, bu 65 hastadan 21'i, 28 ayl�k (aral�k 13-67 ay) medyan bir takip s�resiyle tam molek�ler remisyona ula�m��t�r. Bu hastalar�n ya�lar� 25 ile 72 aral���nda olmu�tur. Ek olarak, olgu raporlar�nda ara�t�rmac�lar taraf�ndan semptomatolojide ve di�er organ disfonksiyon anormalliklerindeki geli�meler bildirilmi�tir. Kalp, sinir, deri/derialt� doku, solunum/g���s/mediastinal, kas-iskelet/ba� dokusu/vask�ler ve gastrointestinal organ sistemlerinde geli�meler bildirilmi�tir. HES/CEL'li pediyatrik hastalarda kontroll� �al��ma yoktur. 3 yay�nda PDGFR gen yeniden d�zenlemeleri ile ili�kili HES ve CEL'li �� (3) hasta bildirilmi�tir. Bu hastalar�n ya�lar� 2 ila 16 y�l aras�nda de�i�mi�tir ve imatinib g�nde 300 mg/m2 veya g�nl�k 200 ila 400 mg aras�nda de�i�en dozlarda verilmi�tir. T�m hastalarda tam hematolojik yan�t, tam sitogenetik yan�t ve/veya tam molek�ler yan�t elde edilmi�tir. Rezeke edilemeyen ve/veya metastatik GIST'de yap�lan klinik �al��malarRezeke edilemeyen veya metastatik malign gastrointestinal stromal t�m�rleri (GIST) olan hastalarda faz II, a��k etiketli, randomize, kontrols�z �ok uluslu bir �al��ma y�r�t�lm��t�r. Bu �al��maya 147 hasta kaydedilmi� ve 36 ay boyunca g�nde bir kez oral olarak 400 mg veya 600 mg kullan�m�na randomize edilmi�tir. Bu hastalar�n ya�lar�, 18 ila 83 aras�ndad�r ve patolojik olarak rezeke edilemeyen ve/veya metastatik Kit-pozitif malign G�ST tan�s�na sahiptir. �mm�nohistokimya Kit antikoru ile (A-4502, tav�an poliklonal antiserumu, 1:100; DAKO Corporation, Carpinteria, CA) antijen geri kazan�m� sonras� avidin-biotin-peroksidaz kompleksi y�ntemi ile analize g�re rutin olarak y�r�t�lm��t�r. Birincil etkililik kan�t�, objektif yan�t oranlar�n� temel alm��t�r. T�m�rlerin en az bir hastal�k b�lgesinde �l��lebilir olmas� gerekmi� olup, yan�t karakterizasyonu G�neybat� Onkoloji Grubu (SWOG) kriterlerini temel alm��t�r. Bulgular, Tablo 8'de sunulmaktad�r. Tablo 8: STIB2222 kodlu GIST �al��mas�nda en iyi t�m�r yan�t�
�ki doz grubu aras�nda yan�t oranlar� bak�m�ndan farkl�l�klar s�z konusu olmam��t�r. Ara analiz tarihinde �nemli say�da stabil hastal��a sahip hasta, daha uzun s�reli tedavi ile k�smi yan�ta ula�m��t�r (medyan takip s�resi 31 ay). Yan�ta kadar ge�en medyan s�re, 13 hafta olmu�tur (%95 GA 12-23). Yan�t veren olgularda tedavi ba�ar�s�zl���na kadar ge�en medyan s�re 122 hafta (%95 GA 106-147), genel �al��ma pop�lasyonunda ise 84 hafta (%95 GA 71-109) bulunmu�tur. Medyan genel sa�kal�m noktas�na ula��lamam��t�r. 36 ayl�k izlem sonras�nda Kaplan-Meier sa�kal�m tahmini %68'dir. �ki klinik �al��mada (�al��ma B2222 ve gruplar aras� �al��ma S0033), g�nl�k GL�VEC dozu, 400 mg veya 600 mg daha d���k g�nl�k dozlar�nda progrese olan hastalarda 800 mg'a y�kseltilmi�tir. Doz, toplam 103 hastada 800 mg'a ��kar�lm��t�r; doz y�kseltildikten sonra 6 hasta k�smi yan�ta ve 21 hasta hastal�k stabilizasyonuna ula�arak %26'l�k genel klinik yarar sonucunu vermi�tir. Eldeki g�venlilik verilerinden yola ��k�larak, 400 mg veya 600 mg daha d���k g�nl�k dozlar�nda progrese olan hastalarda dozun g�nde 800 mg'a ��kar�lmas�n�n, GL�VEC'in g�venlilik profilini etkilemedi�i g�r�lmektedir. Adjuvan GIST i�in klinik �al��malarAdjuvan tedavi ko�ullar�nda GL�VEC, 773 hasta ile y�r�t�len �ok merkezli, �ift k�r, uzun s�reli, plasebo kontroll� bir faz III �al��mada (Z9001) ara�t�r�lm��t�r. Bu hastalar�n ya�lar�, 18-91 aral���nda olmu�tur. �mm�nhistokimya ile Kit proteini eksprese eden primer GIST y�n�nde histolojik tan�s� bulunan ve en geni� yerinde ≥3 cm t�m�r b�y�kl���ne sahip olan, �al��maya kay�t �ncesindeki 14-70 g�n i�erisinde primer GIST'i tam gross rezeksiyon ile al�nan hastalar dahil edilmi�tir. Primer GIST rezeke edildikten sonra hastalar, �u iki koldan birine randomize edilmi�tir: bir y�l s�reyle GL�VEC 400 mg/g�n veya plasebo. �al��man�n birincil sonlanma noktas�, randomizasyon tarihinden rek�rense ya da herhangi bir nedene ba�l� �l�me kadar ge�en s�re �eklinde tan�mlanan rek�renssiz sa�kal�m (RFS) olmu�tur. GL�VEC RFS'de anlaml� uzama sa�lam��, GL�VEC grubunda hastalar�n %75'i 38. ayda rek�renssiz iken plasebo grubundaki hastalar�n %75'i 20. ayda rek�renssiz kalm��t�r (s�ras�yla %95 GA [30-hesaplanamaz]; [14-hesaplanamaz]); (tehlike oran� = 0,398 [0,259-0,610], p<0,0001). Bir y�l sonunda genel RFS, plasebo (%82,3) kar��s�nda GL�VEC i�in anlaml� d�zeyde daha iyi bulunmu�tur (%97,7) (p<0,0001). Bu �ekilde rek�rens riski plaseboya oranla %89 azalt�lm��t�r (tehlike oran� = 0,113 [0,049-0,264]). Primer GIST'lerine y�nelik ameliyatlar� sonras�nda hastalardaki rek�rens riski, �u prognoz fakt�rleri esas al�narak retrospektif �ekilde de�erlendirilmi�tir: t�m�r b�y�kl���, mitotik indeks, t�m�r yeri. Mitotik indeks verileri, tedavi ama�l� (ITT) pop�lasyonu olu�turan 713 hastan�n 556's� i�in mevcuttu. Birle�ik Devletler Ulusal Sa�l�k Enstit�leri (NIH) ve Silahl� Kuvvetler Patoloji Enstit�s� (AFIP) risk s�n�fland�rmalar�na g�re yap�lan alt grup analizlerinin sonu�lar� Tablo 9'da g�sterilmektedir. D���k ve �ok d���k risk gruplar�nda herhangi bir fayda g�zlenmemi�tir. Genel bir sa�kal�m faydas� g�zlenmemi�tir. Tablo 9: NIH ve AFIP risk s�n�fland�rmas�na g�re Z9001 �al��mas� RFS analiz �zeti
*Full takip periyodu- NE-Tahmin edilebilir de�il �kinci bir �ok merkezli, a��k etiketli faz III �al��mada (SSG XVIII/AIO), cerrahi GIST rezeksiyonu sonras�nda olan ve a�a��daki durumlardan birinin bulundu�u hastalarda 400 mg/g�n GL�VEC ile 36 ay kar��s�nda 12 ayl�k tedavi kar��la�t�r�lm��t�r: t�m�r �ap� > 5 cm ve mitotik say�m > 5/50 y�ksek g�� alan� (HPF); veya t�m�r �ap� > 10 cm ve herhangi bir mitotik say�m veya mitotik say�m� > 10/50 HPF olan herhangi bir b�y�kl�kteki t�m�r ya da periton bo�lu�una do�ru r�pt�re olan t�m�rler. Toplam 397 hastadan olur al�nm�� ve bu hastalar �al��maya randomize edilmi�tir (199 hasta 12 ay kolunda ve 198 hasta 36 ay kolunda) medyan ya� 61 idi [aral�k 22 ila 84 ya�]). Medyan takip s�resi 54 ay olup (randomizasyondan veri kesme tarihine kadar) ilk hastan�n randomize edili�inden veri kesme tarihine kadar ge�en medyan s�re 83 ayd�r. �al��man�n birincil sonlanma noktas�, randomizasyon tarihinden n�kse ya da herhangi bir nedene ba�l� �l�me kadar ge�en s�re �eklinde tan�mlanan n�kss�z sa�kal�m (RFS) olmu�tur. 36 ayl�k GL�VEC tedavisi, 12 ayl�k GL�VEC tedavisi ile kar��la�t�r�ld���nda RFS'de anlaml� �l��de uzama sa�lam��t�r (genel tehlike oran� (HR) = 0,46 [0,32, 0,65], p<0,0001) (Tablo 8, �ekil 1). Buna ek olarak, 36 ayl�k GL�VEC tedavisi, 12 ayl�k GL�VEC tedavisi ile kar��la�t�r�ld���nda genel sa�kal�m (OS) s�resini anlaml� �l��de uzatm��t�r (HR = 0,45 [0,22, 0,89], p=0,0187) (Tablo 8, �ekil 2). Daha uzun s�reli tedavi (> 36 ay) yeni rek�renslerin olu�umunu geciktirebilmektedir; ancak, bu bulgunun genel sa�kal�m �zerindeki etkisi halen bilinmemektedir. Toplam �l�m say�s� 12 ayl�k tedavi kolu i�in 25 ve 36 ayl�k tedavi kolu i�in 12 �eklinde olmu�tur. �matinib ile 36 ay s�reli tedavi, ITT analizinde, yani t�m �al��ma pop�lasyonun dahil edildi�i analizde, 12 ayl�k tedaviden daha �st�n bulunmu�tur. Mutasyon tipine g�re yap�lan planl� bir alt grup analizinde, ekson 11 mutasyonlar� olan hastalarda 36 ayl�k tedavide RFS i�in tehlike oran� 0,35 olmu�tur [%95 GA: 0,22, 0,56]. G�zlemlenen olay say�s�n�n d���k olmas� sebebiyle, daha az yayg�n olan mutasyon alt gruplar� i�in herhangi bir sonu� ��kart�lamamaktad�r.
