IMBRUVICA 140 mg 90 kaps�l K�sa �r�n Bilgisi
{ Ibrutinib }
1. BE�ER� TIBB� �R�N�N ADI
IMBRUVICA 140 mg Film Kapl� Tablet
2. KAL�TAT�F VE KANT�TAT�F B�LE��M
Etkin madde
Her bir film kapl� tablet 140 mg ibrutinib i�erir.
Yard�mc� maddeler
Laktoz monohidrat (s���r s�t� kaynakl�) 28 mg Yard�mc� maddeler i�in B�l�m 6.1'e bak�n�z.
3. FARMAS�T�K FORMU
Film kapl� tablet
Bir y�z�nde “ibr” di�er y�z�nde “140 mg” bask�s� bulunan sar�-ye�il ile ye�il aras� yuvarlak tablet (9 mm)
4. KL�N�K �ZELL�KLER
4.1. Terap�tik endikasyonlar
IMBRUVICA,
Mantle H�creli Lenfoma (MHL):
En az 3 k�r rituksimab ve alkilleyici ajan kombinasyonu sonras� n�ks eden veya diren�li olan veya otolog k�k h�cre nakli sonras� n�ks eden mantle h�creli lenfomada (MHL) endikedir.
Kronik Lenfositik L�semi (KLL):
4.2. Pozoloji ve uygulama �ekli
Bu ila� ile tedavi, anti-kanser ila�lar�n kullan�m�nda deneyimli bir hekim taraf�ndan
ba�lat�lmal� ve g�zlem alt�nda tutulmal�d�r.
Pozoloji uygulama s�kl��� ve s�resi:
Mantle H�creli Lenfoma (MHL)
MHL tedavisi i�in �nerilen doz g�nde bir kez 560 mg'd�r.
Kronik Lenfositik L�semi (KLL) ve Waldenstr�m Makrogloblunemisi (WM)
KLL ve WM i�in �nerilen doz, monoterapi veya kombinasyon halinde g�nde bir kez a��zdan al�nan 420 mg'd�r (kombinasyon rejimlerine ili�kin detayl� bilgi i�in bkz. B�l�m 5.1).
IMBRUVICA ile tedavi hastal�k progresyonuna ya da hasta taraf�ndan art�k tolere edilemez hale gelinceye kadar s�rd�r�lmelidir. KLL tedavisi i�in venetoklaks ile kombinasyon halinde IMBRUVICA, 3 k�r (1 k�r 28 g�nd�r) i�in tek bir ajan olarak uygulanmal�, ard�ndan 12 k�r IMBRUVICA ve venetoklaks uygulanmal�d�r. Venetoklaks dozaj bilgisi i�in, Venetoklaks K�sa �r�n Bilgisi'ne (K�B) bak�n�z.
IMBRUVICA, anti-CD20 tedavisiyle kombinasyon halinde uygulanaca�� zaman, IMBRUVICA'n�n ayn� g�n verilecek anti-CD20 tedavisinden �nce uygulanmas� �nerilmektedir.
Doz ayarlamalar�
Orta g��te ya da g��l� CYP3A4 inhibit�rleri ibrutinib maruziyetini art�r�r (bkz. B�l�m 4.4 ve 4.5).
Orta g��te CYP3A4 inhibit�rleri ile e� zamanl� kullan�ld���nda ibrutinib dozu g�nde 280 mg'a d���r�lmelidir.
G��l� CYP3A4 inhibit�rleri ile e� zamanl� kullan�ld���nda ibrutinib dozu g�nde 140 mg'a d���r�lmeli ya da tedaviye 7 g�ne kadar ara verilmelidir.
Herhangi bir yeni ba�layan ya da k�t�le�en Derece 2 kalp yetmezli�i, Derece 3 kardiyak aritmiler, Derece 3 ya da �zerindeki non-hematolojik toksisite, ate� veya enfeksiyon ile birlikte olan Derece 3 ya da �zerindeki n�tropeni veya Derece 4 hematolojik toksisite meydana geldi�inde IMBRUVICA tedavisi durdurulmal�d�r. Toksisite semptomlar� Derece 1 veya ba�lang�� seviyesine d�nd���nde (d�zelme), IMBRUVICA tedavisine a�a��daki tablolara g�re �nerilen dozda devam edilmelidir.
