HEMLIBRA 30 mg/ 1 ml SC enjeksiyonluk çözelti Kısa Ürün Bilgisi
{ Emisizumab }
1. BEŞERİ TIBBİ ÜRÜNÜN ADI
HEMLIBRA 30 mg/1 mL S.C. enjeksiyonluk çözelti Steril
2. KALİTATİF VE KANTİTATİF BİLEŞİM
Etkin madde
1 mL'lik her bir flakon, 30 mg/mL konsantrasyonda 30 mg emicizumab içermektedir.
Emicizumab; Çin Hamsteri Over (CHO) hücrelerinde rekombinant DNA teknolojisi ile üretilmiş, faktör IXa ve faktör X'u birleştiren, bispesifik antikor yapısına sahip bir monoklonal, hümanize, modifiye edilmiş immünoglobulin G4 (IgG4) antikorudur.
Yardımcı maddeler
Yardımcı maddeler için Bölüm 6.1'e bakınız.
3. FARMASÖTİK FORMU
Enjeksiyonluk çözelti içeren flakon. Renksiz ile hafif sarı arası renkte çözelti.
4. KLİNİK ÖZELLİKLER
4.1. Terapötik endikasyonlar
HEMLIBRA, faktör VIII inhibitörlü ya da inhibitörsüz hemofili A (konjenital faktör VIII eksikliği) hastalarında rutin profilakside endikedir.
HEMLIBRA tüm yaş gruplarında kullanılabilir.
4.2. Pozoloji ve uygulama şekli
Başka bir biyolojik tıbbi ürünle değiştirilmesi, reçeteyi yazan hekimin onayını gerektirmektedir.
Tedavi, hemofili ve/veya kanama bozukluklarının tedavisinde deneyimli bir hekimin gözetiminde başlatılmalıdır.
Bypass ajanlarıyla (örn. aPCC ve rFVIIa) tedavi, (rutin profilaksi dahil) HEMLIBRA tedavisi başlatılmadan önceki gün durdurulmalıdır (bkz. Bölüm 4.4).
Faktör VIII (FVIII) profilaksisine HEMLIBRA tedavisinin ilk 7 gününde devam edilebilir.
Pozoloji/uygulama sıklığı ve süresi:
Önerilen doz, subkutan enjeksiyon yolu ile ilk 4 hafta boyunca haftada 1 kez 3 mg/kg (yükleme dozu) ve bunu takiben idame dozu olarak haftada 1 kez 1,5 mg/kg, 2 haftada 1 kez 3 mg/kg veya 4 haftada 1 kez 6 mg/kg'dır ve tüm dozlar subkutan enjeksiyon olarak verilir.
Yükleme dozu, idame dozundan bağımsız olarak aynıdır.
Uyuncu desteklemek adına, idame doz rejimi hekim ve hasta/hasta bakımını üstlenen kişinin tercihine göre seçilmelidir.
Uygulama şekli:
Hasta dozu (mg cinsinden) ve hacmi (mL cinsinden) aşağıdaki şekilde hesaplanmalıdır:
İlk 4 hafta boyunca haftada 1 kez yükleme dozu (3 mg/kg):
Hasta vücut ağırlığı (kg) x doz (3 mg/kg) = uygulanacak toplam emicizumab miktarı (mg)
Sonrasında idame dozu olarak 5. haftadan itibaren haftada 1 kez 1,5 mg/kg, 2 haftada 1 kez 3 mg/kg veya 4 haftada bir kez 6 mg/kg:
Hasta vücut ağırlığı (kg) x doz (1,5; 3 veya 6 mg/kg) = uygulanacak toplam emicizumab miktarı (mg)
Subkutan olarak enjekte edilecek toplam HEMLIBRA hacmi aşağıdaki şekilde hesaplanır:
Uygulanacak toplam emicizumab miktarı (mg) ÷ flakon konsantrasyonu (mg/mL) = enjekte edilecek toplam HEMLIBRA hacmi (mL).
Uygulanacak toplam hacim hazırlanırken farklı HEMLIBRA konsantrasyonları (30 mg/mL ve 150 mg/mL) birleştirilmemelidir.
Enjeksiyon başına 2 mL'nin üzerinde hacim uygulanmamalıdır. Örnekler:
Hastanın vücut ağırlığı 16 kg, hastaya uygulanan idame doz rejimi haftada 1 kez 1,5 mg/kg:
Yükleme dozu (ilk 4 hafta) örneği: 16 kg x 3 mg/kg = 48 mg yükleme dozu için gerekli emicizumab miktarı.
Uygulanacak hacmi hesaplamak üzere hesaplanmış doz 48 mg, 150 mg/mL'ye bölünür; 48 mg emicizumab ÷ 150 mg/mL = 0,32 mL enjekte edilecek 150 mg/mL HEMLIBRA konsantrasyonu.
4.3. Kontrendikasyonlar
Etkin madde
4.4. Özel kullanım uyarıları ve önlemleri
İzlenebilirlik
Biyolojik tıbbi ürünlerin izlenebilirliğini arttırmak üzere, uygulanan ürünün adı ve seri numarası net olarak kaydedilmelidir.
HEMLIBRA ve aktive protrombin kompleksi konsantresi (aPCC) ile bağlantılı trombotik mikroanjiyopati
HEMLIBRA profilaksisi alan hastalarda yürütülen bir klinik çalışmada, 24 saat veya daha uzun süreyle >100U/kg/24 saat ortalama kümülatif miktarda aktive protrombin kompleks konsantresi (aPCC) uygulandığı durumda trombotik mikroanjiopati (TMA) vakaları bildirilmiştir (bkz. Bölüm 4.8). TMA olaylarına yönelik tedavi, plazmaferez veya hemodiyaliz ile birlikte ya da tek başına destekleyici tedavi uygulanmasını kapsamaktadır. HEMLIBRA tedavisine ara verilmesi ve aPCC'nin kesilmesinin ardından bir hafta içerisinde TMA'nın iyileştiği görülmüştür. Bu hızlı klinik iyileşme, trombotik trombositopenik purpura ve atipik hemolitik üremik sendrom gibi klasik TMA'larda gözlemlenen olağan klinik seyirden farklıdır (bkz. Bölüm 4.8). Bir hasta TMA'nın düzelmesini takiben HEMLIBRA'ya yeniden başlamış ve tedavisi güvenli bir şekilde sürdürülmüştür.
HEMLIBRA profilaksisi alan hastalara aPCC uygulanırken TMA gelişimi açısından hastaların izlenmesi gereklidir. Hekim, TMA ile tutarlı klinik semptomların ve/veya laboratuvar bulgularının gözlenmesi durumunda aPCC'yi acilen bırakmalı, HEMLIBRA tedavisine ara vermeli ve klinik olarak endike olduğu şekilde tedavi uygulamalıdır. Hekimler ve hastalar/bakım verenler, her bir vaka için ayrı ayrı olmak üzere, TMA'nın ortadan kalkmasını takiben HEMLIBRA profilaksisini yeniden başlatmanın faydalarını ve risklerini değerlendirmelidir. HEMLIBRA profilaksisi uygulanan bir hastada bir bypass edici ajanın endike olduğu durumda, aşağıda bulunan bypass edici ajanların kullanımına yönelik doz uygulaması kılavuzuna bakınız.
Yüksek TMA riski (örn. TMA geçmişi veya ailesel TMA geçmişi) olan veya TMA gelişme risk faktörü bulunan kombine ilaç (örn. siklosporin, kinin, takrolimus) kullanan hastaların tedavisinde dikkatli olunmalıdır.
HEMLIBRA ve aktive protrombin kompleksi konsantresi (aPCC) ile bağlantılı tromboembolizm
HEMLIBRA profilaksisi alan hastalarda yürütülen bir klinik çalışmada, 24 saat veya daha uzun süre >100U/kg/24 saat ortalama kümülatif miktarda aktive protrombin kompleksi konsantresi (aPCC) uygulandığında trombotik olayların (TE) ortaya çıktığı bildirilmiştir (bkz. Bölüm 4.8). Hiçbir vaka antikoagülan tedavisini gerektirmemiştir. aPCC'nin bırakılması ve HEMLIBRA tedavisinin kesilmesini takiben, iyileşme veya düzelme kanıtı bir ay içinde görülmüştür (bkz. Bölüm 4.8). Bir hasta trombotik olayın düzelmesini takiben HEMLIBRA'ya yeniden başlamış ve tedavi güvenli bir şekilde sürdürülmüştür.
HEMLIBRA profilaksisi alırken aPCC uygulanan hastalar tromboemboli gelişimi açısından izlenmelidirler. Hekim, trombotik olaylarla tutarlı klinik semptomlar, görüntüleme ve/veya laboratuvar bulguları ortaya çıkarsa, aPCC'yi acilen bırakmalı, HEMLIBRA tedavisine ara vermeli ve klinik olarak endike olduğu şekilde tedavi uygulamalıdır. Hekimler ve hastalar/bakım verenler, her bir vaka için ayrı ayrı olmak üzere, trombotik olayların ortadan kalkmasını takiben HEMLIBRA profilaksisini yeniden başlatmanın faydalarını ve risklerini değerlendirmelidir. HEMLIBRA profilaksisi uygulanan bir hastada bir bypass edici ajanının endike olduğu durumda, aşağıda bulunan bypass edici ajanlarının kullanımına yönelik doz uygulaması kılavuzuna bakınız.
HEMLIBRA profilaksisi alan hastalarda bypass edici ajanların kullanımı ile ilgili kılavuz Bypass edici ajanlarla tedavi HEMLIBRA tedavisinin başlatıldığı günden önce kesilmelidir.
Hekimler, HEMLIBRA profilaksisi uygulanırken, gerekmesi durumunda, tüm hastaları ve/ veya bakım verenleriyle, bypass edici ajanların kesin dozları ve uygulama sıklıklarını tartışmalıdır.
aPCC'nin 50 U/kg'a kadar olan başlangıç dozu ile kontrol edilemezse, tıbbi kılavuzluk veya gözetim altında, TMA veya tromboembolizm teşhisi için laboratuvar izlemesi ve dozun tekrarlanmasından önce kanama kontrolü de göz önünde bulundurularak, ek aPCC dozları uygulanmalıdır. Toplam aPCC dozu, tedavinin ilk 24 saatinde 100 U/kg'ı geçmemelidir. Tedaviyi uygulayan hekimler, ilk 24 saat içinde 100 U/kg'u aşan aPCC tedavisini düşünüyorlarsa, TMA ve TE riskinin kanama riskine karşı dikkatli bir şekilde tartılması gereklidir.
Klinik çalışmalarda, HEMLIBRA profilaksisi uygulanan hastalarda tek başına aktive rekombinant FVII (rFVIIa) kullanımı ile TMA ya da TE vakaları gözlenmemiştir.