Tablo 10: 12 ayl�k ve 36 ayl�k GL�VEC Tedavisi (SSGXVIII/AIO �al��mas�)
�ekil 1 Primer rek�renssiz sa�kal�m sonlan�m noktas� i�in Kaplan-Meier tahminleri (ITT pop�lasyonu)
�ekil 2 Genel sa�kal�m i�in Kaplan-Meier tahminleri (ITT pop�lasyonu)
C-Kit pozitif GIST olan pediyatrik hastalarda kontroll� �al��ma bulunmamaktad�r. 7 yay�nda GIST'li (Kit ve PDGFR mutasyonlar� olan veya olmayan) onyedi (17) hasta bildirilmi�tir. Bu hastalar�n ya��, 8 ila 18 aral���nda olmu�tur ve imatinib, hem adjuvan hem de metastatik ko�ullarda g�nde 300 ile 800 mg aras�nda de�i�en dozlarda verilmi�tir. GIST tedavisi g�ren pediyatrik hastalar�n �o�unda c-kit veya PDGFR mutasyonlar�n� do�rulayan veriler bulunmamakta olup bu durum kar���k klinik sonu�lara yol a�m�� olabilir. DFSP'de klinik �al��malarG�nl�k 800 mg GL�VEC ile tedavi edilen DFSP'li 12 hastay� i�eren faz II, a��k etiketli, �ok merkezli bir klinik �al��ma (�al��ma B2225) y�r�t�lm��t�r. DFSP'li hastalar�n ya��, 23 ile 75 aras�nda de�i�mi�tir; DFSP metastatiktir, ilk rezektif cerrahiyi takiben lokal olarak n�ksetmi�tir ve �al��maya giri� s�ras�nda daha fazla rezektif cerrahiye uygun g�r�lmemi�tir. Etkilili�in birincil kan�t�, objektif yan�t oranlar�na dayanm��t�r. Biri tamamen ve 8'i k�smen olmak �zere, kay�tl� 12 hastadan 9'u yan�t vermi�tir. K�smi yan�t verenlerden ���, daha sonra ameliyatla hastal�ks�z hale getirilmi�tir. B2225 �al��mas�nda medyan tedavi s�resi, 6,2 ayd�r ve maksimum s�re 24,3 ayd�r. Yay�nlanm�� 5 vaka raporunda GL�VEC ile tedavi edilen 6 DFSP hastas� daha bildirilmi�tir ve ya�lar� 18 ay ile 49 y�l aras�nda de�i�mektedir. Yay�nlanm�� literat�rde bildirilen eri�kin hastalar, g�nl�k 400 mg (4 vaka) veya 800 mg (1 vaka) GL�VEC ile tedavi edilmi�tir. ��� tamamen ve 2'si k�smen olmak �zere be� (5) hasta yan�t vermi�tir. Yay�nlanan literat�rde medyan tedavi s�resi 4 hafta ile >20 ay aras�nda de�i�mektedir. GL�VEC tedavisine yan�t verenlerin neredeyse tamam�nda translokasyon t(17:22)[(q22:q13)] veya bunun gen �r�n� mevcuttur. DFSP'li pediatrik hastalarda kontroll� �al��ma bulunmamaktad�r. �� yay�nda DFSP ve PDGFR gen yeniden d�zenlemelerine sahip be� (5) hasta bildirilmi�tir. Bu hastalar�n ya�� yenido�an ile 14 ya� aras�nda de�i�mektedir ve imatinib g�nde 50 mg dozda veya g�nde 400 ila 520 mg/m2 aras�nda de�i�en dozlarda verilmi�tir. T�m hastalar, k�smi ve/veya tam yan�t elde etmi�tir. 5. FARMAKOLOJ�K �ZELL�KLER5.1. Farmakodinamik �zelliklerFarmakoterap�tik grup: Antineoplastik ajanlar, BCR-ABL tirozin kinaz inhibit�rleri ATC kodu: L01EA01 Etki mekanizmas�: �matinib k���k bir molek�l yap�s�na sahip bir protein-tirozin kinaz inhibit�r�d�r; Bcr- Abl tirozin kinaz (TK) aktivitesini ve bir�ok resept�r TK'y� kuvvetli bir �ekilde inhibe etmektedir: KIT, c-KIT proto-onkogen taraf�ndan kodlanan k�k h�cre fakt�r� (Stem cell factor – SCF) resept�r�, diskoidin etki b�lgesine ait resept�rler (DDR1 ve DDR2), koloni uyar�c� fakt�r resept�r� (CSF-1R), trombosit k�kenli b�y�me fakt�r� (Platelet derived growth factor – PDGF) resept�rleri alfa ve beta (PDGFR-alfa ve PDGFR-beta). �matinib ayn� zamanda bu resept�r kinazlar�n aktivasyonunun arac�l�k etti�i h�cresel olaylar� da inhibe edebilmektedir. Farmakodinamik etkiler: �matinib, Bcr-Abl tirozin kinaz� in vitro, h�cresel ve in vivo seviyelerde g��l� bir �ekilde inhibe eden bir protein-tirozin kinaz inhibit�r�d�r. Bile�ik, Philadelphia kromozomu pozitif KML ve akut lenfoblastik l�semi (ALL) hastalar�ndan al�nan taze l�semik h�crelerin yan� s�ra Bcr-Abl pozitif h�cre dizilerinde proliferasyonu se�ici olarak inhibe eder ve apoptozu ind�kler. Bile�ik, in vivo olarak, Bcr-Abl pozitif t�m�r h�creleri kullan�lan hayvan modellerinde tek ajan olarak anti-t�m�r aktivite g�sterir. �matinib, ayn� zamanda trombosit t�revi b�y�me fakt�r� (Platelet derived growth factor – PDGF), PDGF-R ve k�k h�cre fakt�r� (Stem cell factor – SCF), c-KIT tirozin kinazlar�n resept�r� i�in bir inhibit�rd�r ve PDGF- ve SCF- arac�l� h�cresel olaylar� inhibe eder. In vitro olarak, imatinib, aktive edici bir KIT mutasyonunu ekspres eden GIST h�crelerinde proliferasyonu inhibe eder ve apopitozu uyar�r. PDGF resept�r�n�n veya Abl protein tirozin kinazlar�n �e�itli ortak proteinlere f�zyonunun veya yap�sal PDGF �retiminin bir sonucu olan yap�sal aktivasyonun, MDS/MPD, HES/CEL ve DFSP'nin patojenezinde rol oynad��� �ne s�r�lm��t�r. Ayr�ca, c-KIT ya da PDGFR'nin konstit�tif aktivasyonu SM'nin patojenezinde rol oynayan muhtemel nedendir. �matinib, d�zensiz PDGFR veya Abl kinaz aktivitesinin y�nlendirdi�i sinyalizasyonu ve h�cre proliferasyonunu inhibe eder. Kronik Miyeloid L�semide Klinik �al��malarGL�VEC'in etkinli�i, bir b�t�n olarak elde edilen hematolojik ve sitogenetik yan�t oranlar�n� ve hastal�ks�z sa�kal�m s�resini temel al�r. Yeni tan� alm�� kronik faz KML harici, hastal�k ili�kili semptomlar�n iyile�mesi veya sa�kal�m s�resinin artmas� gibi klinik faydalar�n oldu�unu g�steren kontroll� �al��ma yoktur. �leri evre, blast veya h�zland�r�lm�� faz hastal�kta Philadelphia kromozomu pozitif (Ph +) KML, di�er Ph + l�semiler veya kronik fazda KML'si olan fakat daha �nce interferon- alfa (IFN) tedavide ba�ar�s�z olunan hastalarda �� b�y�k, uluslararas�, a��k etiketli, kontroll� olmayan Faz II �al��ma yap�lm��t�r. Yeni tan� alm�� Ph + KML hastalar�nda b�y�k, a��k etiketli, �ok merkezli, uluslararas�, randomize bir Faz III �al��ma y�r�t�lm��t�r. Ek olarak, iki Faz I �al��mada ve bir Faz II �al��mada �ocuklar tedavi edilmi�tir. T�m klinik �al��malarda hastalar�n %38-40'� ≥ 60 ya��nda ve hastalar�n %10-12'si ≥ 70 ya��ndad�r. Kronik faz, yeni tan� konulmu�: Eri�kin hastalarda yap�lan bu faz III �al��mada, tek ajan GL�VEC veya interferon-alfa (IFN) art� sitarabin (Ara-C) kombinasyonu ile tedavi kar��la�t�r�lm��t�r. Yan�ts�zl�k (6 ayda tam hematolojik yan�t (THY) olmamas�, artan WBC, 24 ayda maj�r sitogenetik yan�t (MSY) olmamas�), yan�t kayb� (THY veya MSY kayb�) veya tedaviye �iddetli intolerans g�steren hastalar�n alternatif tedavi koluna ge�melerine izin verilmi�tir. GL�VEC kolunda hastalar, g�nl�k 400 mg ile tedavi edilmi�tir. IFN grubunda, hastalar 10 g�n / ay boyunca subkutan Ara-C 20 mg / m2 / g�n ile kombinasyon halinde subkutan olarak 5 MIU / m2 / g�n hedef IFN dozu ile tedavi edilmi�tir. Toplam 1106 (her grupta 553) hasta, randomize edilmi�tir. �ki kol aras�nda �al��ma ba�lang�c� �zellikleri iyi d�zeyde dengelenmi�tir. Medyan ya� 51 y�l (aral�k 18-70 y�l) olup, hastalar�n % 21,9'u 60 ya��nda veya �zerindedir. % 59'u erkek ve % 41'i kad�n; % 89,9'u beyaz ve % 4,7'si siyahi hastalardan olu�mu�tur. Son hastan�n �al��maya al�nmas�ndan yedi y�l sonra, GL�VEC ve IFN kollar�nda medyan birinci basamak tedavi s�resi s�ras�yla 82 ve 8 ay olmu�tur. GL�VEC ile ikinci basamak tedavinin medyan s�resi 64 ayd�r. Genel olarak, birinci basamak olarak GL�VEC alan hastalarda verilen ortalama g�nl�k doz, 406±76 mg'd�r. �al��man�n primer etkililik sonlan�m noktas�, progresyonsuz sa�kal�md�r. Progresyon, a�a��daki olaylardan herhangi biri olarak tan�mlanm��t�r: h�zlanm�� faz veya blast krizine progresyon, �l�m, THY veya MSY kayb� ya da uygun terap�tik tedaviye ra�men bir CHR'ye ula�amayan hastalarda WBC art���. Maj�r sitogenetik yan�t, hematolojik yan�t, molek�ler yan�t (minimal rezid�el hastal���n de�erlendirilmesi), h�zland�r�lm�� faza veya blast krizine kadar ge�en s�re ve hayatta kalma, ana sekonder sonlan�m noktalard�r. Yan�t verileri Tablo 4'te g�sterilmektedir.