Kardiyak olmayan olaylar i�in �nerilen doz modifikasyonlar� a�a��daki gibidir:
Olaylar | Toksisite Meydana Gelmesi | �yile�me Sonras� MHL Doz Modifikasyonu | �yile�me Sonras� KLL/WM Doz Modifikasyonu |
Derece 3 veya 4 hematolojik olmayan toksisiteler | Birinci | 560 mg/g�n dozunda yeniden ba�lan�r. | 420 mg/g�n dozunda yeniden ba�lan�r. |
| 420 mg/g�n dozunda yeniden ba�lan�r. | 280 mg/g�n dozunda yeniden ba�lan�r. | |
Enfeksiyon veya ate�le birlikte Derece 3 veya 4 n�tropeni
4. Derece hematolojik toksisiteler | �kinci | ||
���nc� | 280 mg/g�n dozunda yeniden ba�lan�r. | 140 mg/g�n dozunda yeniden ba�lan�r. | |
D�rd�nc� | IMBRUVICA'y� kesin | IMBRUVICA'y� kesin |
![]()
Olaylar | Toksisite Meydana Gelmesi | �yile�me Sonras� MHL Doz Modifikasyonu | �yile�me Sonras� KLL/WM Doz Modifikasyonu |
Derece 2 kalp yetmezli�i | Birinci | 420 mg/g�n dozunda yeniden ba�lan�r. | 280 mg/g�n dozunda yeniden ba�lan�r. |
�kinci | 280 mg/g�n dozunda yeniden ba�lan�r. | 140 mg/g�n dozunda yeniden ba�lan�r. | |
���nc� | IMBRUVICA'y� kesin | ||
Derece 3 kardiyak aritmiler | Birinci | 420 mg/g�n dozunda yeniden ba�lan�r. | 280 mg/g�n dozunda yeniden ba�lan�r. |
�kinci | IMBRUVICA'y� kesin | ||
Derece 3 veya 4 kalp yetmezli�i
Derece 4 kardiyak aritmiler |
Birinci |
IMBRUVICA'y� kesin | |
Kalp yetmezli�i veya kardiyak aritmi olaylar� i�in �nerilen doz modifikasyonlar� a�a��daki gibidir:
E�er bir doz planlanan zamanda al�nmazsa, ayn� g�n i�erisinde m�mk�n olan en k�sa zamanda al�nabilir ve sonraki g�n normal uygulama plan�na devam edilir. Ka��r�lan dozu telafi etmek i�in fazladan tablet al�nmamal�d�r.
Uygulama �ekli:
IMBRUVICA her g�n yakla��k ayn� saatte, g�nde bir kere a��zdan bir bardak su ile al�nmal�d�r. Tabletler b�t�n olarak yutulmal�, k�r�lmamal� ya da �i�nenmemelidir. IMBRUVICA greyfurt suyu veya turun�giller ile al�nmamal�d�r (bkz. B�l�m 4.5).
�zel pop�lasyonlara ili�kin ek bilgiler:
B�brek yetmezli�i:
B�brek yetmezli�i olan hastalarda �zel klinik �al��malar yap�lmam��t�r. Hafif ya da orta dereceli b�brek yetmezli�i olan hastalar IMBRUVICA klinik �al��malar�nda tedavi edilmi�tir. Hafif ya da orta dereceli b�brek yetmezli�i olan (kreatinin klerensi 30 ml/dakikadan b�y�k) hastalarda doz ayarlamas� gerekli de�ildir. Hidrasyon sa�lanmal� ve serum kreatinin d�zeyleri periyodik olarak takip edilmelidir. �iddetli b�brek yetmezli�i (kreatinin klerensi < 30 ml/dak) hastalar�nda, IMBRUVICA ancak fayda riskten a��r bast��� takdirde kullan�lmal� ve hastalar toksisite belirtileri a��s�ndan yak�ndan izlenmelidir. �iddetli b�brek yetmezli�i hastalar�nda ya da diyaliz hastalar�nda veri mevcut de�ildir (bkz. B�l�m 5.2).
Karaci�er yetmezli�i:
�brutinib karaci�erde metabolize edilir. Bir karaci�er yetmezli�i �al��mas�nda, veriler ibrutinib maruziyetinde art�� ortaya koymu�tur (bkz. B�l�m 5.2). Hafif dereceli karaci�er
yetmezli�i olan hastalarda(Child-Pughs�n�f� A),�nerilen doz g�nde 280 mg'd�r. Orta
![]()
dereceli karaci�er yetmezli�i olan hastalarda (Child-Pugh s�n�f� B), �nerilen doz g�nde 140
mg'd�r. Hastalar IMBRUVICA toksisitesi belirtileri a��s�ndan izlenir ve gerekti�i gibi doz modifikasyon k�lavuzlar� takip edilir. �iddetli karaci�er yetmezli�i olan hastalarda IMBRUVICA kullan�lmas� �nerilmez (Child-Pugh s�n�f� C).