Bypass edici ajanlara ilişkin doz uygulama kılavuzu, HEMLIBRA profilaksisinin bırakılmasını takiben en az 6 ay boyunca takip edilmelidir (bkz. Bölüm 5.2).
İmmünojenisite
Klinik çalışmalarda, emicizumab konsantrasyonunun azalmasına yol açan ve etkinlik kaybına yol açan nötralize edici anti-emicizumab antikorlarının gelişimi nadiren gözlenmiştir (bkz. Bölüm 4.8 ve 5.1). Klinik etkililik kaybı belirtileri olan hastalar (örn. ani kanama olaylarında artış), etiyolojiyi belirlemek için derhal değerlendirilmeli ve nötralize edici anti-emicizumab antikorlarından şüpheleniliyorsa diğer terapötik seçenekler düşünülmelidir.
HEMLIBRA'nın koagülasyon testlerine etkisi
HEMLIBRA eksik olan aktive faktör VIII'in (FVIIIa) tenaz kofaktörü aktivitesini yerine koyar. İntrinsik pıhtılaşma temelli koagülasyon laboratuvar testleri, aktive edilmiş pıhtılaşma zamanı (ACT), aktive edilmiş parsiyel tromboplastin zamanı (örn., aPTT) dahil, trombin yoluyla FVIII'in FVIIIa'ya aktive edilmesi için gerekli zaman dahil olmak üzere toplam pıhtılaşma süresini ölçer. Bu tip intrinsik yolağa dayalı testler, trombin ile aktivasyonu gerekmeyen HEMLIBRA ile aşırı derecede kısalmış pıhtılaşma süreleri verecektir. Aşırı kısalmış intrinsik pıhtılaşma süresi, aPTT'ye dayanan, tek aşamalı FVIII aktivite tayini gibi tek faktörlü tayinlerin tümünü bozacaktır (bkz. Bölüm 4.4, Tablo 1). Bununla birlikte, kromojenik veya immüno-bazlı yöntemleri kullanan tek faktörlü analizler HEMLIBRA'dan etkilenmez ve aşağıda tarif edildiği gibi FVIII kromojenik aktivite tayinleri için özel hususlar ile birlikte tedavi esnasında koagülasyon parametrelerini izlemek için kullanılabilir.
Kromojenik faktör VIII aktivite testleri, insan veya bovin koagülasyon proteinleri ile birlikte üretilebilir. İnsan koagülasyon faktörlerini içeren tayinler HEMLIBRA'ya duyarlıdır; fakat HEMLIBRA'nın klinik hemostatik potansiyelini olduğundan daha yüksek gösterebilir. Buna karşılık, bovin koagülasyon faktörleri içeren tayinler HEMLIBRA'ya duyarlı değildir (aktivite ölçülmez) ve endojen ya da infüze faktör VIII aktivitesinin izlenmesi ya da anti- FVIII inhibitörlerinin ölçülmesi için kullanılabilirler.
HEMLIBRA, faktör VIII'e karşı gelişen inhibitörlerin varlığında aktif kalmaktadır ve bu nedenle faktör VIII inhibitörleri için pıhtılaşma bazlı Bethesda analizlerinde yanlış-negatif bir sonuç oluşturacaktır. Bunun yerine, HEMLIBRA'ya duyarsız olan bovin temelli bir kromojenik faktör VIII tayini uygulanan bir kromojenik Bethesda tayini kullanılabilir.
Bu iki farmakodinamik belirteç in vivo HEMLIBRA'nın gerçek hemostatik etkisini yansıtmamakla birlikte (aPTT aşırı kısalır ve bildirilen faktör VIII aktivitesi olduğundan fazla hesaplanabilir), HEMLIBRA'nın pro-koagülan etkisine ilişkin göreceli bir gösterge sağlar.
Özetle, HEMLIBRA ile tedavi edilen hastalarda intrinsik yolak pıhtılaşma bazlı laboratuar testi bulguları aktivitesinin izlenmesi, faktör replasmanı veya anti-koagülasyon için doz uygulama tayini veya faktör VIII inhibitörleri titrelerinin ölçülmesinde kullanılmamalıdır. İntrinsik pıhtılaşma bazlı laboratuar testleri kullanılırsa, bulgularının yanlış yorumlanması kanama epizotları yaşayan hastaların yetersiz tedavisine yol açabileceğinden, bu da potansiyel olarak şiddetli veya hayati risk taşıyan kanamalarla sonuçlanabileceğinden dikkat edilmelidir.
HEMLIBRA'dan etkilenen ve etkilenmeyen laboratuvar testleri aşağıdaki Tablo 1'de gösterilmektedir. Uzun yarılanma ömründen dolayı, koagülasyon analizleri üzerindeki bu etkiler son dozu takiben 6 aya kadar sürebilir (bkz. Bölüm 5.2).
Tablo 1. HEMLIBRA'dan Etkilenen ve Etkilenmeyen Koagülasyon Test Sonuçları
HEMLIBRA'dan Etkilenen Sonuçlar | HEMLIBRA'dan Etkilenmeyen Sonuçlar |
Aktive parsiyel tromboplastin süresi (aPTT)
4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri
HEMLIBRA ile yeterli ya da iyi kontrollü ilaç-ilaç etkileşim çalışmaları yürütülmemiştir.
Klinik deneyim, HEMLIBRA ve aPCC arasında bir ilaç etkileşimi olduğuna işaret etmektedir (bkz. Bölüm 4.4 ve 4.8).
Klinik öncesi deneylere dayalı olarak, HEMLIBRA ile rFVIIa veya FVIII için bir hiperkoagülabilite olasılığı mevcuttur. HEMLIBRA koagülasyon potansiyelini arttırmaktadır, bu nedenle hemostaza erişmek için gerekli rFVIIa veya FVIII dozu, HEMLIBRA profilaksisi kullanılmadığı zamana göredahadüşükolabilir.
Trombotik komplikasyon durumlarında hekim klinik endikasyonuna göre rFVIIa veya FVIII kullanımını kesmeyi ve HEMLIBRA profilaksisine ara vermeyi değerlendirmelidir. Tedavinin daha ileri yönetimi için bireysel klinik duruma göre çözüm üretilmelidir.
Doz ayarlanması yapılırken, diğer ilaçların yarı ömrü düşünülmelidir; özellikle HEMLIBRA kullanımının kesilmesi durumunda hemen etki görülmeyebilir.
FVIII kromojenik testlerinin kullanılması koagülasyon faktörlerinin uygulanmasını yönlendirebilir ve trombofilik özelliklerin test edilmesi de düşünülebilir.
HEMLIBRA profilaksisi alan hastalarda anti fibrinolitiklerin aPCC veya rFVIIa ile birlikte kullanımına dair tecrübe sınırlıdır. Ancak HEMLIBRA alan hastalarda, sistemik anti fibrinolitiklerin aPCC veya rFVIIa ile birlikte kullanıldığında trombotik olayların gelişme ihtimali göz önünde bulundurulmalıdır.
Özel popülasyonlara ilişkin ek bilgiler Herhangi bir etkileşim çalışması yapılmamıştır. Pediyatrik popülasyon:
Listelenmiş etkileşimler hem yetişkinler hem de çocuklar için geçerlidir.
4.6. Gebelik ve laktasyon
Gebelik kategorisi: C
Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon)
HEMLIBRA kullanmakta olan ve çocuk doğurma potansiyeline sahip kadınlar, HEMLIBRA tedavisi süresince ve tedavinin kesilmesini takiben en az 6 ay boyunca etkili doğum kontrol yöntemleri kullanmalıdırlar (bkz. Bölüm 5.2).
Gebelik dönemi
Hayvanlar üzerinde yapılan çalışmalar, gebelik /ve-veya/ embriyonal/fetal gelişim /ve-veya/ doğum /ve-veya/ doğum sonrası gelişim üzerindeki etkiler bakımından yetersizdir (bkz. Bölüm 5.3). İnsanlara yönelik potansiyel risk bilinmemektedir.
HEMLIBRA gerekli olmadıkça gebelik döneminde kullanılmamalıdır.
Gebe kadınlarda HEMLIBRA kullanımı ile ilgili herhangi bir klinik çalışma bulunmamaktadır. HEMLIBRA ile hayvanlarda üreme çalışmaları yapılmamıştır. HEMLIBRA'nın gebe bir kadına uygulandığında fetüse zarar verip vermeyeceği veya üreme kapasitesini etkileyip etkilemeyeceği bilinmemektedir. HEMLIBRA, hamilelik sırasında ve doğumdan sonra tromboz riskinin arttığı ve çeşitli gebelik komplikasyonlarının artan Yaygın Damar içi Pıhtılaşma (YDP) riski le ilişkili olduğu göz önüne alınarak, yalnızca anneye
yönelik potansiyel yararın fetusa yönelik potansiyel riskten daha fazla olması durumunda kullanılmalıdır.
Laktasyon dönemi
Emicizumabın anne sütüne geçip geçmediği bilinmemektedir. Emicizumabın süt üretimi üzerindeki etkisi ya da anne sütündeki varlığı ile ilgili çalışma yürütülmemiştir. İnsan IgG'nin insan sütünde bulunduğu bilinmektedir. Bebek için emzirmenin faydası ve kadın için tedavinin faydası göz önüne alınarak emzirmeyi bırakma veya HEMLIBRA'yı bırakma/uzak durma kararı verilmelidir.
Üreme yeteneği/Fertilite
Hayvan çalışmaları, üreme toksisitesi açısından doğrudan veya dolaylı zararlı etkilere işaret etmemektedir (bkz. Bölüm 5.3). İnsanlarda fertilite verileri mevcut değildir. Dolayısıyla emicizumabın erkek ve dişi fertilitesi üzerindeki etkisi bilinmemektedir.
4.7. Araç ve makine kullanımı üzerindeki etkiler
HEMLIBRA'nın araç ve makine kullanımı üzerinde etkisi yoktur.
4.8. İstenmeyen etkiler
Güvenlilik profilinin özeti
HEMLİBRA'nın genel güvenlilik profili, klinik araştırmalardan ve pazarlama sonrası gözetimden elde edilen verilere dayanmaktadır. HEMLIBRA ile gerçekleştirilen klinik çalışmalardan bildirilen en ciddi advers ilaç reaksiyonları (AİR'ler), kavernöz sinüs trombozu (CST) ve deri nekrozu ile eşzamanlı olarak gerçekleşen yüzeyel ven trombozu dahil olmak üzere trombotik olaylar ve trombotik mikroanjiyopatidir (TMA) (bkz. aşağıdaki kısım ve Bölüm 4.4).
En az bir doz HEMLIBRA ile tedavi edilmiş hastaların ≥%10'unda bildirilen en yaygın AİR'ler: enjeksiyon yeri reaksiyonları (%20), artralji (%15) ve baş ağrısıdır (%14).