Tablo 4: Yeni tan� konulan KML �al��mas�ndaki yan�tlar (84 ayl�k veri)
Birinci basamak tedavide tam hematolojik yan�t, maj�r sitogenetik yan�t ve tam sitogenetik yan�t oranlar�, son muayene tarihinde yan�ts�zl�klar�n sans�rlendi�i Kaplan- Meier yakla��m� kullan�larak hesaplanm��t�r. Bu yakla��m kullan�ld���nda, GL�VEC ile birinci basamak tedavi i�in hesaplanan k�m�latif yan�t oranlar� 12 ayl�k tedaviden 84 ayl�k tedaviye �u �ekilde d�zelme g�stermi�tir: THY %96,4'ten %98,4'e ve TSY %69,5'ten %87,2'ye. 7 y�ll�k takipte, GL�VEC grubunda 93 (%16,8) progresif olay olmu�tur: 37 (%6,7) h�zlanm�� faz/blastik kriz (AF/BK) ilerleme, 31 (%5,6) major sitogenetik yan�t (MSY) kayb�, 15 (%2,7) tam hematolojik yan�t (THY) kayb� ya da WBC (beyaz kan h�cresi) art��� ve 10 (%1,8) KML ile ili�kisiz �l�m. Buna kar��l�k IFN+Ara-C grubunda 165 (%29,8) olay olmu� ve bunlar�n 130'u birinci se�enek IFN+Ara-C tedavisi s�ras�nda meydana gelmi�tir. 84 ayda akselere faz veya blast krizine ilerlemeyen hastalar�n tahmini oran�, IFN koluna k�yasla GL�VEC kolunda �nemli �l��de daha y�ksekti (%92,5'e kar�� %85, p<0.001). Tedavide ge�en s�re ile birlikte h�zland�r�lm�� faza veya blast krizine y�ll�k progresyon oran� azalm�� ve d�rd�nc� ve be�inci y�llarda y�ll�k % 1'den az olmu�tur. 84 ayda progresyonsuz sa�kal�m tahmini oran� GL�VEC grubunda % 81,2 ve kontrol grubunda % 60,6 bulunmu�tur (p <0,001). GL�VEC i�in herhangi bir t�rdeki y�ll�k progresyon oranlar� da zamanla azalm��t�r. GL�VEC ve IFN+Ara-C gruplar�nda, s�ras�yla, toplam 71 (%12,8) ve 85 (%15,4) hasta �lm��t�r. 84 ayda randomize GL�VEC ve IFN+Ara-C gruplar�nda tahmin edilen genel sa�kal�m, s�ras�yla %86,4'e (83, 90) kar�� %83,3 (80, 87) d�zeyindedir (p=0,073, log- rank testi). Bu olaya kadar ge�en zaman sonlan�m noktas�, IFN + Ara-C'den GL�VEC'e y�ksek ge�i� oran�ndan b�y�k �l��de etkilenir. GL�VEC tedavisinin kronik fazdaki, yeni tan� konulmu� KML'deki sa�kal�m etkisi, ayn� rejimde IFN+Ara-C (n=325) kullan�lan ba�ka bir Faz III �al��madan elde edilen birincil verilerle birlikte yukar�da belirtilen GL�VEC verilerinin retrospektif analizinde ayr�nt�l� olarak incelenmi�tir. Bu retrospektif analizde, genel sa�kal�m bak�m�ndan GL�VEC'in IFN+Ara-C kar��s�ndaki �st�nl��� kan�tlanm��t�r (p<0,001); 42 ay i�inde 47 (%8,5) GL�VEC hastas� ve 63 (%19,4) IFN+Ara-C hastas� �lm��t�r. GL�VEC tedavisindeki hastalarda sitogenetik yan�t ve molek�ler yan�t derecesi, uzun d�nem sonu�lar �zerinde a��k bir etkiye sahip olmu�tur. 12 ayda TSY'si (KSY) olan hastalar�n tahmini %96's�nda (%93) 84 ayda akselere faza/blast krizine progresyon olmazken 12 ayda MSY'si olmayan hastalar�n sadece %81'inde 84 ayda ilerlemi� KML'ye progresyon olmad��� g�r�lm��t�r (genel p<0,001, TSY ile KSY aras�nda p=0,25). 12 ayda Bcr-Abl transkriptlerinde en az 3 logaritmal�k azalmas� olan hastalarda akselere faza/blast krizine progresyonsuz kalma olas�l��� 84 ayda %99 bulunmu�tur. 18 ayl�k d�n�m noktas� analizine dayan�larak benzer bulgular tespit edilmi�tir. Bu �al��mada g�nde 400 mg'dan 600 mg'a, ard�ndan g�nde 600 mg'dan 800 mg'a doz art�r�mlar�na izin verilmi�tir. 42 ayl�k izlem sonras�nda 11 hasta sitogenetik yan�tlar�nda do�rulanm�� bir kay�p (4 hafta i�inde) deneyimlemi�tir. Bu 11 hastan�n 4'�nde doz g�nde 800 mg'a art�r�lm�� olup hastalar�n 2'si sitogenetik yan�t� tekrar elde etmi� (1'inde k�smi, 1'inde tam; tam yan�t elde eden ayr�ca molek�ler yan�ta da ula�m��t�r), di�er yandan dozlar� art�r�lmayan 7 hastan�n sadece biri tam sitogenetik yan�t� tekrar elde etmi�tir. Doz art�r�m� �ncesindeki hasta pop�lasyonu (n=551) ile kar��la�t�r�ld���nda, dozun g�nde 800 mg'a y�kseltildi�i 40 hastada baz� advers reaksiyonlar�n y�zdesi daha y�ksek olmu�tur. Daha s�k g�r�len advers reaksiyonlar gastrointestinal hemorajileri, konjonktivit ve transaminazlar veya bilirubinde y�kselmeyi i�ermi�tir. Di�er advers olaylar daha d���k ya da e�it s�kl�kla bildirilmi�tir. Kronik faz, interferon ba�ar�s�zl���: 532 hasta, 400 miligraml�k ba�lang�� dozuyla tedavi edilmi�tir. Bu hastalar; hematolojik ba�ar�s�zl�k (%29), sitogenetik ba�ar�s�zl�k (%35) veya interferon intolerans� (%36) olmak �zere ba�l�ca 3 gruba ayr�lm��t�r. Hastalar daha �nce medyan 14 ay boyunca ≥25 x 106 IU/hafta dozlarda IFN tedavisi g�rm��t�r ve hepsi de ge� kronik fazdad�r; tan�dan itibaren ge�en medyan s�re 32 ayd�r. �al��man�n birincil etkililik de�i�keni maj�r sitogenetik yan�t oran�d�r (tam yan�t art� k�sm� yan�t, kemik ili�inde %0 ila %35 Ph+ metafaz). Bu �al��mada hastalar�n %65'i bir maj�r sitogenetik yan�ta ula�m��t�r; hastalar�n %53'�nde (do�rulanm�� %43) yan�t tamd�r (Tablo 3). Hastalar�n %95'inde tam hematolojik yan�ta ula��lm��t�r. H�zlanm�� faz: H�zlanm�� faz hastal��� olan 235 yeti�kin hasta kaydedilmi�tir. �lk 77'sinde tedaviye g�nde 400 mg ile ba�lanm��t�r; daha sonra �al��ma protokol�, daha y�ksek GL�VEC dozlar�n�n kullan�lmas�na olanak tan�yacak �ekilde d�zenlenmi� ve geriye kalan 158 hasta, ba�lang��ta 600 mg GL�VEC kullanm��t�r. Birincil etkililik de�i�keni, ya tam hematolojik yan�t, l�semi kan�t� olmamas� (yani ilik ve kandan blastlar�n temizlenmesi, fakat tam yan�tlar i�in t�m periferik kan parametrelerinde iyile�me olmamas�) ya da kronik faz KML'ye geri d�n�� olarak rapor edilen hematolojik yan�t oran� olmu�tur. Do�rulanm�� hematolojik yan�t, hastalar�n % 71,5'inde elde edilmi�tir (Tablo 3). �nemli olarak, hastalar�n %27,7'si ayn� zamanda maj�r bir sitogenetik yan�t elde etmi� olup bunlar�n %20,4'�nde (%16 do�rulanm��t�r) yan�t tam olmu�tur. 600 mg ile tedavi edilen hastalar i�in, medyan progresyonsuz sa�kal�m ve genel sa�kal�m i�in mevcut tahminler s�ras�yla 22,9 ve 42,5 ayd�r. Miyeloid blast krizi: Miyeloid blast krizi olan 260 hasta kaydedilmi�tir. Bu hastalar�n 95'i (%37'si), h�zlanm�� faz veya yine blast krizi nedeniyle daha �nce de kemoterapi g�rm��t�r (“�nceden tedavi edilmi� olan hastalar”), 165 (%63) hastada ise daha �nce kemoterapi uygulanmam��t�r (“�nceden tedavi edilmemi� olan hastalar”). �lk 37 hastaya 400 mg ile ba�lanm��, daha sonra protokol daha y�ksek doza izin verecek �ekilde de�i�tirilmi� ve geri kalan 223 hastaya 600 mg ba�lanm��t�r. Primer etkililik de�i�keni, h�zlanm�� faz �al��mas�nda oldu�u gibi ayn� kriterler kullan�larak tam hematolojik yan�t, l�semi kan�t�n�n mevcut olmamas� veya kronik faza d�n�� olarak tan�mlanan, hematolojik yan�t oran� olmu�tur. Bu �al��mada hastalar�n %31'inde hematolojik yan�t elde edilmi�tir (daha �nce tedavi g�rmemi� hastalarda %36, daha �nce tedavi g�rm�� hastalarda %22). 600 mg GL�VEC kullanan hastalardaki hematolojik yan�t oran�, 400 mg GL�VEC kullanm�� olanlara k�yasla daha y�ksektir (%16'ya kar��l�k %33, p=0,0220). Daha �nceden tedavi edilmemi� ve tedavi edilmi� hastalar�n mevcut medyan ortalama sa�kal�m� s�ras�yla 7,7 ve 4,7 ayd�r. Lenfoid blast krizi: Faz I �al��malara s�n�rl� say�da hasta kaydedilmi�tir (n=10). Hematolojik yan�t oran�, 2-3 ayl�k s�re ile %70 bulunmu�tur. Tablo 5: Yeti�kin KML �al��malar�nda elde edilen yan�tlar