�iddetli kalp hastal���:
�iddetli kalp hastal��� olan hastalar IMBRUVICA'n�n klinik �al��malar�ndan d��lanm��t�r.
Pediyatrik pop�lasyon:
IMBRUVICA'n�n 0-18 ya� aras� �ocuklarda ve adolesanlarda etkilili�i ortaya konmad���ndan kullan�m� tavsiye edilmemektedir. Mat�r B h�creli non-Hodgkin lenfoml� hastalara ili�kin mevcut veriler B�l�m 4.8, 5.1 ve 5.2'de a��klanmaktad�r.
Geriyatrik pop�lasyon:
Ya�l� hastalarda (65 ya� ve �st�) doz ayarlamas� gerekli de�ildir.
4.3. Kontrendikasyonlar
Etkin madde
St. John's Wort (Sar� kantaron) i�eren �r�nler ile birlikte IMBRUVICA kullan�m� kontrendikedir.
4.4. �zel kullan�m uyar�lar� ve �nlemleri
Kanama ili�kili olaylar
IMBRUVICA ile tedavi edilen hastalarda, trombositopeninin e�lik etti�i ve etmedi�i kanama olay bildirimleri olmu�tur. Bunlar aras�nda kont�zyon, burun kanamas� ve pete�i gibi minor kanama olaylar� ve gastrointestinal kanama, intrakraniyal kanama ve hemat�ri gibi baz�lar� �l�mc�l olan, maj�r kanama olaylar� yer al�r.
Varfarin ya da di�er K vitamini antagonistleri IMBRUVICA ile e� zamanl� olarak kullan�lmamal�d�r.
IMBRUVICA ile birlikte antikoag�lan veya trombosit fonksiyonunu inhibe eden (antiplatelet ajanlar) t�bbi �r�nlerin e� zamanl� kullan�lmas� maj�r kanama riskini artt�r�r. Antikoag�lanlarda, antiplatelet ajanlardan daha fazla maj�r kanama riski g�zlendi. IMBRUVICA ile birlikte uyguland���nda, antikoag�lan veya antiplatelet tedavisinin risklerini ve yararlar�n� g�z �n�nde bulundurun. Kanama bulgu ve semptomlar� i�in hasta izlenmelidir.
Bal�k ya�� ve E vitamini gibi g�da takviyelerinin kullan�m�ndan ka��n�lmal�d�r.
IMBRUVICA tedavisi cerrahinin tipine ve kanama riskine ba�l� olarak, cerrahi �ncesinde ve sonras�nda en az 3 ila 7 g�n kesilmelidir.
Kanama ile ili�kili olaylar�n mekanizmas� tam olarak anla��lamam��t�r. Konjenital kanama diyatezi olan hastalar incelenmemi�tir.
L�kostaz
![]()
tedavisinin ge�ici olarak ara verilmesi d���n�lmelidir. Hastalar yak�ndan izlenmelidir.
Belirtildi�i �ekilde hidrasyon ve/veya sitored�ksiyon dahil, destekleyici bak�m uygulanmal�d�r.
Dalak r�pt�r�
IMBRUVICA tedavisinin kesilmesinin ard�ndan dalak r�pt�r� vakalar� bildirilmi�tir. IMBRUVICA tedavisine ara veya son verildi�inde, hastal�k durumu ve dalak boyutu dikkatle izlenmelidir (�rn. klinik muayene, ultrason). Sol �st kar�n veya omuz ucu a�r�s� geli�en hastalar de�erlendirilmeli ve dalak r�pt�r� tan�s� d���n�lmelidir.
Enfeksiyonlar
IMBRUVICA ile tedavi edilen hastalarda enfeksiyonlar g�zlenmi�tir (sepsis, n�tropenik sepsis, bakteri, vir�s ya da mantar enfeksiyonlar� dahil). Bu enfeksiyonlar�n baz�lar� hastaneye yatma ve �l�mle sonu�lanm��t�r. �l�mc�l enfeksiyonlar� olan hastalar�n �o�unda n�tropeni de vard�r. Hastalar ate�, anormal karaci�er fonksiyon testleri, n�tropeni ve enfeksiyonlar a��s�ndan izlenmeli ve endike oldu�u gibi uygun anti-enfektif tedavi ba�lat�lmal�d�r. F�rsat�� enfeksiyon a��s�ndan artm�� risk alt�nda olan hastalar i�in standart tedaviye g�re profilaksi d���n�lmelidir.