HEMLIBRA profilaksisi uygulanan klinik çalışmalarda toplamda üç hasta (%0,8) AİR'ler nedeniyle tedaviden çekilmiştir; bunlar TMA, yüzeyel tromboflebit ile eş zamanlı deri nekrozu ve baş ağrısıdır.
Advers ilaç reaksiyonlarının tablo haline getirilmiş listesi
Aşağıdaki advers ilaç reaksiyonları (AİR'ler), toplam 373 erkek hemofili A hastasının rutin profilaksi olarak en az bir HEMLIBRA dozu aldığı pazarlama sonrası gözetimden elde edilen verilere ve dört faz III klinik çalışmanın (yetişkin ve ergen çalışmaları [BH29884 – HAVEN 1, BH30071 – HAVEN 3 ve BO39182 – HAVEN 4] ve pediyatrik çalışma BH29992 – HAVEN 2) birleştirilmiş verilerini temel alır. Klinik araştırma katılımcılarından 266 hasta (%71) yetişkindi, 47 hasta (%13) ergen (>12 ila <18 yaş), 55 hasta (%15) çocuk (>2 ila < 12 yaş) ve 5 hasta (%1) bebektir (1 ay ila <2 yaş). Çalışmalar arasında medyan maruziyet süresi 33 haftadır (aralık: 0,1 ila 94,3 hafta).
HEMLIBRA almış hastalar üzerinde gerçekleştirilen Faz III klinik çalışmalardan ve pazarlama sonrası gözetimdene ldee dilenAİR'ler,MedDRA sistem organ sınıfına göre
listelenmektedir (Tablo 2). Her bir AİR için ilgili sıklık kategorileri aşağıdaki sınıflandırmayı temel almaktadır: çok yaygın (≤ 1/10), yaygın (≥1/100 ila <1/10), yaygın olmayan (≥1/1000 ila <1/100), seyrek (≥1/10.000 ila <1/1000), çok seyrek (<1/10.000) ve bilinmiyor (mevcut verilere dayalı olarak tahmin edilemez).
Tablo 2 HEMLIBRA ile Yapılan Birleştirilmiş HAVEN Klinik Çalışmalarında ve Pazarlama Sonrası Deneyimde Gözlenen Advers İlaç Reaksiyonlarının Özeti
Sistem Organ Sınıfı | Advers reaksiyonlar (tercih edilen terim, MedDRA) | Sıklık |
Kan ve lenf sistemi hastalıkları | Trombotik mikroanjiyopati | Yaygın olmayan |
Sinir sistemi hastalıkları | Baş ağrısı | Çok yaygın |
Vasküler hastalıklar | Yüzeyel tromboflebit | Yaygın olmayan |
Kavernöz sinüs trombozu | Yaygın olmayan | |
Gastrointestinal hastalıklar | İshal | Yaygın |
Deri ve deri altı doku hastalıkları | Deri nekrozu | Yaygın olmayan |
Anjiyoödem | Yaygın olmayan | |
Ürtiker | Yaygın | |
Döküntü | Yaygın | |
İskelet-kas ve bağ doku hastalıkları | Artralji | Çok yaygın |
Miyalji | Yaygın | |
Genel rahatsızlıklar ve uygulama bölgesine ilişkin hastalıklar | Enjeksiyon yeri reaksiyonu | Çok yaygın |
Ateş | Yaygın | |
Terapötik yanıt azalması | Yaygın olmayan |
Seçilmiş advers ilaç reaksiyonlarının tanımı Trombotik mikroanjiyopati
Havuzlanmış faz III klinik çalışmalarında, trombotik mikroanjiyopati (TMA) olayları, hastaların %1'inden az (3/373) ve HEMLIBRA ile tedavi edilirken en az bir doz aPCC almış hastaların %9,7'sinde (3/31) bildirilmiştir. Üç TMA'nın tümü, 24 saat veya daha uzun süre
>100 U/Kg/24 saat ortalama kümülatif miktarda aPCC uygulandığında meydana gelmiştir (bkz. Bölüm 4.4). Hastalar trombositopeni, mikroanjiyopatik hemolitik anemi ve ADAMTS13 aktivitesinde şiddetli eksiklik olmaksızın akut böbrek hasarı ile başvurmuştur. Bir hasta, TMA olaylarının tekrarlamadan düzelmesini takiben HEMLIBRA kullanımına devam etmiştir.
Trombotik olaylar
Havuzlanmış faz III klinik çalışmalarında ciddi trombotik olaylar hastaların %1'inden az (2/373) ve HEMLIBRA ile tedavi edilirken en az bir aPCC dozu almış hastaların %6,5'inde (2/31) bildirilmiştir. Her iki ciddi trombotik olay da 24 saat veya daha uzun süre >100 U/Kg/24 saat ortalama kümülatif miktarda aPCC uygulandığında meydana gelmiştir. Bir hasta, trombotik olayların tekrarlamadan düzelmesini takiben HEMLIBRA kullanımına devam etmiştir (bkz. bölüm 4.4).
Pivotal klinik çalışmalarda HEMLIBRA ve aPCC tedavisi arasındaki etkileşimin karakterizasyonu
HEMLIBRA profilaksisi alan hastalarda 82 aPCC tedavisi* uygulanmış olup, bunların sekizinde (%10) 24 saat veya daha uzun süre >100 U/Kg/24 saat ortalama kümülatif miktarda aPCC uygulanmıştır, bu sekiz örneğin ikisi trombotik olaylar ve sekizden üçü TMA ile ilişkilidir (Tablo 3). Geri kalan aPCC tedavileri ile ilişkili bir TMA veya trombotik olay söz konusu olmamıştır. Tüm aPCC tedavilerinden %68'i <100 U/kg'lık tek bir infüzyondan ibarettir.
Tablo 3 Havuzlanmış faz III klinik çalışmalarında aPCC tedavisinin* karakterizasyonu
aPCC tedavisinin süresi | 24 saatte ortalama kümülatif aPCC miktarı (U/kg/24 saat) | ||
<50 | 50-100 | >100 | |
<24 saat | 9 | 47 | 13 |
24-48 saat | 0 | 3 | 1 |
>48 saat | 1 | 1 | 7 |
Enjeksiyon yeri reaksiyonları
Enjeksiyon yeri reaksiyonları (EYR'ler) klinik çalışmalarda çok yaygın (%20) olarak bildirilmiştir. HEMLIBRA klinik çalışmalarında gözlenen tüm EYR'ler, ciddi olmayan olaylar olarak bildirilmiştir ve hafif ila orta şiddettedirler. EYR'lerin %95'i tedavi uygulanmadan ortadan kalkmıştır. En yaygın şekilde bildirilen EYR semptomları, enjeksiyon yerinde kızarıklık (%11), enjeksiyon yerinde ağrı (%4) ve enjeksiyon yerinde kaşıntı (pruritus) (%3) olmuştur.
İmmünojenisite
Hemlibra ile havuzlanmış Faz III klinik çalışmalarda, emicizumab konsantrasyonunun azalmasıyla ilişkili nötralize edici anti-emicizumab antikorlarının gelişimi yaygın değildir (bkz. Bölüm 5.1). Emicizumab konsantrasyonunun azalmasıyla nötralize edici anti- emicizumab antikorları geliştiren bir hasta, beş haftalık tedaviden sonra etkililik kaybı yaşamıştır (ara kanama olarak kendini gösterdi) ve daha sonra Hemlibra tedavisini bırakmıştır (bkz. Bölüm 4.4 ve 5.1).
Pediyatrik popülasyon:
Araştırılan pediyatrik popülasyon, 5'i (%5) bebek (1 ay ila 2 yaş altı), 55'i (%51) çocuk (2 yaş sonrası ila 12 yaş altı) ve 47'si (%44) ergen (12 yaş ila 18 yaş altı) olan toplamda 107 hastadan oluşur.
HEMLIBRA'nın güvenlilik profili bebekler, çocuklar, ergenler ve yetişkinler arasında genel olarak tutarlı bulunmuştur.
Şüpheli advers reaksiyonların raporlanması
Ruhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar/risk dengesinin sürekli olarak izlenmesine olanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu Türkiye Farmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e- posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks: 0312 218 35 99).
4.9. Doz aşımı ve tedavisi
HEMLIBRA doz aşımı ile ilgili deneyim kısıtlıdır. Semptomlar
Kazara doz aşımı hiperkoagülabiliteye yol açabilir. Tedavi
Kazara doz aşımının olduğu hastalar acilen hekimleriyle iletişime geçmeli ve yakından izlenmelidir.
5. FARMAKOLOJİK ÖZELLİKLER
5.1. Farmakodinamik özellikler
Farmakoterapötik grup: Antihemorajikler, diğer sistemik hemostatikler ATC kodu: B02BX06
Etki mekanizması
Emicizumab, bispesifik antikor yapısı olan hümanize monoklonal modifiye edilmiş immünoglobülin G4 (IgG4) antikorudur.
Emicizumab, etkili hemostaz için gerekli olan eksik aktive faktör VIII'in fonksiyonunun yerine konması için aktive faktör IX ve faktör X'u birleştirir.
Emicizumabın faktör VIII ile yapısal ilişkisi veya sekans homolojisi yoktur ve bu haliyle faktör VIII'e karşı direkt antikor gelişimini indüklemez ya da arttırmaz.
Farmakodinamik
HEMLIBRA ile profilaksi tedavisi, aPTT'yi kısaltır ve testlerde bildirilen faktör VIII aktivitesini (insan koagülasyon faktörleri ile bir kromojenik tayin kullanılarak) arttırır. Bu iki farmakodinamik belirteç emicizumabın gerçek in vivo hemostatik etkisini yansıtmamaktadır (aPTT aşırı derecede kısalmıştır ve bildirilen faktör VIII aktivitesi olduğundan daha yüksek çıkabilir); ancak emicizumabın prokoagülan etkisi ile ilgili bağıl bir gösterge sağlamaktadır.
Klinik etkililik ve güvenlilik
HEMLIBRA'nın FVIII inhibitörlü veya inhibitörsüz hemofili A hastalarının rutin profilaksisinde etkililiği dört klinik çalışmada (üç erişkin ve ergen çalışması [HAVEN 3, HAVEN 1 ve HAVEN 4] ve bir pediyatrik çalışmada [HAVEN 2]) değerlendirilmiştir.