Pediyatrik pop�lasyon: Kronik faz KML'si (n=11) veya blast krizi a�amas�nda KML'si ya da Ph+ akut l�semileri (n=15) olan, 18 ya� alt� toplam 26 pediyatrik hasta bir faz I doz y�kseltme �al��mas�na kaydedilmi�tir. Bu, yo�un �n tedavi g�rm�� hastalardan olu�an bir pop�lasyondur: hastalar�n %46's� �nceden BMT ve %73'� �nceden �oklu ajanl� kemoterapi g�rm��t�r. Hastalar 260 mg/m2/g�n (n=5), 340 mg/m2/g�n (n=9), 440 mg/m2/g�n (n=7) ve 570 mg/m2/g�n (n=5) GL�VEC dozlar� ile tedavi edilmi�tir. Kronik faz KML'si ve mevcut sitogenetik verileri olan 9 hastadan 4'� (%44) ve 3'� (%33) %77'lik bir MSY oran�yla s�ras�yla tam ve k�smi sitogenetik yan�t elde etmi�tir. Yeni tan� alm�� ve tedavi edilmemi�, kronik fazda KML'si olan toplam 51 pediyatrik hasta a��k-etiketli, �ok merkezli, tek kollu bir faz II �al��maya kaydedilmi�tir. Hastalar 340 mg/m2/g�n GL�VEC ile tedavi edilmi�, doz s�n�rlay�c� toksisitesi hari� ara verilmemi�tir. GL�VEC tedavisi yeni tan� konmu� pediyatrik KML hastalar�nda, 8 haftal�k tedavi sonras�nda %78 THY oran� ile h�zl� yan�t sa�lamaktad�r. Y�ksek THY oran�na, hastalar�n %65'inde tam sitojenik yan�t (TSY) geli�imi e�lik etmi� olup bu oran, eri�kinlerde g�zlenen sonu� ile kar��la�t�r�labilir niteliktedir. Ek olarak, hastalar�n %16's�nda k�sm� sitojenik yan�t (KSY) g�zlenmi�, bu da %81 MSY de�erini vermi�tir. TSY'ye ula�an hastalar�n b�y�k �o�unlu�u, Kaplan-Meier tahmine dayal� 5,6 ayl�k yan�ta kadar ge�en medyan s�re ile TSY'ye 3 ila 10'uncu aylar aras�nda ula�m��t�r. Avrupa �la� Ajans�, Philadelphia kromozomu (bcr-abl translokasyon) pozitif kronik faz kronik miyeloid l�semide pediyatrik pop�lasyonun t�m alt k�melerinde GL�VEC ile �al��malar�n sonu�lar� sunma zorunlulu�unu iptal etmi�tir (pediyatrik kullan�m ile ilgili bilgi i�in bkz. B�l�m 4.2). Ph+ ALL i�in klinik �al��malar Yeni te�his edilen Ph+ ALL: �matinibin, 55 ya� ve �zeri yeni tan� alm�� 55 hastada kemoterapi ind�ksiyonuyla kar��la�t�r�ld��� kontroll� bir �al��mada (ADE10), tek ajan olarak kullan�lan imatinib, kemoterapiye k�yasla anlaml� derecede daha y�ksek tam hematolojik yan�t oran� ile sonu�lanm��t�r (%96,3'e kar��l�k %50, p=0,0001). Kemoterapiye yan�t vermeyen veya zay�f yan�t veren hastalarda imatinib kurtarma tedavisi olarak kullan�ld���nda, 11 hastan�n 9'unda (%81,8) tam hematolojik yan�t elde edilmi�tir. Bu klinik etki, 2 haftal�k tedaviden sonra, kemoterapi kolu ile kar��la�t�r�ld���nda imatinib ile tedavi edilen hastalarda, bcr-abl transkriptlerinde daha b�y�k bir azalmayla ili�kilendirilmi�tir (p=0,02). T�m hastalar ind�ksiyon sonras�nda imatinib ve konsolidasyon kemoterapisi alm�� (bkz. Tablo 4) ve bcr-abl transkriptlerinin d�zeyleri sekizinci haftada iki kolda ayn� olmu�tur. �al��ma tasar�m� do�rultusunda beklendi�i �zere, iki grup aras�nda remisyon s�resi, hastal�ks�z sa�kal�m veya genel sa�kal�m a��s�ndan herhangi bir fark g�zlenmemi�, ancak tam molek�ler yan�t elde edilen ve minimal rezid�el hastal�k d�zeyinde kalan hastalarda gerek remisyon s�resi (p=0,01) gerekse hastal�ks�z sa�kal�m (p=0,02) bak�m�ndan sonu�lar daha iyi olmu�tur. Kontrol gruplar�na yer verilmeyen d�rt klinik �al��mada (AAU02, ADE04, AJP01 ve AUS01) yeni tan� alm�� 211 Ph+ ALL hastas�ndan olu�an bir pop�lasyonda g�zlenen sonu�lar, yukar�da tarif edilen sonu�lar ile uyumludur. Kemoterapi ind�ksiyonu ile kombinasyon halindeki imatinib (bkz. Tablo 4) %93'l�k bir tam hematolojik yan�t oran� (de�erlendirilebilir 158 hastan�n 147'si) ve %90'l�k bir maj�r sitogenetik yan�t oran� (de�erlendirilebilir 21 hastan�n 19'u) sonu�lar�n� vermi�tir. Tam molek�ler yan�t oran� %48 bulunmu�tur (de�erlendirilebilir 102 hastan�n 49'u). Hastal�ks�z sa�kal�m (DFS) ve genel sa�kal�m (OS) her durumda 1 y�l� ge�mi�tir ve iki �al��madaki (AJP01 ve AUS01) ge�mi� kontrolden �st�n olmu�tur (DFS p<0,001; OS p<0,0001). Tablo 6: �matinible kombinasyon halinde kullan�lan kemoterapi rejimi
Pediyatrik pop�lasyon: I2301 �al��mas�nda, Ph+ ALL'si olan toplam 93 pediyatrik, ergen ve gen� yeti�kin hasta (1 ila 22 ya�lar� aras�nda) a��k etiketli, �ok merkezli, s�ral� gruplu, randomize olmayan bir faz III �al��maya kaydedilmi� ve ind�ksiyon tedavisinden sonra yo�un kemoterapi ile kombinasyon halinde GL�VEC (340 mg/m2/g�n) ile tedavi edilmi�tir. GL�VEC, 1-5 aras� kohortlarda aral�kl� olarak uygulanm��, kohorttan kohorta s�re artm�� ve GL�VEC daha erken ba�lam��t�r: en d���k yo�unlukta GL�VEC'i grup 1 ve en y�ksek yo�unlukta GL�VEC'i grup 5 alm��t�r (ilk kemoterapi k�rleri s�ras�nda s�rekli g�nl�k GL�VEC dozlamas� ile g�n cinsinden en uzun s�re). Kohort 5 hastalar�nda (n=50) kemoterapi ile kombinasyon halinde tedavi k�r�n�n erken d�nemlerinde GL�VEC'e s�rekli g�nl�k maruziyet, GL�VEC'siz standart kemoterapinin uyguland��� tarihsel kontrollerle (n=120) kar��la�t�r�ld���nda 4 y�ll�k olays�z sa�kal�m� (EFS) art�rm��t�r (s�ras�yla %69,6'ya kar��l�k %31,6). Kohort 5 hastalar�nda tahmini 4 y�ll�k GS, tarihsel kontrollerdeki %44,8 de�eri ile kar��la�t�r�ld���nda %83,6 olmu�tur. Kohort 5'teki 50 hastadan 20'si (% 40) hematopoietik k�k h�cre nakli alm��t�r. Tablo 7: �al��ma I2301'de imatinib ile kombinasyon halinde kullan�lan kemoterapi rejimi
G-CSF = gran�losit koloni uyar�c� fakt�r, VP-16 = etoposid, MTX = metotreksat, IV = intraven�z, SC = subkutan, IT = intratekal, PO = oral, IM = intram�sk�ler, ARA-C = sitarabin, CPM = siklofosfamid, VCR = vinkristin, DEX = deksametazon, DAUN = daunorubisin, 6-MP = 6-merkaptopurin, E.Coli L-ASP = L- asparaginaz, PEG-ASP = PEG asparaginaz, MESNA = 2-merkaptoetan s�lfonat sodyum, iii = veya MTX d�zeyi <0,1 µM olana kadar, q6h= her 6 saatte bir, Gy = Gray �al��ma AIT07, kemoterapi ile kombinasyon halinde imatinib ile tedavi edilen 128 hastay� (1 ila <18 ya�) i�eren �ok merkezli, a��k etiketli, randomize, Faz II / III bir �al��mad�r. Bu �al��madan elde edilen g�venlilik verilerinin, imatinibin Ph + ALL hastalar�nda g�venlilik profili ile uyumlu oldu�u g�r�lmektedir. N�ksetmi�/tedaviye refrakter Ph+ ALL�matinib, yineleyen/refrakter Ph+ ALL hastalar�nda tek ajan olarak kullan�ld���nda, 411 hastan�n 53'�nde yan�t de�erlendirilebilmi�, hematolojik yan�t oran� %30 (%9'u tam) ve maj�r sitogenetik yan�t oran� ise %23 olarak bulunmu�tur (Not: 411 hastan�n 353'�, primer yan�t verileri toplanmaks�z�n geni�letilmi� eri�im �al��mas�nda tedavi edilmi�tir). 411 yineleyen/refrakter Ph+ ALL hastas�ndan olu�an toplam pop�lasyonda progresyona kadar ge�en medyan s�re 2,6 ile 3,1 ay aral���nda olurken, de�erlendirilebilir 401 hastada medyan genel sa�kal�m 4,9 ile 9 ay aral���nda bulunmu�tur. Bu veriler, sadece 55 ya� ve �zeri hastalar dahil edilecek �ekilde yeniden analiz yap�ld���nda da benzer olmu�tur. MDS/MPD'de klinik �al��malarBu endikasyonda GL�VEC ile deneyim �ok s�n�rl�d�r ve hematolojik ve sitogenetik yan�t oranlar�na dayanmaktad�r. Klinik bir fayda veya artan sa�kal�m� g�steren kontroll� �al��ma yoktur. Abl, Kit veya PDGFR protein tirozin kinazlarla ili�kili ya�am� tehdit eden hastal�klardan muzdarip �e�itli hasta pop�lasyonlar�nda GL�VEC'in test edildi�i bir a��k etiketli, �ok merkezli, faz II klinik �al��ma (�al��ma B2225) y�r�t�lm��t�r. Bu �al��ma, g�nde 400 mg GL�VEC ile tedavi edilen MDS/MPD'li 7 hastay� i�ermektedir. �� hasta tam hematolojik yan�t (THY) ve bir hasta k�smi hematolojik yan�t (KHY) vermi�tir. Orjinal analiz s�ras�nda, PDGFR gen yeniden d�zenlemeleri saptanan d�rt hastadan ���nde hematolojik yan�t (2 THY ve 1 KHY) geli�mi�tir. Bu hastalar�n ya�lar� 20 ile 72 aras�nda de�i�mektedir. PDGFR-β yeniden d�zenlemesi olan ve GL�VEC ile tedavi edilmi� miyeloproliferatif neoplazmalardan muzdarip hastalarda uzun vadeli g�venlilik ve etkililik verilerini toplamak i�in g�zlemsel bir kay�t �al��mas� (�al��ma L2401) yap�lm��t�r. Bu kay�t �al��mas�nda yer alan 23 hasta, medyan 7,2 y�l (0,1 ile 12,7 y�l) boyunca medyan g�nl�k 264 mg (aral�k: 100 ile 400 mg) dozda GL�VEC alm��t�r. Bu kay�t �al��mas�n�n g�zlemsel yap�s� nedeniyle, kay�tl� 23 hastan�n s�ras�yla 22, 9 ve 17'si i�in hematolojik, sitogenetik ve molek�ler de�erlendirme verileri mevcuttur. Konservatif olarak, eksik verileri olan hastalar�n yan�t vermeyenler oldu�u varsay�ld���nda, 20/23 (%87) hastada THY, 9/23 (%39,1) hastada TSY ve 11/23 (%47,8) hastada MY g�zlemlenmi�tir. En az bir ge�erli de�erlendirmesi olan hastalardan yan�t oran� hesapland���nda, THY, TSY ve MY i�in yan�t oran� s�ras�yla 20/22 (%90,9), 9/9 (%100) ve 11/17 (%64,7) olmu�tur. Ayr�ca 13 yay�nda MDS/MPD'li 24 hasta daha bildirilmi�tir. 21 hasta g�nl�k 400 mg GL�VEC ile tedavi edilirken, di�er 3 hasta daha d���k dozlar alm��t�r. On bir hastada PDGFR gen yeniden d�zenlemeleri tespit edilmi�tir, bunlardan 9'u bir THY ve 1 KHY elde etmi�tir. Bu hastalar�n ya�lar� 2 ile 79 aras�nda de�i�mi�tir. Yak�n tarihli bir yay�nda, bu 11 hastan�n 6's�ndan al�nan g�ncellenmi� bilgiler, t�m bu hastalar�n sitogenetik remisyonda (32-38 ay aral���nda) kald���n� ortaya koymu�tur. Ayn� yay�nda, PDGFR gen yeniden d�zenlemeleri olan 12 MDS/MPD hastas�ndan (B2225 �al��mas�ndan 5 hasta) uzun s�reli takip verileri bildirilmi�tir. Bu hastalar medyan 47 ay (24 g�n - 60 ay aral���nda) GL�VEC alm��t�r. Bu hastalar�n 6's�nda takip s�resi, art�k 4 y�l�n �zerindedir. On bir hasta h�zl� THY elde etmi�tir; RT-PCR ile �l��ld���nde 10'unda sitogenetik anormallikler tamamen d�zelmi� ve f�zyon transkriptleri azalm�� ya da kaybolmu�tur. Hematolojik ve sitogenetik yan�tlar, s�ras�yla medyan 49 ay (19-60 aral���) ve 47 ay (16-59 aral���) boyunca s�rd�r�lm��t�r. Genel sa�kal�m tan�dan itibaren 65 ayd�r (aral�k 25-234). Genetik translokasyonu olmayan hastalara GL�VEC uygulamas�, genellikle herhangi bir iyile�me sa�lamamaktad�r. MDS/MPD'li pediatrik hastalarda kontroll� �al��ma yoktur. D�rt yay�nda PDGFR gen yeniden d�zenlemeleriyle ili�kili MDS/MPD'li be� (5) hasta bildirilmi�tir. Bu hastalar�n ya�lar� 3 ay ile 4 y�l aras�nda de�i�mi�tir ve imatinib g�nde 50 mg dozda veya g�nde 92,5 ila 340 mg/m2 aras�nda de�i�en dozlarda verilmi�tir. T�m hastalarda tam hematolojik yan�t, sitogenetik yan�t ve/veya klinik yan�t elde edilmi�tir. HES/CEL ile �lgili Klinik �al��malarAbl, KIT ya da PDGFR protein tirozin kinazlarla ili�kili ya�am� tehdit edici hastal�klar� olan farkl� hasta pop�lasyonlar�nda GL�VEC'in test edildi�i a��k-etiketli, �ok merkezli bir faz II klinik �al��ma (�al��ma B2225) y�r�t�lm��t�r. Bu �al��mada HES/CEL'i olan 14 hasta, g�nde 100 mg ila 1000 mg dozda GL�VEC ile tedavi edilmi�tir. Yay�nlanm�� 35 vaka raporu ve vaka serisinde bildirilen HES/CEL'li 162 hasta daha g�nl�k 75 mg ila 800 mg dozlar�nda GL�VEC alm��t�r. 176 hastadan olu�an toplam pop�lasyonun 117'sinde sitogenetik anormallikler de�erlendirilmi�tir. Bu 117 hastan�n 61'inde FIP1L1- PDGFRα f�zyon kinaz tan�mlanm��t�r. Di�er 3 yay�nlanm�� raporda d�rt HES hastas�n�n daha FIP1L1-PDGFRα pozitif oldu�u bulunmu�tur. 65 FIP1L1-PDGFRα f�zyon kinaz pozitif hastan�n t�m�, aylarca s�rd�r�len bir THY elde etmi�tir (raporlama s�ras�nda sans�rlenen 1+ ila 44+ ay aras�nda). Yak�n tarihli bir yay�nda bildirildi�i gibi, bu 65 hastadan 21'i, 28 ayl�k (aral�k 13-67 ay) medyan bir takip s�resiyle tam molek�ler remisyona ula�m��t�r. Bu hastalar�n ya�lar� 25 ile 72 aral���nda olmu�tur. Ek olarak, olgu raporlar�nda ara�t�rmac�lar taraf�ndan semptomatolojide ve di�er organ disfonksiyon anormalliklerindeki geli�meler bildirilmi�tir. Kalp, sinir, deri/derialt� doku, solunum/g���s/mediastinal, kas-iskelet/ba� dokusu/vask�ler ve gastrointestinal organ sistemlerinde geli�meler bildirilmi�tir. HES/CEL'li pediyatrik hastalarda kontroll� �al��ma yoktur. 3 yay�nda PDGFR gen yeniden d�zenlemeleri ile ili�kili HES ve CEL'li �� (3) hasta bildirilmi�tir. Bu hastalar�n ya�lar� 2 ila 16 y�l aras�nda de�i�mi�tir ve imatinib g�nde 300 mg/m2 veya g�nl�k 200 ila 400 mg aras�nda de�i�en dozlarda verilmi�tir. T�m hastalarda tam hematolojik yan�t, tam sitogenetik yan�t ve/veya tam molek�ler yan�t elde edilmi�tir. Rezeke edilemeyen ve/veya metastatik GIST'de yap�lan klinik �al��malarRezeke edilemeyen veya metastatik malign gastrointestinal stromal t�m�rleri (GIST) olan hastalarda faz II, a��k etiketli, randomize, kontrols�z �ok uluslu bir �al��ma y�r�t�lm��t�r. Bu �al��maya 147 hasta kaydedilmi� ve 36 ay boyunca g�nde bir kez oral olarak 400 mg veya 600 mg kullan�m�na randomize edilmi�tir. Bu hastalar�n ya�lar�, 18 ila 83 aras�ndad�r ve patolojik olarak rezeke edilemeyen ve/veya metastatik Kit-pozitif malign G�ST tan�s�na sahiptir. �mm�nohistokimya Kit antikoru ile (A-4502, tav�an poliklonal antiserumu, 1:100; DAKO Corporation, Carpinteria, CA) antijen geri kazan�m� sonras� avidin-biotin-peroksidaz kompleksi y�ntemi ile analize g�re rutin olarak y�r�t�lm��t�r. Birincil etkililik kan�t�, objektif yan�t oranlar�n� temel alm��t�r. T�m�rlerin en az bir hastal�k b�lgesinde �l��lebilir olmas� gerekmi� olup, yan�t karakterizasyonu G�neybat� Onkoloji Grubu (SWOG) kriterlerini temel alm��t�r. Bulgular, Tablo 8'de sunulmaktad�r. Tablo 8: STIB2222 kodlu GIST �al��mas�nda en iyi t�m�r yan�t�
�ki doz grubu aras�nda yan�t oranlar� bak�m�ndan farkl�l�klar s�z konusu olmam��t�r. Ara analiz tarihinde �nemli say�da stabil hastal��a sahip hasta, daha uzun s�reli tedavi ile k�smi yan�ta ula�m��t�r (medyan takip s�resi 31 ay). Yan�ta kadar ge�en medyan s�re, 13 hafta olmu�tur (%95 GA 12-23). Yan�t veren olgularda tedavi ba�ar�s�zl���na kadar ge�en medyan s�re 122 hafta (%95 GA 106-147), genel �al��ma pop�lasyonunda ise 84 hafta (%95 GA 71-109) bulunmu�tur. Medyan genel sa�kal�m noktas�na ula��lamam��t�r. 36 ayl�k izlem sonras�nda Kaplan-Meier sa�kal�m tahmini %68'dir. �ki klinik �al��mada (�al��ma B2222 ve gruplar aras� �al��ma S0033), g�nl�k GL�VEC dozu, 400 mg veya 600 mg daha d���k g�nl�k dozlar�nda progrese olan hastalarda 800 mg'a y�kseltilmi�tir. Doz, toplam 103 hastada 800 mg'a ��kar�lm��t�r; doz y�kseltildikten sonra 6 hasta k�smi yan�ta ve 21 hasta hastal�k stabilizasyonuna ula�arak %26'l�k genel klinik yarar sonucunu vermi�tir. Eldeki g�venlilik verilerinden yola ��k�larak, 400 mg veya 600 mg daha d���k g�nl�k dozlar�nda progrese olan hastalarda dozun g�nde 800 mg'a ��kar�lmas�n�n, GL�VEC'in g�venlilik profilini etkilemedi�i g�r�lmektedir. Adjuvan GIST i�in klinik �al��malarAdjuvan tedavi ko�ullar�nda GL�VEC, 773 hasta ile y�r�t�len �ok merkezli, �ift k�r, uzun s�reli, plasebo kontroll� bir faz III �al��mada (Z9001) ara�t�r�lm��t�r. Bu hastalar�n ya�lar�, 18-91 aral���nda olmu�tur. �mm�nhistokimya ile Kit proteini eksprese eden primer GIST y�n�nde histolojik tan�s� bulunan ve en geni� yerinde ≥3 cm t�m�r b�y�kl���ne sahip olan, �al��maya kay�t �ncesindeki 14-70 g�n i�erisinde primer