Adölesan ve erişkinlerde yapılan klinik çalışmalar
FVIII inhibitörsüz hemofili A hastaları (≥ 12 yaş ve > 40 kg) (Çalışma BH30071 – HAVEN 3)
HAVEN 3 çalışması, daha önce FVIII ile kanadıkça (“ihtiyaç halindeâ€) veya profilaksi tedavi almış olan, FVIII inhibitörsüz hemofili A hastası 152 adölesan ve erişkin erkekte (> 12 yaş ve
> 40 kg) yapılmış olan randomize, çok merkezli, açık etiketli bir faz III klinik çalışmadır. Hastalar ilk dört hafta boyunca haftada bir kez 3 mg/kg ve bunu takiben haftada bir kez 1,5 mg/kg (Kol A ve D) veya iki haftada bir kez 3 mg/kg (Kol B) subkutan HEMLIBRA almış veya profilaksi almamıştır (Kol C). Kol C'deki hastalar profilaksi uygulanmaksızın en az 24 haftayı tamamladıktan sonra HEMLIBRA'ya (iki haftada bir 3 mg/kg) geçebilmiştir. Kol A ve B'de, iki veya daha fazla anlamlı kanama (kararlı durumda iken meydana gelen spontan ve klinik olarak anlamlı kanamalar) geçiren hastalar için haftada bir kez 3 mg/kg doz titrasyonuna izin verilmiştir. Kol D hastalarında, ikinci anlamlı kanamadan sonra doz titrasyonu yapılabilmiştir. Ara analiz yapıldığı sırada, beş hastanın idame dozunda yukarı titrasyonun yapılmış olduğu saptanmıştır.
Daha önce kanadıkça (“ihtiyaç halindeâ€) FVIII ile tedavi edilmiş olan 89 hasta, haftada bir kez HEMLIBRA almak üzere (Kol A; N = 36), iki haftada bir kez HEMLIBRA almak üzere (Kol B; N = 35) veya hiç profilaksi almamak üzere (Kol C; N = 18) 2:2:1 oranda randomize edilmiş, önceki 24 haftalık kanama oranına göre (< 9 veya ≥ 9) katmanlama yapılmıştır. Daha önce FVIII profilaksisi ile tedavi edilmiş olan 63 hasta, HEMLIBRA (haftada bir kez 1,5 mg/kg) almak üzere Kol D'ye kaydedilmiştir.
Çalışmanın birincil amacı, daha önce kanadıkça FVIII ile tedavi edilmiş olan hastalarda haftada bir kez (Kol A) veya iki haftada bir kez (Kol B) uygulanan HEMLIBRA profilaksisinin, hiç profilaksi uygulanmamasına (Kol C) kıyasla etkililiğini, koagülasyon faktörleri ile tedavi gerektiren kanamaların sayısına dayanarak değerlendirmek olmuştur (bkz. Tablo 4). Çalışmanın diğer amaçları, Kol A veya B ile Kol C'nin randomize karşılaştırmasının HEMLIBRA profilaksisinin tüm kanamaların, spontan kanamaların, eklem kanamalarının ve hedef eklem kanamalarının sayısını azaltmadaki etkililiği açısından değerlendirilmesini (bkz. Tablo 4) ve hasta bildirimli sağlık ile ilgili yaşam kalitesinin (HRQoL) ölçülmesini içermiştir. Ayrıca, bir tercih anketi kullanılarak hastaların tedavi tercihi de değerlendirilmiştir.
HEMLIBRA profilaksisinin etkililiği, çalışmaya kaydedilmeden önce girişimsel olmayan bir çalışmaya katılmış olan hastalarda önceki profilaktik FVIII tedavisi (Kol D) ile de karşılaştırılmıştır (bkz. Tablo 5). Bu karşılaştırmaya yalnızca girişimsel olmayan çalışmadan gelen hastalar dahil edilmiştir; çünkü kanama ve tedavi verileri HAVEN 3'teki ile aynı veri derinliği düzeyinde toplanmıştır.
Girişimsel olmayan çalışma gözlemsel bir çalışma olup, temel amacı hemofili A hastalarında girişimsel çalışma düzeni dışında kanama epizotları ve hemofili ilaçlarının kullanımı hakkında detaylı klinik verilerin yakalanması olmuştur.
Faktör VIII inhibitörlü (>12 yaş) hemofili A hastaları (Çalışma BH29884 – HAVEN 1)
HAVEN 1 çalışması, daha önce bypass edici ajanlar (aPCC ve rFVIIa) ile kanadıkça tedavi ya da profilaksi almakta olan, faktör VIII inhibitörlü hemofili A hastası 109 adölesan ve erişkin erkek (yaşları ≥ 12) üzerinde gerçekleştirilen bir randomize, çok merkezli, açık etiketli klinik çalışmadır. Çalışmada, hastalara haftalık HEMLIBRA profilaksisi (Grup A, C ve D) - 4 hafta boyunca haftada bir kez 3 mg/kg, ardından haftada bir kez 1,5 mg/kg - ya da kanadıkça tedavi uygulanmıştır (Grup B). Grup B'deki randomize hastalar, profilaksi olmadan en az 24 haftayı tamamladıktan sonra HEMLIBRA profilaksisine geçebilmiştir. 2 ya da daha fazla anlamlı kanama geçirmiş hastalarda (kararlı halde spontan ve klinik olarak kanıtlanmış anlamlı kanamanın olması durumunda) HEMLIBRA profilaksisinde 24 hafta geçirildikten sonra haftada bir kez 3 mg/kg'a çıkarılacak şekilde doz titrasyonuna izin verilmiştir. Primer analiz süresi boyunca, iki hastanın idame dozları haftada bir kez 3 mg/kg'a yükseltilmiştir.
Daha önce bypass edici ajanlarla kanadıkça tedavi almakta olan 53 hasta, çalışmadan önceki
24 haftalık kanama sayılarına (<9 veya ≥ 9) göre yapılan sınıflandırmayla birlikte HEMLIBRA profilaksisi (Grup A) ya da kanadıkça tedavi alacak (Grup B) şekilde 2:1 oranında randomize edilmişlerdir.
Daha önce bypass edici ajan profilaksisi ile tedavi edilen 49 hasta, HEMLIBRA profilaksisi almak için Grup C'ye kaydedilmiştir. Kayıttan önce NIS'e (Girişimsel olmayan çalışma) katılan, ancak A ve B gruplarının kapatılmasından önce HAVEN 1'e kaydolamamış, kanadıkça bypass edici ajan tedavisi almakta olan 7 hasta, HEMLIBRA profilaksisi almak için Grup D'ye kaydedilmişlerdir.
Çalışmanın temel amacı, öncesinde kanadıkça bypass edici ajan tedavisi almakta olan hastalarda, kanadıkça tedavi ile karşılaştırmalı olarak, haftalık HEMLIBRA profilaksisinin, zaman içinde (en az 24 hafta veya kesilme tarihi) (Bkz. Tablo 6), koagülasyon faktörleri ile tedaviyi gerektiren kanama sayısı üzerindeki etkisini değerlendirmektir (Grup A'ya karşılık Grup B). A ve B gruplarının randomize olarak karşılaştırılmasının diğer ikincil hedefleri, tüm kanamaların, spontan kanamaların, eklem kanamalarının ve hedef eklem kanamalarının (Bkz. Tablo 6) sayısının azaltılmasında HEMLIBRA profilaksisinin etkililiğini ve bunun yanı sıra hastaların sağlıkla ilişkili yaşam kaliteleri ile sağlık durumlarını değerlendirmektir (Bkz. Tablo 9 ve 10). Çalışmadaki tüm hastalar için ortalama maruziyet süresi (+SD) 21,38 haftadır (12,01). Her bir tedavi kolu için ortalama maruziyet süreleri (+SD) Kol A için 28,86 hafta (8,37), Kol B için 8,79 (3,62), Kol C için 21,56 (11,85) ve Kol D için 7,08 haftadır (3,89). Kol A'daki bir hasta HEMLIBRA başlatılmadan çalışmadan çekilmiştir.
Çalışma aynı zamanda, kayıt öncesi NIS'e katılan hastalarda (sırasıyla Grup A ve C) (Bkz. Tablo 7) daha önce uygulanmış olan bypass edici ajanlar ile kanadıkça tedavi ya da profilaksi karşısında (ayrı karşılaştırmalar) haftalık HEMLIBRA profilaksisinin etkililiğini de değerlendirmiştir.
Faktör VIII inhibitörlü ya da inhibitörsüz hemofili A hastaları (≥ 12 yaş) (Çalışma BO39182 – HAVEN 4)
HEMLIBRA, daha önce bypass edici ajanları veya FVIII ile kanadıkça (“ihtiyaç halindeâ€) ya da profilaksi almış olan, FVIII inhibitörlü veya inhibitörsüz 41 erişkin ve adölesan hemofili A hastası erkekte (≥ 12 yaş ve > 40 kg) yapılan tek kollu, çok merkezli bir faz III klinik çalışmada araştırılmıştır. Hastalar, dört hafta boyunca haftada bir kez 3 mg/kg ve bunu takiben dört haftada bir 6 mg/kg HEMLIBRA profilaksisi almışlardır.
Çalışmanın birincil amacı, dört haftada bir verilen HEMLIBRA profilaksisinin yeterli kanama kontrolünün sürdürülmesindeki etkililiğini, tedavi edilen kanamalara dayanarak değerlendirmek olmuştur. Diğer amaçlar, HEMLIBRA profilaksisinin tüm kanamalar, tedavi edilen spontan kanamalar, tedavi edilen eklem kanamaları ve tedavi edilen hedef eklem kanamaları üzerindeki klinik etkililiğini değerlendirmek olmuştur (bkz. Tablo 8). Ayrıca, bir tercih anketi kullanılarak hastaların tedavi tercihi de değerlendirilmiştir.
Erişkinlerde ve Adölesanlarda Etkililik Bulguları HAVEN 3
Tüm kanamalar, tedavi edilen spontan kanamalar, tedavi edilen eklem kanamaları ve tedavi edilen hedef eklem kanamaları için kanama oranı açısından HEMLIBRA profilaksisinin hiç profilaksi uygulanmamasına kıyasla etkililik bulguları Tablo 4'te gösterilmektedir.