GIST'i tam gross rezeksiyon ile al�nan hastalar dahil edilmi�tir. Primer GIST rezeke edildikten sonra hastalar, �u iki koldan birine randomize edilmi�tir: bir y�l s�reyle GL�VEC 400 mg/g�n veya plasebo. �al��man�n birincil sonlanma noktas�, randomizasyon tarihinden rek�rense ya da herhangi bir nedene ba�l� �l�me kadar ge�en s�re �eklinde tan�mlanan rek�renssiz sa�kal�m (RFS) olmu�tur. GL�VEC RFS'de anlaml� uzama sa�lam��, GL�VEC grubunda hastalar�n %75'i 38. ayda rek�renssiz iken plasebo grubundaki hastalar�n %75'i 20. ayda rek�renssiz kalm��t�r (s�ras�yla %95 GA [30-hesaplanamaz]; [14-hesaplanamaz]); (tehlike oran� = 0,398 [0,259-0,610], p<0,0001). Bir y�l sonunda genel RFS, plasebo (%82,3) kar��s�nda GL�VEC i�in anlaml� d�zeyde daha iyi bulunmu�tur (%97,7) (p<0,0001). Bu �ekilde rek�rens riski plaseboya oranla %89 azalt�lm��t�r (tehlike oran� = 0,113 [0,049-0,264]). Primer GIST'lerine y�nelik ameliyatlar� sonras�nda hastalardaki rek�rens riski, �u prognoz fakt�rleri esas al�narak retrospektif �ekilde de�erlendirilmi�tir: t�m�r b�y�kl���, mitotik indeks, t�m�r yeri. Mitotik indeks verileri, tedavi ama�l� (ITT) pop�lasyonu olu�turan 713 hastan�n 556's� i�in mevcuttu. Birle�ik Devletler Ulusal Sa�l�k Enstit�leri (NIH) ve Silahl� Kuvvetler Patoloji Enstit�s� (AFIP) risk s�n�fland�rmalar�na g�re yap�lan alt grup analizlerinin sonu�lar� Tablo 9'da g�sterilmektedir. D���k ve �ok d���k risk gruplar�nda herhangi bir fayda g�zlenmemi�tir. Genel bir sa�kal�m faydas� g�zlenmemi�tir. Tablo 9: NIH ve AFIP risk s�n�fland�rmas�na g�re Z9001 �al��mas� RFS analiz �zeti
*Full takip periyodu- NE-Tahmin edilebilir de�il �kinci bir �ok merkezli, a��k etiketli faz III �al��mada (SSG XVIII/AIO), cerrahi GIST rezeksiyonu sonras�nda olan ve a�a��daki durumlardan birinin bulundu�u hastalarda 400 mg/g�n GL�VEC ile 36 ay kar��s�nda 12 ayl�k tedavi kar��la�t�r�lm��t�r: t�m�r �ap� > 5 cm ve mitotik say�m > 5/50 y�ksek g�� alan� (HPF); veya t�m�r �ap� > 10 cm ve herhangi bir mitotik say�m veya mitotik say�m� > 10/50 HPF olan herhangi bir b�y�kl�kteki t�m�r ya da periton bo�lu�una do�ru r�pt�re olan t�m�rler. Toplam 397 hastadan olur al�nm�� ve bu hastalar �al��maya randomize edilmi�tir (199 hasta 12 ay kolunda ve 198 hasta 36 ay kolunda) medyan ya� 61 idi [aral�k 22 ila 84 ya�]). Medyan takip s�resi 54 ay olup (randomizasyondan veri kesme tarihine kadar) ilk hastan�n randomize edili�inden veri kesme tarihine kadar ge�en medyan s�re 83 ayd�r. �al��man�n birincil sonlanma noktas�, randomizasyon tarihinden n�kse ya da herhangi bir nedene ba�l� �l�me kadar ge�en s�re �eklinde tan�mlanan n�kss�z sa�kal�m (RFS) olmu�tur. 36 ayl�k GL�VEC tedavisi, 12 ayl�k GL�VEC tedavisi ile kar��la�t�r�ld���nda RFS'de anlaml� �l��de uzama sa�lam��t�r (genel tehlike oran� (HR) = 0,46 [0,32, 0,65], p<0,0001) (Tablo 8, �ekil 1). Buna ek olarak, 36 ayl�k GL�VEC tedavisi, 12 ayl�k GL�VEC tedavisi ile kar��la�t�r�ld���nda genel sa�kal�m (OS) s�resini anlaml� �l��de uzatm��t�r (HR = 0,45 [0,22, 0,89], p=0,0187) (Tablo 8, �ekil 2). Daha uzun s�reli tedavi (> 36 ay) yeni rek�renslerin olu�umunu geciktirebilmektedir; ancak, bu bulgunun genel sa�kal�m �zerindeki etkisi halen bilinmemektedir. Toplam �l�m say�s� 12 ayl�k tedavi kolu i�in 25 ve 36 ayl�k tedavi kolu i�in 12 �eklinde olmu�tur. �matinib ile 36 ay s�reli tedavi, ITT analizinde, yani t�m �al��ma pop�lasyonun dahil edildi�i analizde, 12 ayl�k tedaviden daha �st�n bulunmu�tur. Mutasyon tipine g�re yap�lan planl� bir alt grup analizinde, ekson 11 mutasyonlar� olan hastalarda 36 ayl�k tedavide RFS i�in tehlike oran� 0,35 olmu�tur [%95 GA: 0,22, 0,56]. G�zlemlenen olay say�s�n�n d���k olmas� sebebiyle, daha az yayg�n olan mutasyon alt gruplar� i�in herhangi bir sonu� ��kart�lamamaktad�r.
Tablo 10: 12 ayl�k ve 36 ayl�k GL�VEC Tedavisi (SSGXVIII/AIO �al��mas�)
�ekil 1 Primer rek�renssiz sa�kal�m sonlan�m noktas� i�in Kaplan-Meier tahminleri (ITT pop�lasyonu)
�ekil 2 Genel sa�kal�m i�in Kaplan-Meier tahminleri (ITT pop�lasyonu)
C-Kit pozitif GIST olan pediyatrik hastalarda kontroll� �al��ma bulunmamaktad�r. 7 yay�nda GIST'li (Kit ve PDGFR mutasyonlar� olan veya olmayan) onyedi (17) hasta bildirilmi�tir. Bu hastalar�n ya��, 8 ila 18 aral���nda olmu�tur ve imatinib, hem adjuvan hem de metastatik ko�ullarda g�nde 300 ile 800 mg aras�nda de�i�en dozlarda verilmi�tir. GIST tedavisi g�ren pediyatrik hastalar�n �o�unda c-kit veya PDGFR mutasyonlar�n� do�rulayan veriler bulunmamakta olup bu durum kar���k klinik sonu�lara yol a�m�� olabilir. DFSP'de klinik �al��malarG�nl�k 800 mg GL�VEC ile tedavi edilen DFSP'li 12 hastay� i�eren faz II, a��k etiketli, �ok merkezli bir klinik �al��ma (�al��ma B2225) y�r�t�lm��t�r. DFSP'li hastalar�n ya��, 23 ile 75 aras�nda de�i�mi�tir; DFSP metastatiktir, ilk rezektif cerrahiyi takiben lokal olarak n�ksetmi�tir ve �al��maya giri� s�ras�nda daha fazla rezektif cerrahiye uygun g�r�lmemi�tir. Etkilili�in birincil kan�t�, objektif yan�t oranlar�na dayanm��t�r. Biri tamamen ve 8'i k�smen olmak �zere, kay�tl� 12 hastadan 9'u yan�t vermi�tir. K�smi yan�t verenlerden ���, daha sonra ameliyatla hastal�ks�z hale getirilmi�tir. B2225 �al��mas�nda medyan tedavi s�resi, 6,2 ayd�r ve maksimum s�re 24,3 ayd�r. Yay�nlanm�� 5 vaka raporunda GL�VEC ile tedavi edilen 6 DFSP hastas� daha bildirilmi�tir ve ya�lar� 18 ay ile 49 y�l aras�nda de�i�mektedir. Yay�nlanm�� literat�rde bildirilen eri�kin hastalar, g�nl�k 400 mg (4 vaka) veya 800 mg (1 vaka) GL�VEC ile tedavi edilmi�tir. ��� tamamen ve 2'si k�smen olmak �zere be� (5) hasta yan�t vermi�tir. Yay�nlanan literat�rde medyan tedavi s�resi 4 hafta ile >20 ay aras�nda de�i�mektedir. GL�VEC tedavisine yan�t verenlerin neredeyse tamam�nda translokasyon t(17:22)[(q22:q13)] veya bunun gen �r�n� mevcuttur. DFSP'li pediatrik hastalarda kontroll� �al��ma bulunmamaktad�r. �� yay�nda DFSP ve PDGFR gen yeniden d�zenlemelerine sahip be� (5) hasta bildirilmi�tir. Bu hastalar�n ya�� yenido�an ile 14 ya� aras�nda de�i�mektedir ve imatinib g�nde 50 mg dozda veya g�nde 400 ila 520 mg/m2 aras�nda de�i�en dozlarda verilmi�tir. T�m hastalar, k�smi ve/veya tam yan�t elde etmi�tir. 5.2. Farmakokinetik �zelliklerGenel �zelliklerGL�VEC'in farmakokineti�i 25 - 1000 mg'l�k bir doz aral���nda de�erlendirilmi�tir. Plazma farmakokinetik profilleri, 1. g�nde ve plazmada kararl� d�zeylerin elde edildi�i 7. ya da 28. g�nde analiz edilmi�tir. Emilim: �matinibin ortalama mutlak biyoyararlan�m�, % 98'dir. Bir oral dozu takiben plazma imatinib e�ri alt�nda kalan alan (EAA) de�erlerinde, y�ksek oranda hastalar aras� de�i�kenlik g�r�lm��t�r. Y�ksek ya� i�eren bir g�da ile birlikte verildi�inde, imatinibin emilim oran� minimal d�zeyde azalm�� (C'da %11 azalma ve t'da 1,5 saatlik uzama), a�l�k ko�ullar�na g�re EAA de�erinde k���k bir azalma (% 7,4) olmu�tur. Ge�irilmi� gastrointestinal cerrahinin ila� emilimi �zerindeki etkisi ara�t�r�lmam��t�r. Da��l�m: Klinik a��dan uygun konsantrasyonlarda kullan�lan imatinibin plazma proteinlerine ba�lanmas� yakla��k % 95 olmu�, in vitro deneyler temelinde, daha �ok alb�min ve alfa- asit-glikoproteine, az miktarda da lipoproteine ba�lanm��t�r. Biyotransformasyon: �nsanlarda dola��mdaki ana metabolit, ana ilaca benzer in vitro etki g�steren N-demetile edilmi� piperazin t�revidir. Bu metabolitin plazma EAA de�erinin imatinibin EAA de�erinin sadece % 16's� oldu�u bulunmu�tur. N-demetile metabolitin plazma proteinlerine ba�lanmas�, as�l bile�i�inkine benzerdir. �matinib ve N-demetil metaboliti birlikte, dola��mdaki radyoaktivitenin yakla��k %65'ini olu�turmu�tur (EAA (0-48saat)). Dola��mdaki radyoaktivitenin kalan k�sm�, bir dizi min�r metabolitten olu�mu�tur. �n vitro sonu�lar CYP3A4'�n, imatinib biyotransformasyonunu katalize eden ba�l�ca P450 enzimi oldu�unu g�stermi�tir. Potansiyel e�zamanl� ila�lardan (asetaminofen, asiklovir, allopurinol, amfoterisin, sitarabin, eritromisin, flulonazol, hidroksi�re, norfloksasin, penisilin V) olu�an bir panelde sadece eritromisin (IC50 50 μM) ve flukonazol (IC50 118 μM) imatinib metabolizmas�nda klinik a��dan anlaml� olabilecek inhibisyon g�stermi�tir. �n vitro ko�ullarda imatinibin CYP2C9, CYP2D6 ve CYP3A4/5'in mark�r substratlar�n�n kompetitif bir inhibit�r� oldu�u g�sterilmi�tir. �nsan karaci�eri mikrozomlar�nda Ki de�erleri s�ras�yla 27, 7,5 ve 7,9 µmol/l bulunmu�tur. Hastalarda imatinibin maksimal plazma konsantrasyonlar� 2–4 µmol/l'dir, dolay�s�yla bir arada uygulanan ila�lar�n CYP2D6 ve/veya CYP3A4/5 arac�l� metabolizmas�nda inhibisyon olas�d�r. �matinib, 5-fluorourasil biyotransformasyonuna m�dahale etmemi�tir fakat kompetitif CYP2C8 inhibisyonu (Ki = 34,7 μM) sonucu paklitaksel metabolizmas�n� inhibe etmi�tir. Bu Ki de�eri, hastalarda beklenen imatinib plazma d�zeylerinin �ok �zerindedir, dolay�s�yla 5-fluorourasil ya da paklitakselin imatinib ile bir arada uygulanmas� sonucu herhangi bir etkile�im beklenmemektedir. Eliminasyon: Oral C i�aretli bir imatinib dozundan sonra bile�i�in/bile�iklerin tespitine dayal� olarak, dozun yakla��k %81'i 7 g�n i�inde d��k�da (dozun %68'i) ve idrarda (dozun %13'�) tespit edilmi�tir. De�i�memi� durumdaki imatinib, dozun % 25'ini (% 5 idrar, % 20 fe�es) olu�turmu�tur, geriye kalan k�s�m metabolitlerdir. Plazma farmakokineti�i: Sa�l�kl� g�n�ll�lerde oral uygulaman�n ard�ndan, imatinibin tde�eri yakla��k 18 saat olmas� g�nde tek doz �eklindeki pozolojinin uygun oldu�u izlenimini vermektedir. Oral olarak 25-1000 mg imatinib uyguland�ktan sonra artan dozla birlikte ortalama EAA art��� do�rusal bir seyir izlemi�tir. Tekrarlanan dozlarda imatinib kineti�inde de�i�iklik olmam�� ve g�nde bir kez uyguland���nda kararl� durumda birikim, 1,5-2,5 kat� olmu�tur. GIST hastalar�nda farmakokinetikGIST hastalar�nda kararl� durum maruziyeti, ayn� dozajda (400 mg/g�n) KML hastalar� i�in g�zlenenden 1,5 kat daha y�ksek olmu�tur. GIST hastalar�ndaki �n pop�lasyon farmakokineti�i analizine dayal� olarak, �� de�i�kenin (alb�min, WBC ve bilirubin) imatinib farmakokineti�i ile anlaml� ili�kiye sahip oldu�u bulunmu�tur. Daha d���k alb�min de�erleri daha d���k klirense (CL/f) sebep olmu� ve daha y�ksek WBC d�zeyleri CL/f azalmas�na neden olmu�tur. Ancak bu ili�kiler, doz ayarlamas�n� gerektirecek �l��de anlaml� �ekilde �n plana ��kmam��t�r. Bu hasta pop�lasyonunda hepatik metastazlar�n varl���, potansiyel olarak karaci�er yetmezli�ine ve azalm�� metabolizmaya yol a�abilir. Pop�lasyon farmakokineti�iKML hastalar�ndaki pop�lasyon farmakokineti�i analizlerine g�re ya��n da��l�m hacmi �zerinde k���k bir etkisi olmu�tur (> 65 ya��ndaki hastalarda % 12 art��). Bu de�i�imin klinik a��dan anlaml� olmad��� d���n�lmektedir. V�cut a��rl���n�n imatinib klerensi �zerindeki etkisine bak�ld���nda, 50 kg a��rl���ndaki bir ki�ide klerensin 8,5 l/s olmas� beklenirken, 100 kg a��rl���ndaki bir ki�ideki klerens 11,8 l/s'e y�kselmektedir. Bu de�i�iklikler v�cut a��rl���na g�re bir doz ayarlamas� yap�lmas� i�in yeterli olarak kabul edilmemi�tir. Cinsiyetin imatinib kineti�i �zerinde etkisi olmam��t�r. �ocuklarda farmakokinetikEri�kin hastalarda oldu�u gibi, hem faz I hem de faz II �al��malar�nda pediatrik hastalarda oral uygulamadan sonra imatinib h�zla emilmi�tir. �ocuklarda 260 ve 340 mg/m2 imatinible elde edilen maruziyet de�erleri, eri�kinlerde s�ras�yla 400 ve 600 mg imatinible elde edilenler gibidir. 340 mg/m2 imatinibin birinci ve sekizinci g�nlerdeki EAAde�erleri bu ilac�n, tekrarlanan g�nde bir kez dozlardan sonra 1,7 kat birikti�ini g�stermi�tir. Hematolojik bozukluklar� (KML, Ph+ALL ya da imatinib ile tedavi edilen di�er hematolojik bozukluklar) olan pediyatrik hastalarda birle�tirilmi� pop�lasyon farmakokineti�i analizine dayal� olarak imatinib klerensi, v�cut y�zey alan�n�n (VYA) artmas�na paralel olarak y�kselmektedir. VYA etkisi i�in d�zeltme yap�ld�ktan sonra, ya�, v�cut a��rl��� ve v�cut kitle indeksi gibi di�er demografik fakt�rler, imatinib maruziyeti �zerinde klinik a��dan anlaml� etkiler yapmam��t�r. Yap�lan analiz, g�nde bir kere 260 mg/m2 (g�nde 400 mg'� ge�memek �zere) ya da g�nde bir kere 340 mg/m2 alan (g�nde 600 mg'� ge�memek �zere) pediyatrik hastalarda imatinib maruziyetinin, g�nde bir kere 400 mg ya da 600 mg imatinib alan yeti�kin hastalardakine benzer oldu�unu do�rulam��t�r. Organ fonksiyonu bozuklu�u�matinib ve metabolitleri, b�brek yoluyla anlaml� miktarda at�lmaz. B�brek fonksiyonlar�nda hafif ve orta �iddette bozukluk olan hastalar, b�brek fonksiyonlar� normal hastalardan daha y�ksek plazma de�erlerine sahip g�r�nmektedir. Art��, yakla��k olarak 1,5-2 katt�r ve imatinibin g��l� bir bi�imde ba�land��� plazma alfa asit glikoprotein (AGP) de�erinde 1,5 katl�k bir art��a kar��l�k gelir. B�brek bozuklu�u olan hastalarda imatinibin serbest ila� klerensi muhtemelen b�brek fonksiyonlar� normal hastalardakinin bir benzeridir ��nk� b�brekler yoluyla at�l�m imatinib i�in min�r bir eliminasyon yolunu olu�turmaktad�r (bkz. B�l�m 4.2 ve 4.4). Farmakokinetik analiz sonu�lar�n�n ki�iden ki�iye de�i�ikliklerin s�z konusu oldu�unu g�stermesine ra�men, de�i�ik derecelerde karaci�er yetersizli�i olan hastalardaki imatinibe ortalama maruz kal�m, karaci�er fonksiyonlar� normal olan hastalara k�yasla y�kselmemi�tir (bkz. B�l�m 4.2, 4.4 ve 4.8). 5.3. Klinik �ncesi g�venlilik verileri�matinibin klinik �ncesi g�venlili�i s��anlarda, k�peklerde, maymunlarda ve tav�anlarda de�erlendirilmi�tir. �oklu doz toksisite �al��malar� s��anlarda, k�peklerde ve maymunlarda hafif ile orta dereceli hematolojik de�i�iklikler ortaya koymu�, s��anlarda ve k�peklerde bu de�i�ikliklere kemik ili�i de�i�iklikleri e�lik etmi�tir. Karaci�er, s��anlarda ve k�peklerde hedef organ olmu�tur. �ki t�rde de transaminazlarda hafif ila orta dereceli art��lar ve kolesterol, trigliseritler, total protein ve alb�min d�zeylerinde hafif d����ler g�zlenmi�tir. S��an karaci�erinde herhangi bir de�i�iklik g�r�lmemi�tir. �ki hafta s�reyle tedavi edilen k�peklerde y�kselmi� karaci�er enzimleri, hepatosel�ler nekroz, safra kanal� nekrozu ve safra kanal� hiperplazisi ile �iddetli karaci�er toksisitesi g�zlenmi�tir. �ki hafta s�reyle tedavi edilen maymunlarda fokal mineralizasyon ve renal t�b�llerin dilatasyonu ve t�b�ler nekroz ile renal toksisite g�zlenmi�tir. Bu hayvanlar�n birka��nda kan �re azotunda (BUN) ve kreatininde art�� g�zlenmi�tir. 13 haftal�k �al��mada s��anlarda ≥ 6 mg/kg dozlarda serum veya idrar parametrelerinde de�i�iklikler olmaks�z�n