Tablo 4 HAVEN 3 çalışması: ≥12 yaş inhibitörsüz hastalarda profilaksi uygulanmamasına kıyasla HEMLIBRA profilaksisi ile yıllık kanama sayıları
Sonlanım noktası | C kolu: Profilaksi yok (N=18) | A kolu: Haftalık 1,5 mg/kg HEMLIBRA (N=36) | B kolu: 2 haftada 1 kez 3 mg/kg HEMLIBRA (N=35) |
Tedavi edilen kanamalar | |||
Yıllık kanama sayısı (ABR) (%95 GA) | 38,2(22,9; 63,8) | 1,5 (0,9; 2,5) | 1,3 (0,8; 2,3) |
% azalma (RR), p-değeri | Uygulanabilir değil | %96 (0,04), <0,0001 | %97 (0,03), <0,0001 |
0 kanama izlenen hasta yüzdesi (%95 GA) | 0 (0; 18,5) | 55,6 (38,1; 72,1) | 60 (42,1; 76,1) |
Medyan yıllık kanama sayısı (ABR) (IQR) | 40,4 (25,3; 56,7) | 0 (0; 2,5) | 0 (0; 1,9) |
Tüm kanamalar | |||
Yıllık kanama sayısı (ABR) (%95 GA) | 47,6 (28,5; 79,6) | 2,5 (1,6; 3,9) | 2,5 (1,6; 4,3) |
% azalma (RR), p-değeri | Uygulanabilir değil | %95 (0,05), <0,0001 | %94 (0,06), <0,0001 |
0 kanama izlenen hasta yüzdesi (%95 GA) | 0 (0; 18,5) | 50 (32,9; 67,1) | 40 (23,9; 57,9) |
Tedavi edilen spontan kanamalar | |||
Yıllık kanama sayısı (ABR) (%95 GA) | 15,6 (7,6; 31,9) | 1 (0,5; 1,9) | 0,3 (0,1; 0,8) |
Uygulanabilirdeğil | |||
0 kanama izlenen hasta yüzdesi (%95 GA) | 22,2 (6,4; 47,6) | 66,7 (49; 81,4) | 88,6 (73,3; 96,8) |
Tedavi edilen eklem kanamaları | |||
Yıllık kanama oranı (ABR) (%95 GA) | 26,5 (14,67; 47,79) | 1,1 (0,59; 1,89) | 0,9 (0,44; 1,67) |
% azalma (RR), p-değeri | Uygulanabilir değil | %96 (0,04), <0,0001 | %97 (0,03), <0,0001 |
0 kanama izlenen hasta yüzdesi (%95 GA) | 0 (0; 18,5) | 58,3 (40,8; 74,5) | 74,3 (56,7; 87,5) |
Tedavi edilen hedef eklem kanamaları | |||
Yıllık kanama sayısı (ABR) (%95 GA) | 13 (5,2; 32,3) | 0,6 (0,3; 1,4) | 0,7 (0,3; 1,6) |
% azalma (RR), p-değeri | Uygulanabilir değil | %95 (0,05), <0,0001 | %95 (0,05), <0,0001 |
0 kanama izlenen hasta yüzdesi (%95 GA) | 27,8 (9,7; 53,5) | 69,4 (51,9; 83,7) | 77,1 (59,9; 89,6) |
HAVEN 3 klinik çalışmasında gerçekleştirilen hasta içi analizlerinde, hastalar çalışmaya dahil edilmeden önce girişimsel olmayan çalışmadan toplanan FVIII profilaksisi verileri ile kıyaslandığında, HEMLIBRA profilaksisini tedavi edilen kanama sayısında istatistiksel olarak anlamlı (p<0,0001) bir azalma sağlamıştır (%68) (bkz. Tablo 5).
Tablo 5 HAVEN 3 çalışması: FVIII profilaksisine kıyasla HEMLIBRA profilaksisi ile yıllık kanama sayılarına ait hasta içi değerlendirmesi
Sonlanım noktası | D NIS kolu: Önceki FVIII profilaksisi (N=48) | D kolu: Haftalık 1,5 mg/kg HEMLIBRA (N=48) |
Medyan Etkililik Süresi (hafta) | 30,1 | 33,7 |
Tedavi edilen kanamalar | ||
Yıllık kanama sayısı (ABR) (%95 GA) | 4,8 (3,2; 7,1) | 1,5 (1; 2,3) |
% azalma (%95 GA), p- değeri | % 68 (% 48,6; %80,5), <0,0001 | |
0 kanama izlenen hasta yüzdesi (%95 GA) | 39,6 (25,8; 54,7) | 54,2 (39,2; 68,6) |
Medyan yıllık kanama sayısı (ABR) (IQR) | 1,8 (0; 7,6) | 0 (0; 2,1) |
HAVEN 1
Profilaksi almayan hastalar ile kıyaslandığında HEMLIBRA profilaksisinin tedavi edilen kanamalar, tüm kanamalar, tedavi edilen spontan kanamalar, tedavi edilen eklem kanamaları ve tedavi edilen hedef eklem kanamaları için oranlar açısından etkililik sonuçları Tablo 6'da gösterilmektedir.
Tablo 6 HAVEN 1 çalışması: ≥12 yaş FVIII inhibitörlü hastalarda profilaksi uygulanmamasına kıyasla HEMLIBRA profilaksisi ile yıllık kanama sayıları
Sonlanım Noktası | Grup B: profilaksi yok | Grup A: Haftalık 1,5 mg/kg HEMLIBRA |
| N=18 | N=35 |
Tedavi Edilen Kanamalar |
| |
ABR (%95 GA) | 23,3 (12,33; 43,89) | 2,9 (1,69; 5,02) |
% azalma (%95 GA), p-değeri | %87 (0,13), < 0,0001 | |
0 kanama izlenen hasta %'si (%95 GA) | 5,6 (0,1; 27,3) | 62,9 (44,9; 78,5) |
Medyan ABR (IQR) | 18,8 (12,97; 35,08) | 0 (0; 3,73) |
Tüm Kanamalar | ||
ABR (%95 GA) | 28,3 (16,79; 47,76) | 5,5 (3,58; 8,60) |
% azalma (%95 GA), p-değeri | %80 (0,20), < 0,0001 | |
0 kanamanın olduğu izlenen hasta %'si (%95 GA) | 5,6 (0,1; 27,3) | 37,1 (21,5; 55,1) |
Tedavi Edilen Spontan Kanamalar |
| |
ABR (%95 GA) | 16,8 (9,94; 28,30) | 1,3 (0,73; 2,19) |
% azalma (%95 GA), p-değeri | %92 (%0,08), < 0,0001 | |
0 kanamanın olduğu izlenen hasta %'si (%95 GA) | 11,1 (1,4; 34,7) | 68,6 (50,7; 83,1) |
Tedavi Edilen Eklem Kanamaları |
| |
ABR (%95 GA) | 6,7 (21,99; 22,42) | 0,8 (0,26; 2,20) |
% azalma (%95 GA), p-değeri | %89 (%0,11), 0,0050 | |
0 kanamanın olduğu izlenen hasta %'si (%95 GA) | 50 (26; 74) | 85,7 (69,7; 95,2) |
Tedavi Edilen HedefEklem | ||
Kanamaları |
| |
ABR (%95 GA) | 3 (0,96; 9,13) | 0,1 (0,03; 0,58) |
% azalma (%95 GA), p-değeri | %95 (%0,05), 0,0002 | |
0 kanama izlenen hasta %'si (%95 GA) | 50 (26; 74) | 94,3 (80,8; 99,3) |
Güven aralığı (GA), negatif binomiyal regresyon (NBR) modelinden ve p-değeri Katmanlaştırılmış Wald testinden alınmış olup, belirtilmiş kollar arasında ABR'yi karşılaştırmaktadır. Grup B: yalnızca profilaksi uygulanmayan dönemi kapsar. Kanama tanımları ISTH kriterlerine dayalıdır. Tedavi edilmiş kanamalar: bypass edici ajanlar ile tedavi edilmiş kanamalar. Tüm kanamalar: bypass edici ajanlar ile tedavi edilmiş ve tedavi edilmemiş kanamalar Dozu yukarı titre edilmiş hastalar için yalnızca yukarı titrasyondan önceki verileri içerir. Emicizumaba maruz kalmış hastalar, tedaviye 4 hafta boyunca haftada 1 kez 3 mg/kg/gün yükleme dozuyla başlamışlardır. ABR = Yıllık Kanama Sayısı; GA = güven aralığı; IQR = çeyrekler açıklığı, 25. ile 75. yüzdelik dilim arası | ||
HAVEN 1 hasta içi analizinde, öncesinde girişimsel olmayan çalışmada bypass edici ajan profilaksisi ile elde edilen sonuçlara kıyasla, HEMLIBRA profilaksisi ile tedavi edilen kanama sayılarında istatistik (p=0,003) ve klinik olarak anlamlı (%79) azalma ortaya çıkmıştır (bkz. Tablo 7).
Tablo 7 HAVEN 1: HEMLIBRA Profilaksisine karşı öncesinde bypass edici ajanla profilaksi almış hastaların (NIS Hastaları) Yıllık Kanama Sayısı için kendi içerisinde karşılaştırması
Sonlanım Noktası | Cgrubu: Daha önce bypass edici ajan profilaksisi | Grup C: haftada bir 1,5 mg/kg HEMLIBRA |
| N = 24 | N = 24 |
Tedavi Edilen Kanamalar | ||
ABR (%95 GA) | 15,7 (11,08; 22,29) | 3,3 (1,33; 8,08) |
0 kanama izlenen hasta yüzdesi (%95 GA) | 12,5 (2,7; 32,4) | 70,8 (48,9; 87,4) |
Ortalama ABR (IQR) | 12 (5,73; 24,22) | 0 (0; 2,23) |
% azalma (RR), p-değeri | %79 (0,21); 0,0003 | |
| ||
HAVEN 4
4 haftada 1 uygulanan HEMLIBRA profilaksisinin tedavi edilen kanamalar, tüm kanamalar, tedavi edilen spontan kanamalar, tedavi edilen eklem kanamaları ve tedavi edilen hedef eklem kanamaları açısından etkililik sonuçları Tablo 8'de gösterilmektedir. ≥12 yaşındaki 41 hasta, medyan 25,6 hafta süresince değerlendirilmiştir (24,1-29,4 aralığı).
Tablo 8 HAVEN 4: ≥12 yaş Faktör VIII inhibitörlü ve inhibitörsüz hastalarda HEMLIBRA profilaksisi ile yıllık kanama sayıları
| 4 haftada 1 kez 6 mg/kg HEMLIBRA | ||
Sonlanım noktaları | 0 kanama yüzdesi (%95 GA) | ||
N | 41 | 41 | 41 |
Tedavi edilen kanamalar | 2,4 (1,4; 4,3) | 0 (0; 2,1) | 56,1 (39,7; 71,5) |
Tüm kanamalar | 4,5 (3,1; 6,6) | 2,1 (0; 5,9) | 29,3 (16,1; 45,5) |
Tedavi edilen spontan kanamalar | 0,6 (0,3; 1,5) | 0 (0; 0) | 82,9 (67,9; 92,8) |
Tedavi edilen eklem kanamaları | 1,7 (0,8; 3,7) | 0 (0; 1,9) | 70,7 (54,5; 83,9) |
Tedavi edilen hedef eklem kanamaları | 1 (0,3; 3,3) | 0 (0; 0) | 85,4 (70,8; 94,4) |
Erişkin ve Adölesanlarda Sağlık İle İlgili Sonuç Ölçütleri
Erişkin ve adölesanların dahil olduğu HAVEN klinik çalışmalarında, hasta bildirimli sonuçlar birkaç ölçüt ile değerlendirilmiştir. Hastalarda hemofili ile ilgili yaşam kalitesi, erişkinler (>18 yaş) için Hemofiliye Özgü Yaşam Kalitesi (Haem-A-QoL) anketi ve bunun ergen versiyonu (Haemo-QoL-SF, 8 ila <18 yaş için) ile değerlendirilmiştir. Haem-A-QoL ve Haemo-QoL-SF için, Fiziksel Sağlık Skoru (yani ağrılı şişlikler, eklem ağrısı varlığı hareket ile ağrı, uzağa yürümede güçlük ve hazırlanmak için gereken sürenin artması) ve Toplam Skor (tüm skorların özeti) protokolde tanımlanmış ilgi konusu sonlanım noktalarıdır. Sağlık durumundaki değişimi ölçmek için, EuroQoL Beş Boyutlu Beş Düzeyli Anketine (EQ-5D- 5L) ait Görsel Analog Skala (VAS) ve İndeks Yararlılık Skoru (IUS) incelenmiştir.
HAVEN 1 Sağlıkla İlişkili Sonuçlar
Bu çalışmada, ≥18 yaş hastalar için sağlıkla ilişkili sonuçlar, 25. haftada erişkinlere yönelik (Bkz. Tablo 9) Haem-A-QoLanketiiledeğerlendirilmiştir. Başlangıç Toplam Skorları
(ortalama = sırasıyla 41,14 ve 44,58) ve Fiziksel Sağlık ölçeği skorları (ortalama = sırasıyla 52,41 ve 57,19) HEMLIBRA profilaksisi ve kanadıkça tedavi grubu için benzerdir. Tablo 9'da HEMLIBRA profilaksisi kolu (Kol A) ve profilaksi almayan kol (Kol B) arasında 24 haftalık tedavi sonrası Haem-A-QoL Toplam Skoru ve Fiziksel Sağlık ölçeğine ilişkin karşılaştırmanın bir özeti sunulmaktadır. Haftalık HEMLIBRA profilaksisi önceden belirlenmiş sonlanım noktaları olan 25. Hafta değerlendirmesinde Haem-A-QoL Toplam Skoru ve Fiziksel Sağlık Ölçeği skorunda kanadıkça tedaviye kıyasla istatistiksel ve klinik anlamlı bir iyileşme göstermiştir.
Tablo 9 HAVEN 1: ≥18 yaş Faktör VIII inhibitörlü hastalarda profilaksi uygulanmasına kıyasla HEMLIBRA profilaksisi ileHaem-A-QoL skorlarındaki değişim
25. haftada Haem-A-QoL Skorları | Grup B: profilaksi yok (N=14) | Grup A: 1,5 mg/kg Haftalık HEMLIBRA (N=25) |
Toplam Skor (0 ila 100 aralığı) | ||
Düzeltilmiş ortalama | 54,17 | 32,61 |
Düzeltilmiş ortalamalarda farklılık (%95 GA) | 21,55 (7,89; 35,22) | |
p-değeri | 0,0029 | |
Fiziksel Sağlık skoru (0 ila 100 aralığı) | ||
Düzeltilmiş ortalama | 43,21 | 29,2 |
Düzeltilmiş ortalamalarda farklılık (%95 GA) | 14,01 (5,56; 22,45) | |
Grup B: yalnızca profilaksi uygulanmayan dönemi kapsar. Dozu yukarı titre edilmiş hastalar için yalnızca yukarı titrasyondan önceki verileri içerir. Emicizumaba maruz kalmış hastalar, tedaviye 4 hafta boyunca haftada 1 kez 3 mg/kg/gün yükleme dozuyla başlamışlardır. Haem-A_QoL ölçekleri 0 ila 100 arasında değişir, düşük skorlar daha iyi HRQoL'ye işaret etmektedir. Klinik olarak anlamlı farklılık: Toplam skor: 7 puan; Fiziksel Sağlık: 10 puan. Analizler hem başlangıç noktasında, hem de 25.Hafta değerlendirmesinde yanıt veren bireylerden elde edilen verilere dayanmaktadır. | ||
HAVEN 1 Sağlık Durumu Sonuçları
Tablo 10'da HEMLIBRA profilaksisi kolu (Kol A) ve profilaksi almayan kol (Kol B) arasında 24 haftalık tedavi sonrası EQ-5D-5L indeksi fayda ölçeği ve görsel analog ölçeğe ilişkin karşılaştırmanın bir özeti sunulmaktadır.
![]()
Tablo 10 HAVEN 1 : 25. haftada ≥12 yaşındaki hastalarda EQ-5D-5L skorları
24 haftadan sonra EQ-5D- 5L | Grup B: profilaksi yok (N=16) | Grup A: 1,5 mg/kg Haftalık HEMLIBRA (N=29) |
Görsel Analog Ölçek |
| |
Düzeltilmiş ortalama | 74,36 | 84,08 |
Düzeltilmiş ortalamalarda farklılık (%95 GA) | -9,72 (-17,62; -1,82) | |
İndeks Fayda Skoru |
| |
0,81 | ||
Düzeltilmiş ortalamalarda farklılık (%95 GA) | -0,16 (-0,25; -0,07) |
Grup B: yalnızca profilaksi uygulanmayan dönemi kapsar. Dozu yukarı titre edilmiş hastalar için yalnızca yukarı titrasyondan önceki verileri içerir. Emicizumaba maruz kalmış hastalar, tedaviye 4 hafta boyunca haftada 1 kez 3 mg/kg/gün yükleme dozuyla başlamışlardır. Daha yüksek skorlar daha iyi yaşam kalitesine işaret etmektedir. Klinik açıdan önemli farklılık: VAS: 7 puan; İndeks fayda skoru: 0,07 puan Analizler hem başlangıç noktasında, hem de 25.Hafta değerlendirmesinde yanıt veren bireylerden elde edilen verilere dayanmaktadır. | |
Pediyatrik hastalarda klinik çalışma
Faktör VIII inhibitörlü pediyatrik hemofili A hastaları (yaş <12 yaş veya <40 kg ağırlığında 12 ila 17 yaş) (Çalışma BH29992 – HAVEN 2)
Haftalık HEMLIBRA profilaksisi, faktör VIII inhibitörlü pediyatrik hemofili A hastalarında (12 yaş altı veya 12 ila 17 yaş arasında, <40 kg ağırlığında), tek kollu, çok merkezli, açık etiketli bir klinik çalışmada değerlendirilmiştir. Hastalar, ilk 4 hafta boyunca haftada bir kez 3 mg/kg ve daha sonra haftada bir kez 1,5 mg/kg olacak şekilde HEMLIBRA profilaksisi almıştır.
Çalışmada, kayıttan önce NIS'e katılan hastalarda (kendi içerisinde değerlendirilen hastalar) daha önceki kanadıkça ya da profilaktik bypass edici ajan tedavisi ile karşılaştırmalı olarak, haftalık HEMLIBRA profilaksisinin etkililiği farmakokinetik özellikleri ve güvenliliği değerlendirmiştir.
HAVEN 2 Çalışmasının Pediyatrik Etkililik Bulguları (Ara Analiz)
Ara analizler sırasında < 2 yaşında olan 4 hasta, 2 ila < 6 yaş arasında olan 17 hasta ve 6 ila < 12 yaş arasında olan 38 hasta dahil olmak üzere, 12 yaşından küçük olan ve en az 12 hafta boyunca haftada 1 kez HEMLIBRA profilaksisi alan 59 hastada etkililik değerlendirilmiştir. 59 hasta için, yıllık olarak hesaplanmış kanama sayısı ve sıfır kanama gözlenen hastaların yüzdesi tespit edilmiştir (bkz. Tablo 11). Bu hastalar için medyan gözlem süresi 29,6 haftadır (aralık: 18,4 ila 63,0 hafta).
Tablo 11 HAVEN 2: Etkililik genel özeti (Ara Analizler)
Sonlanım noktası | % Sıfır Kanama (%95 GA) | ||
Tedavi edilen kanamalar | 0,3 (0,1; 0,5) | 0 (0; 0) | 86,4 (75; 94) |
Tüm kanamalar | 3,8 (2,2; 6,5) | 0 (0; 3,4) | 55,9 (42,4; 68,8 |
Tedavi edilen spontan kanamalar | 0 (0; 0,2) | 0 (0; 0) | 98,3 (90,9; 100) |
Tedavi edilen eklem kanamaları | 0,2 (0,1; 0,4) | 0 (0; 0) | 89,8 (79,2; 96,2) |
Tedavi edilen hedef eklem kanamaları | 0,1 (0; 0,7) | 0 (0; 0) | 96,6 (88,3; 99,6) |
Kendi içerisinde analiz edilen hastalarda, haftalık HEMLIBRA profilaksisi, kayıttan önce NIS'da toplanmış kanama sayıları ile karşılaştırıldığında en az 12 haftalık tedavi alan 18 pediyatrik hastada tedavi edilmiş kanama oranında klinik olarak anlamlı azalma ile sonuçlanmıştır (%98) (Tablo 12).
Tablo 12 HAVEN 2: HEMLIBRA profilaksisi uygulanan hastaların önceki bypass edici ajan profilaksisine kıyasla Yıllık Kanama sayılarına ait hasta içi karşılaştırması
Sonlanım noktası | Önceki bypass ajanı tedavisi* (N=18) | Hemlibra profilaksisi (N = 18) |
Tedavi edilen kanamalar |
|
|
ABR (%95 GA) | 19,8 (15,3; 25,7) | 0,4 (0,15; 0,88) |
% azalma (RR) | %98 (0,02) | |
Sıfır kanama izlenen hasta yüzdesi (%95 GA) | 5,6 (0,1; 27,3) | 77,8 (52,4; 93,6) |
Medyan ABR (IQR) | 16,2 (11,49; 25,78) | 0 (0; 0) |
| ||
Pediyatrik Hastalarda Sağlık ile ilgili Sonuçlara Ait Bulgular HAVEN 2 Çalışmasında SağlıkileİlgiliSonuçlar
HAVEN 2'de, ≥ 8 ila < 12 yaşındaki hastalar için HRQoL değerlendirmesi 25. haftada çocuklar için Haemo-QoL-SF anketine dayanılarak değerlendirilmiştir (bkz. Tablo 13). Haemo-QoL-SF, HRQoL için geçerli ve güvenilir bir ölçüttür.
<12 yaş hastalar için HRQoL değerlendirmesi de 25. haftada hasta bakımını verenler tarafından doldurulan Bakımveren Yükü Yönlerini İçeren Adapte Edilmiş InhibQoL anketi ile gerçekleştirilmiştir (bkz. Tablo 13). Adapte Edilmiş InhibQoL, HRQoL için geçerli ve güvenilir bir ölçüttür.