mesane ve renal papilla transisyonel epitelyum hiperplazisi g�zlenmi�tir. Kronik imatinib tedavisi ile f�rsat�� enfeksiyonlar�n oran�nda art�� g�zlenmi�tir. 39 haftal�k maymun �al��mas�nda NOAEL (advers etkinin g�r�lmedi�i d�zey), 15 mg/kg olan en d���k dozda saptanm�� olup bu doz, v�cut y�zeyi baz�nda 800 mg'l�k maksimum insan dozunun yakla��k ��te biridir. Tedavi, bu hayvanlarda normalde bask�lanm�� olan malaryal enfeksiyonlarda k�t�le�me ile sonu�lanm��t�r. �matinib, bir in vitro bakteriyel h�cre testinde (Ames test), bir in vitro memeli h�cre testinde (fare lenfomas�) ve bir in vivo s��an mikron�kleus testinde test edildi�inde genotoksik etki g�stermemi�tir. Metabolik aktivasyon varl���nda klastojenisiteye (kromozom aberasyonu) y�nelik bir in vitro memeli h�cre testinde (�in hamster� overi) imatinib i�in pozitif genotoksik etkiler elde edilmi�tir. �retim prosesinin, son �r�nde de bulunan iki ara �r�n� Ames testinde mutajenisite a��s�ndan pozitiftir. Bu ara �r�nlerden biri ayr�ca fare lenfoma testinde de pozitif sonu� vermi�tir. Bir fertilite �al��mas�nda �iftle�meden �nce 70 g�n s�reyle dozlar uygulanan erkek s��anlarda testik�ler ve epididimal a��rl�klar ve hareketli sperm y�zdesi, beden y�zey alan� baz�nda 800 mg/g�n maksimum klinik dozu ile yakla��k olarak e�it olan 60 mg/kg dozunda azalm��t�r. Bu etki, ≤ 20 mg/kg dozlarda g�r�lmemi�tir. K�pekte ≥ 30 mg/kg oral dozlarda spermatogenezde de hafif ila orta dereceli bir azalma g�zlenmi�tir. Di�i s��anlar, �iftle�meden �nce 14 g�n s�reyle ve 6. gestasyon g�n�ne kadar dozlar uyguland���nda �iftle�me ya da gebe hayvan say�s�nda herhangi bir etki s�z konusu olmam��t�r. 60 mg/kg dozunda di�i s��anlarda �nemli �l��de implantasyon sonras� fetal kay�p ve canl� fet�s say�s�nda azalma olmu�tur. Bu etki ≤ 20 mg/kg dozlarda g�r�lmemi�tir. S��anlarla y�r�t�len bir oral prenatal ve postnatal geli�im �al��mas�nda 45 mg/kg/g�n grubunda gestasyonun 14 ya da 15. g�n�nde k�rm�z� vajinal ak�nt� kaydedilmi�tir. Ayn� dozda �l� do�an yavru say�s� ve ayr�ca do�um sonras� 0-4 g�nler aras�nda �len yavru say�s� da artm��t�r. F1 yavrularda ayn� doz d�zeyinde ortalama v�cut a��rl�klar� do�umdan �ld�r�lene kadar ge�en s�rede azalm��t�r ve prepusyal ayr�lma kriterine ula�an yavru say�s� da hafif azalm��t�r. 45 mg/kg/g�n dozunda F1 fertilitesi etkilenmemi�, di�er yandan rezorpsiyon say�s�nda art�� ve canl� fet�s say�s�nda azalma tespit edilmi�tir. Gerek anne hayvanlar gerekse F1 nesil i�in etkinin g�zlenmedi�i d�zey (NOEL) 15 mg/kg/g�n (800 mg'l�k maksimum insan dozunun d�rtte biri) olmu�tur. �matinib organogenez s�ras�nda, v�cut y�zey alan� baz�nda 800 mg/g�n maksimum klinik doz ile yakla��k olarak e�it olan ≥ 100 mg/kg dozlarda s��anlara uyguland���nda teratojen etki g�stermi�tir. Teratojenik etkiler eksensefali veya ensefalosel, frontal kemiklerin olmamas�/eksik olmas� ve parietal kemiklerin olmamas�n� i�ermi�tir. Bu etkiler ≤ 30 mg/kg dozlarda g�r�lmemi�tir. S��anlarda juvenil geli�im toksikolojisi �al��mas�nda (do�um sonras� 10 ile 70. g�n) bilinen hedef organlara g�re yeni hedef organ tan�mlanmam��t�r. Juvenil toksikoloji �al��mas�nda, ortalama pediyatrik maruziyet olarak �nerilen en y�ksek doz olan 340 mg/m2 d�zeyinin yakla��k 0,3 ila 2 kat� d�zeylerde, b�y�me �zerinde ge�ici etkiler ve vaginal a��lma ve prepusyal ayr�lmada gecikme g�zlenmi�tir. Ayr�ca, ortalama pediyatrik maruziyet olarak �nerilen en y�ksek doz olan 340 mg/m2 d�zeyinin yakla��k 2 kat� d�zeylerde, juvenil hayvanlarda (yakla��k olarak s�tten kesilme d�neminde) mortalite g�zlemlenmi�tir. 2 y�ll�k s��an karsinojenisite �al��mas�nda 15, 30 ve 60 mg/kg/g�n olarak imatinib uygulanmas�, erkeklerde 60 mg/kg/g�n dozunda ve di�ilerde ≥ 30 mg/kg/g�n dozunda ya�am s�resi �zerinde istatistiksel a��dan anlaml� azalmaya neden olmu�tur. �lenlerde yap�lan histopatolojik inceleme, �l�m�n temel nedeni ya da �ld�r�lme nedeni olarak kardiyomiyopati (her iki cinsiyet), kronik ilerleyici nefropati (di�iler) ve prepusyal bez papillomunu ortaya koymu�tur. Neoplastik de�i�iklikler a��s�ndan hedef organlar b�brekler, mesane, �retra, prepusyal ve klitoral bez, ince ba��rsak, paratiroid bezleri, adrenal bezler ve gland�ler-olmayan mide olmu�tur. Prepusyal/klitoral bezde papilloma/karsinoma s�ras�yla 400 mg/g�n veya 800 mg/g�n dozlar�nda insan g�nl�k maruziyetinin 0,5 veya 0,3 kat�na (EAA baz�nda) ya da �ocuklarda 340 mg/m2/g�n dozunda g�nl�k maruziyetin 0,4 kat�na kar��l�k gelen (EAA baz�nda) 30 mg/kg/g�n dozundan itibaren g�zlenmi�tir. Etki g�zlenmeyen d�zey (NOEL) 15 mg/kg/g�n olmu�tur. 400 mg/g�n veya 800 mg/g�n dozlar�nda insan g�nl�k maruziyetinin s�ras�yla 1,7 veya 1 kat�na (EAA baz�nda) ya da �ocuklarda 340 mg/m2/g�n dozunda g�nl�k maruziyetin 1,2 kat�na kar��l�k gelen (EAA baz�nda) 60 mg/kg/g�n dozunda renal adenoma/karsinoma, mesane ve �retra papillomas�, ince ba��rsak adenokarsinomalar�, paratiroit bezleri adenomalar�, adrenal bezlerde benign ve malign med�ller t�m�rler ve gland�ler olmayan mide papillomalar�/karsinomalar� g�r�lm��t�r. Etki g�zlenmeyen d�zey (NOEL), 30 mg/kg/g�n olmu�tur. �nsanlar i�in s��an karsinojenisite �al��mas�ndaki bu bulgular�n mekanizmas� ve �nemi, hen�z a��kl��a kavu�turulmam��t�r. Erken d�nem klinik �al��malarda tan�mlanmayan non-neoplastik lezyonlar kardiyovask�ler sistem, pankreas, endokrin organlar ve di�lerle ilgili olmu�tur. En �nemli de�i�iklikler baz� hayvanlarda kalp yetmezli�i belirtilerine yol a�an kardiyak hipertrofi ve dilatasyonu i�ermi�tir. Etkin maddeimatinib, tortul tabaka organizmalar� i�in �evresel risk olu�turur.6. FARMAS�T�K �ZELL�KLER6.1. Yard�mc� maddelerin listesiMikrokristalin sel�loz, krospovidon, hidroksipropil metilsel�loz, kolloidal silikon dioksid, magnezyum stearat, k�rm�z� demir oksit, sar� demir oksit, polietilen glikol 4000 ve talk i�erir. 6.2. Ge�imsizliklerGe�erli de�ildir. 6.3. Raf �mr�36 ay 6.4. Saklamaya y�nelik �zel tedbirler30 C'nin alt�ndaki oda s�cakl���nda saklay�n�z. 6.5. Ambalaj�n niteli�i ve i�eri�iPVC/PE/PVDC-ALU folyo blister ambalaj 6.6. Be�eri t�bbi �r�nden arta kalan maddelerin imhas� ve di�er �zel �nlemlerKullan�lmam�� olan �r�nler ya da at�k materyaller “T�bbi At�klar�n Kontrol� Y�netmeli�i” ve “Ambalaj At�klar�n�n Kontrol� Y�netmeli�i”'ne uygun olarak imha edilmelidir.
�LA� E�DE�ERLER�
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
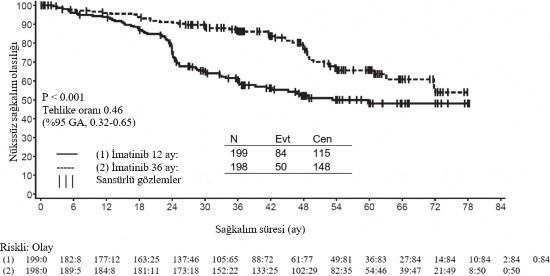
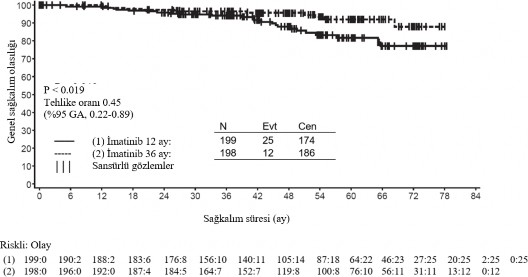

| E�de�er �la� Ad� | Barkodu | �la� Fiyat� |
|---|---|---|
| GLIMATINE | 8680030190111 | 13,060.59TL |
| GLIOTIN | 8699514151012 | 14,589.10TL |
| GLITINIB | 8699262010340 | |
| GLIVEC | 8699504091038 | |
| GLIVON | 8699540037724 | 17,642.21TL |
| Di�er E�de�er �la�lar |
 |
Do�um Sonras� Depresyonu Do�um sonras� depresyonu, do�umdan sonra her on kad�ndan biri taraf�ndan tecr�be edilen stresli bir durumdur. |
 |
Travma Sonras� Bunal�m� Travmatik bir olay, g�nl�k ola�an olaylar�n d���nda olan ve ki�iyi derinden rahats�z eden bir olayd�r.Bir�ok olay b�yle bir etki g�sterebilir. |
 |
Artrit Artrit, olduk�a yayg�n bir hastal�kt�r ancak iyi anla��lamam��t�r. Asl�nda �artrit� tek bir hastal���n ad� de�ildir; eklem a�r�s� veya eklem hastal�klar�n� adland�rman�n gayri resmi yoludur. |