Tablo 13 HAVEN 2: Hasta bakımını verenler tarafından raporlanan HEMLIBRA profilaksisi ile hastaların (<12 yaş) başlangıçtan 25. haftaya kadar Haemo- QoL-SF fiziksel sağlık skorlarında ortaya çıkan değişim
| Haemo-QoL-SF |
Fiziksel sağlık skoru (0 ila 100 aralığı) | |
Ortalama başlangıç skoru (%95 GA) (n=18) | 29,5 (16,4 - 42,7) |
Başlangıca göre ortalama değişiklik (%95 GA) (n=15) | -21,7 (-37,1 - -6,3) |
| Bakım veren Yükü Yönlerini İçeren Adapte Edilmiş InhibQoL |
Fiziksel sağlık skoru (0 ila 100 aralığı) |
|
Ortalama başlangıç skoru (%95 GA) (n=54) | 37,2 (31,5-42,8) |
Başlangıca göre ortalama değişiklik (%95 GA) (n=43) | -32,4 (-38,6 – (-26,2)) |
Ameliyatlar ve girişimler sırasında bypass edici ajan veya FVIII kullanımı konusunda deneyim sınırlıdır. Ameliyatlar ve girişimler sırasında bypass edici ajan veya FVIII kullanımı araştırıcı tarafından saptanmıştır.
Ani kanama durumunda, emicizumab profilaksisi alan hastalar mevcut tedaviler ile yönetilmelidir. Bypass ajanları ile ilgili kılavuz için bkz. Bölüm 4.4.
İmmünojenisite
Tüm terapötik proteinlerle olduğu gibi, HEMLIBRA ile tedavi edilen hastalarda da bir immün yanıt potansiyeli mevcuttur. Havuzlanmış klinik çalışmalarında toplam 668 hasta anti-emicizumab antikorlarının varlığı açısından test edilmiştir. Otuz dört hastada (%5,1) anti emicizumab antikorları pozitif çıkmıştır. 18 hastada (%2,7) anti-emicizumab antikorları in vitro olarak nötralize edilmiştir. Bu 18 hastadan nötralize edici anti emicizumab antikorları, 14 hastada HEMLİBRA'nın farmakokinetiği veya etkinliği üzerinde klinik olarak anlamlı bir etkiye sahip değilken, dört hastada (%0,6) emicizumab plazma konsantrasyonlarında düşüş gözlemlenmiştir. Nötralize edici anti-emicizumab antikorları olan ve emicizumab plazma konsantrasyonları azalmış bir hasta (%0,1) beş haftalık tedaviden sonra etkililik kaybı yaşamıştır ve HEMLİBRA'yı bırakmıştır. Genel olarak, HEMLİBRA'nın güvenlik profili, anti-emicizumab antikorları olan (nötralize edici antikorlar dahil) ve olmayan hastalar arasında benzerdir (bkz. Bölüm4.4ve4.8).
Etkililik kaybına ilişkin klinik belirtiler durumunda tedavide değişiklik düşünülmelidir.
Geriyatrik popülasyon
HEMLIBRA'nın 65 yaş ve üstü hemofili A hastalarında kullanımı, erişkin ve ergen çalışmaları HAVEN 1, HAVEN 3 ve HAVEN 4 ile desteklenmektedir. Sınırlı veriler temel alındığında, 65 yaş veya üstü hastalarda etkililik veya güvenlilik açısından farklılık olduğuna dair herhangi bir kanıt yoktur.
Pediyatrik popülasyon
Pediyatrik kullanımla ilgili bilgiler için bkz. Bölüm 4.2.
5.2. Farmakokinetik özellikler
Genel özelliklerEmicizumabın farmakokinetik özellikleri, sağlıklı gönüllülerde ve 389 hemofili A hastasından oluşan bir veritabanında yapılan bir popülasyon farmakokinetik analizleri kullanılarak belirlenmiştir.
Emilim:
Hemofili A hastalarında subkutan uygulamayı takiben emilim yarılanma ömrü 1,6 gündür.
Hemofili A hastalarında ilk 4 hafta boyunca haftada bir kez 3 mg/kg'lık subkutan uygulama sonrası emicizumabın ortalama (± SD) çukur plazma konsantrasyonları 5. haftada 52,6 ± 13,6 mcg/mL'ye ulaşılmıştır.
Tavsiye edilen idame dozları (haftada bir kez 1,5 mg/kg, iki haftada bir kez 3 mg/kg veya dört haftada bir kez 6 mg/kg) için kararlı durumda öngörülen ortalama (± SD) C, Cve C/ Coranları Tablo 14'de gösterilmiştir.
Tablo 14 Ortalama (±SD) kararlı durum emicizumab konsantrasyonları
| İdame dozu | ||
Parametreler | Haftada 1 kez 1,5 mg/kg | 2 haftada 1 kez 3 mg/kg | 4 haftada 1 kez 6 mg/kg |
C(mcg/mL) | 54,9±15,9 | 58,1±16,5 | 66,8±17,7 |
C(mcg/mL) | 53,5±15,7 | 53,5±15,7 | 53,5±15,7 |
C(mcg/mL) | 51,1±15,3 | 46,7±16,9 | 38,3±14,3 |
C/C | 1,08±0,03 | 1,26±0,12 | 1,85±0,46 |
Erişkinlerde/adölesanlarda (≥ 12 yaş) ve çocuklarda (< 12 yaş) haftada bir kez doz uygulamayı (4 hafta boyunca 3 mg/kg/hafta ve bunu takiben 1,5 mg/kg/hafta) takiben benzer farmakokinetik profilleri gözlenmiştir(bkz.Şekil1).
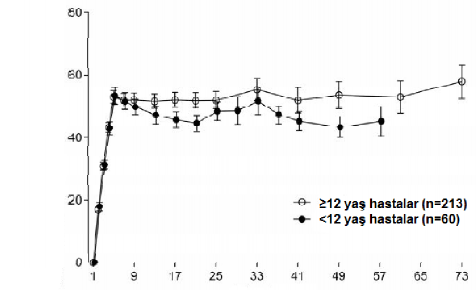
Ortalama (%95 GA) emicizumab konsantrasyonu (mcg/mL)
Şekil 1 ≥12 yaş hastaların ortalama plazma emcizumab konsantrasyonuna karşı zaman profillerinin (HAVEN 1 ve HAVEN 3 çalışmaları) <12 yaş hastalar ile karşılaştırması (HAVEN 2 çalışması)
Zaman (Hafta)
Sağlıklı gönüllülerde, 1 mg/kg subkutan uygulamayı takiben mutlak biyoyararlanım, enjeksiyon bölgesine bağlı olarak %80,4 ile %93,1 arasında değişmiştir. Karın, üst kol ve uyluğa subkutan uygulama sonrasında benzer farmakokinetik profiller gözlenmiştir. Emicizumab bu anatomik bölgelerden uygulanabilir (bkz. Bölüm 4.2).
Dağılım:
Sağlıklı gönüllülerde 0,25 mg/kg emicizumabın tek bir intravenöz dozunun ardından, kararlı durumda dağılım hacmi 106 mL / kg'dır (yani, 70 kg'lık bir yetişkin için 7,4 L).
Emicizumabın ardışık subkutan dozlarını takiben hemofili A hastalarında popülasyon farmakokinetiği analizlerinden hesaplanmış olan dağılım hacmi (V/F) 10,4 L'dir.
Biyotransformasyon:
Emicizumabın metabolizması incelenmemiştir. IgG antikorları temelde lizozomal proteoliz ile katabolize edilir ve daha sonra vücut tarafından elimine edilir ya da yeniden kullanılır.
Eliminasyon:
Sağlıklı gönüllülerde 0,25 mg / kg'lık intravenöz uygulama sonrasında, emicizumabın toplam klerensi 3,26 mL /kg /gün (yani, 70 kg'lık bir yetişkin için 0,228 L/d) ve ortalama terminal yarılanma ömrü 26,7 gün olarak belirlenmiştir.
Sağlıklı gönüllülerde tek subkutan enjeksiyonun ardından, eliminasyon yarılanma ömrü yaklaşık 4-5 hafta olmuştur.
Hemofili A hastalarında ardışık subkutan enjeksiyonların ardından, görünür klerens 0,272 L/gün ve görünür eliminasyon yarılanma ömrü 26,8 gündür.
Doğrusallık/doğrusal olmayan durum:
Emicizumab, subkutan uygulamayı takiben haftada bir kez 0,3 ila 6 mg /kg doz aralığında hemofili A hastalarında doz bağımlı bir farmakokinetik profil sergilemiştir. Birden fazla doz maruziyeti (C) haftada 1 kez 1,5 mg/kg, 2 haftada 1 kez 3 mg/kg ve 4 haftada 1 kez 6 mg/kg için karşılaştırılabilirdir.
Özel Popülasyonlar:
Pediyatrik popülasyon
Hasta yaşının emicizumabın farmakokinetik özellikleri üzerine etkisi, 5 bebek (≥ 1 ay ila < 2 yaş), 55 çocuk (<12 yaş) ve 50 ergeni (12 ila <18 yaş) içeren bir hemofili A hasta popülasyonunun farmakokinetik analizlerinde değerlendirilmiştir.
Yaş, pediyatrik hastalarda emicizumabın farmakokinetiğini etkilememiştir. Geriyatrik popülasyon
Hasta yaşının emicizumabın farmakokinetik özellikleri üzerine etkisi, 65 yaş ve üzerindeki on üç gönüllüyü (hiçbiri 77 yaşından büyük değildir) kapsayan bir popülasyonun farmakokinetik analizleri ile değerlendirilmiştir. Göreceli biyoyararlanım yaşla birlikte azalmış, ancak emicizumabın farmakokinetik özelliklerinde 65 yaş altı ve ≥ 65 yaş bireyler arasında klinik olarak anlamlı farklılıklar gözlenmemiştir.
Irk
Hemofili A hastalarında gerçekleştirilen popülasyon farmakokinetik analizleri, ırkın emicizumab farmakokinetik özelliklerini etkilemediğini göstermiştir. Bu demografik faktör için doz ayarlaması gerekli değildir.
Böbrek yetmezliği
Böbrek yetmezliğinin emicizumabın farmakokinetik özellikleri üzerindeki etkileri ile ilgili özel çalışmalar yürütülmemiştir.
Popülasyon farmakokinetiği analizinde, hemofili A hastalarının çoğunun böbrek fonksiyonunun normal olduğu (N = 332; kreatinin klerensi [KrKl] ≥ 90 mL/dak) veya hafif böbrek yetmezliği (N = 27; KrKl 60-89 mL/dak) olduğu saptanmıştır. Hafif böbrek yetmezliği emicizumabın farmakokinetiğini etkilememiştir. Orta derece böbrek yetmezliği olan hastalarda HEMLIBRA kullanımına ilişkin veri sınırlıdır (KrKl 30-59 mL/dak olan yalnızca 2 hasta) ve şiddetli böbrek yetmezliği olan hastalara ilişkin veri bulunmamaktadır. Hafif veya orta derece böbrek yetmezliğinin emicizumabın farmakokinetiğine etkisi olup olmadığı belirlenememektedir.
Emicizumab bir monoklonal antikor olup, böbrekle atılımdan ziyade katabolizma ile temizlenir ve böbrek yetmezliği olan hastalar için dozda değişiklik gerekmesi beklenmemektedir.
Karaciğer yetmezliği
Karaciğer yetmezliğinin emicizumabın farmakokinetik özellikleri üzerindeki etkileri ile ilgili özel çalışmalar yürütülmemiştir. Popülasyon farmakokinetik analizlerinde yer alan hemofili A hastalarının çoğunda karaciğer fonksiyonları normaldir (bilirubin ve AST ≤ Normal Üst Sınır (NÜS), n = 300) veya hafif karaciğer yetmezliği mevcuttur (bilirubin ≤ NÜS ve AST> NÜS veya bilirubin <1 ila 1,5 × NÜS ve herhangi bir düzeyde AST, n=51). Sadece 6 hastada orta derece karaciğer yetmezliği (1,5 x NÜS < bilirubin ≤ 3 x NÜS ve herhangi bir düzeyde AST) saptanmıştır. Hafif karaciğer yetmezliği, emicizumabın farmakokinetik özelliklerini etkilememiştir (bkz. Bölüm 4.2). Emicizumabın güvenliliği ve etkililiği karaciğer yetmezliği olan hastalarda özel olarak test edilmemiştir. Hafif ila orta şiddette karaciğer yetmezliği olan hastalar klinik çalışmalara dahil edilmiştir. Şiddetli karaciğer yetmezliği olan hastalarda HEMLIBRA kullanımına ilişkin veri yoktur.
Emicizumab bir monoklonal antikor olup, karaciğer metabolizmasından ziyade katabolizma ile temizlenir ve karaciğer yetmezliği olan hastalar için dozda değişiklik gerekmesi beklenmemektedir.
Diğer özel popülasyonlar
Modelleme çalışması, hipoalbuminemi ve yaşlarına göre düşük vücut ağırlığı olan hastalara daha nadir verilen dozların daha düşük emicizumab maruziyetini göstermektedir; simülasyonlar da bu hastalarda klinik olarak anlamlı kanama kontrolü sağlanabileceğine işaret etmektedir.
5.3. Klinik öncesi güvenlilik verileri
5.3. Klinik öncesi güvenlilik verileri
Fertilite
Emicizumab, 30 mg/kg/haftalık en yüksek test edilen doza (EAA temelinde 3 mg/kg/haftalık en yüksek dozda insan maruziyetinin 11 katına eşdeğer) kadar erkek veya dişi sinomolgus maymunlarının üreme organlarında herhangi bir değişikliğe neden olmamıştır.
Teratojenisite
Emicizumabın embriyo-fötal gelişim üzerindeki potansiyel yan etkileri ile ilgili veri mevcut değildir.
Enjeksiyon yeri reaksiyonları
Subkutan enjeksiyon sonrasında hayvanlarda tersine çevrilebilir hemoraji, perivasküler mononükleer hücre infiltrasyonu, subkutis dejenerasyonu/nekrozu ve subkutiste endotelyum şişmesi bildirilmiştir.
6. FARMASÖTİK ÖZELLİKLER
6.1. Yardımcı maddelerin listesi
L-Arjinin L-Histidin
L-Aspartik asit Poloksamer 188 Enjeksiyonluk su
6.2. Geçimsizlikler
HEMLIBRA ile önerilen enjektörler ve iğneler arasında geçimsizlik gözlenmemiştir (bkz. Bölüm 6.6).
Geçimlilik çalışmaları olmadığından, bu tıbbi ürün diğer tıbbi ürünlerle karıştırılmamalıdır.
6.3. Raf ömrü
Açılmamış flakon 24 ay.Buzdolabından çıkarıldığında, açılmamış flakonlar 7 güne kadar oda sıcaklığında (30°C'nin altında) saklanabilir.
Oda sıcaklığında saklandıktan sonra, açılmamış flakonlar buzdolabına geri konulabilir. Oda sıcaklığında kümülatif saklama süresi 7 günü geçmemelidir. Flakonlar asla 30°C'yi aşan sıcaklıklara maruz bırakılmamalıdır. Oda sıcaklığında 7 günden uzun süre bekleyen veya 30°C'yi aşan sıcaklıklara maruz kalan flakonlar imha edilmelidir.
Delinmiş flakon ve doldurulmuş enjektör
Mikrobiyolojik açıdan, flakondan enjektöre transfer edildikten sonra tıbbi ürün hemen kullanılmalıdır. Hemen kullanılmazsa, kullanacak kişi kullanım sırasındaki saklama zamanından ve koşullarından sorumludur.
6.4. Saklamaya yönelik özel tedbirler
Flakonları 2°C-8°C'de buzdolabında saklayınız. Dondurmayınız. Çalkalamayınız. Flakonu, ışıktan korumak için kutusunda saklayınız.
Tıbbi ürün ilk defa açıldıktan sonraki saklama koşulları için bkz. Bölüm 6.3.
6.5. Ambalajın niteliği ve içeriği
1 mL HEMLIBRA çözeltisi (30 mg/mL) içeren, bir floro-reçine film ile lamine edilmiş ve plastik bir geçme disk oturtulmuş bir alüminyum başlıkla bükülerek kapatılmış bütil lastik tıpalı bir adet 3 mL şeffaf cam tip I flakon. Her kartonda 1 flakon bulunmaktadır.
6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler
HEMLIBRA çözeltisi, seyreltilmesine gerek olmayan, subkutan enjeksiyon için steril, koruyucu içermeyen ve kullanıma hazır bir çözeltidir.
HEMLIBRA, uygulama öncesinde herhangi bir partiküllü madde veya renk değişikliğinin olmadığından emin olmak için görsel olarak incelenmelidir. HEMLIBRA, renksiz ile hafif sarı arası renkte bir çözeltidir. Partiküllü maddeler görülebiliyorsa veya ürünün rengi değişirse HEMLIBRA çözeltisi atılmalıdır.
HEMLIBRA çalkalanmamalıdır.
HEMLIBRA enjeksiyonluk çözelti şişeleri yalnızca tek kullanımlıktır.
HEMLIBRA çözeltisini flakondan çekip subkutan yoldan enjekte etmek için bir enjektör, transfer iğnesi ve bir enjeksiyon iğnesi gereklidir.
1 mL'ye kadar HEMLIBRA çözeltisinin enjekte edilmesi için 1 mL'lik bir enjektör kullanılmalıdır; buna karşılık 1 mL'den fazla ve en fazla 2 mL'ye kadar olan bir enjeksiyon için 2 - 3 mL'lik bir enjektör kullanılmalıdır.
Farklı dozlarda flakonların aynı enjektörde kullanımı için HEMLIBRA “Kullanma Talimatıâ€na bakınız. Reçetelenmiş dozu almak için farklı dozlarda flakonlar birlikte kullanıldığında, farklı HEMLIBRA konsantrasyonları (30 mg/mL ve 150 mg/mL) kullanılmamalıdır.
Uygulama ile ilgili ilave bilgiler için lütfen Bölüm 4.2 ve kullanma talimatına bakınız (Kullanma Talimatı'nın sonunda yer alan ‘Uygulama talimatları'na bakınız).
Kullanılmamış olan ürünler ya da atık materyaller, “Tıbbi Atıkların Kontrolü Yönetmeliği'' ve “Ambalaj Atıklarının Kontrolü Yönetmeliğiâ€ne uygun olarak imha edilmelidir.
 Belsoğukluğu, Chlamydia ve Frengi
Belsoğukluğu, bakterilerin sebep olduğu bir enfeksiyondur. Cinsel ilişki
yoluyla bulaşır ve dölyatağı boynunda, idrar yollarında, anüste, makatta ve
boğazda enfeksyona sebep olabilir.
Belsoğukluğu, Chlamydia ve Frengi
Belsoğukluğu, bakterilerin sebep olduğu bir enfeksiyondur. Cinsel ilişki
yoluyla bulaşır ve dölyatağı boynunda, idrar yollarında, anüste, makatta ve
boğazda enfeksyona sebep olabilir. |
 Travma Sonrası Bunalımı
Travmatik bir olay, günlük olağan olayların dışında olan ve kişiyi derinden
rahatsız eden bir olaydır.Birçok olay böyle bir etki gösterebilir.
Travma Sonrası Bunalımı
Travmatik bir olay, günlük olağan olayların dışında olan ve kişiyi derinden
rahatsız eden bir olaydır.Birçok olay böyle bir etki gösterebilir. |
 |
Lösemi Kan Kanseri Lösemi, kan kanseridir ve vücudunun kan oluşturan dokularının hastalanması anlamına gelir. Birçok lösemi türü vardır; bazı lösemi türleri çocuklarda bazıları da yetişkinlerde sık görülür. |
 |
En Yaygın Alerji Türleri Bağışıklık sistemi, polen, arı zehiri veya evcil hayvan gibi yabancı bir maddeye veya çoğu insanda reaksiyona neden olmayan bir yiyeceğe tepki gösterdiğinde alerjiler meydana gelir. |
 |
Ruh ve Akıl Sağlığımızı Geliştirmek İyi akıl ve ruh sağlığı sahip olmaktan ziyade, yaptığınız şeylerdir. Akıl ve ruhsal olarak sağlıklı olmak için kendinize değer vermeli ve kendinizi kabul etmelisiniz. |
İLAÇ GENEL BİLGİLERİ
Roche Müstahzarları Sanayi A.Ş.
| Satış Fiyatı | 59202.19 TL [ 25 Apr 2025 ] |
| Önceki Satış Fiyatı | 59202.19 TL [ 18 Apr 2025 ] |
| Original / Jenerik | Original İlaç |
| Reçete Durumu | Kırmızı Reçeteli bir ilaçdır. |
| Barkodu | 8699505773544 |
| Etkin Madde | Emisizumab |
| ATC Kodu | B02BX06 |
| Birim Miktar | 30 |
| Birim Cinsi | MG/ML |
| Ambalaj Miktarı | 1 |
| Kan ve Kan Yapıcı Organlar > K Vitamini ve Diğer Hemostatikler |
| İthal ( ref. ülke : Isvicre ) ve Beşeri bir ilaçdır. |