�brutinib kullan�m�n� takiben Aspergillozis, Kriptokokkozis ve Pn�mosistis jiroveci enfeksiyonlar� vakalar� dahil olmak �zere invazif fungal enfeksiyon vakalar� bildirilmi�tir. Rapor edilen invaziv mantar enfeksiyonu vakalar� �l�mc�l sonu�larla ili�kilendirilmi�tir.
Ge�mi�te ya da e�zamanl� al�nan imm�nosupresif tedavi ko�ullar�nda �l�mc�l olanlar� dahil olmak �zere Progresif Multifokal L�koensefalopati (PML) vakalar� rapor edilmi�tir. Hekimler yeni ya da k�t�le�en n�rolojik, bili�sel ya da davran��sal belirti veya semptomlar� olan hastalarda PML'yi ay�r�c� tan�da de�erlendirmelidir. PML'den ��phelenildi�i takdirde, uygun tan�sal de�erlendirmeler ger�ekle�tirilmeli ve PML ekarte edilene kadar tedavi durdurulmal�d�r. Herhangi bir ��phe varsa, hastan�n bir n�roloji uzman�na sevk edilmesi ve PML i�in tercihen kontrastl� MRG, JC Viral DNA i�in beyin-omurilik s�v�s� (BOS) testi ve tekrarlayan n�rolojik de�erlendirmeler gibi uygun tan�sal �l��mler d���n�lmelidir.
Hepatik olaylar
IMBRUVICA ile tedavi edilen hastalarda hepatotoksisite, hepatit B reaktivasyonu ve kronik olabilen hepatit E vakalar� meydana gelmi�tir. IMBRUVICA ile tedavi edilen hastalarda �l�mc�l olaylar dahil karaci�er yetmezli�i meydana gelmi�tir. IMBRUVICA ile tedaviye ba�lamadan �nce karaci�er fonksiyonu ve viral hepatit durumu de�erlendirilmelidir. Hastalar, tedavi s�ras�nda karaci�er fonksiyon parametrelerindeki de�i�iklikler i�in periyodik olarak izlenmelidir. Klinik olarak belirtildi�i gibi, enfeksiy�z hepatit i�in viral y�k ve serolojik testler, yerel t�bbi k�lavuzlara g�re yap�lmal�d�r. Hepatik olay te�hisi konan hastalar i�in, y�netim i�in bir karaci�er hastal��� uzman�na dan��may� d���n�n.
Sitopeniler
IMBRUVICA ile tedavi s�ras�nda ortaya ��kan Derece 3 ya da 4 sitopeniler (n�tropeni, trombositopeni ve anemi) bildirilmi�tir. Ayda bir tam kan say�mlar� izlenmelidir.
�nterstisyel Akci�er Hastal��� (�AH)
IMBRUVICA ile tedavi edilen hastalarda �AH vakalar� bildirilmi�tir. Hastalar, �AH'na i�aret eden akci�er semptomlar� bak�m�ndan izlenmelidir. Semptomlar�n ortaya ��kmas� durumunda, IMBRUVICA'ya ara verilmeli ve �AH uygun �ekilde tedavi edilmelidir. Semptomlar�n devam etmesi halinde, IMBRUVICA tedavisinin risk ve faydalar� hesaplanmal� ve doz modifikasyonu k�lavuzuna uyulmal�d�r.
Kardiyak aritmiler ve kardiyak yetmezlik
IMBRUVICA ile tedavi edilen hastalarda �l�mc�l ve ciddi kardiyak aritmiler ve kalp yetmezli�i meydana gelmi�tir. �leri ya�l�, Eastern Cooperative Oncology Group (ECOG) performans durumu ≥2 olan veya kardiyak komorbiditeleri olan hastalar ani �l�mc�l kardiyak olaylar a��s�ndan daha y�ksek riske sahip olabilirler. �zellikle akut enfeksiyonlar ya da kardiyak risk fakt�rleri, hipertansiyon dahil, diyabet hastal��� ve ge�mi� bir kardiyak aritmi �yk�s� olan hastalarda atriyal fibrilasyon, atriyal flutter ve ventrik�ler ta�iaritmi ve kardiyak yetmezlik vakalar� bildirilmi�tir.
IMBRUVICA'ya ba�lamadan �nce klinik olarak uygun kardiyak �yk� ve fonksiyonun de�erlendirmesi yap�lmal�d�r. Hastalar, tedavi s�ras�nda kardiyak fonksiyonda klinik k�t�ye gitme belirtileri a��s�ndan dikkatle izlenmeli ve klinik olarak de�erlendirilmelidir. Kardiyovask�ler a��dan endi�eler olan hastalar i�in ileri de�erlendirme (�rn., EKG, ekokardiyogram) d���n�lmelidir.
Kardiyak olaylar i�in ilgili risk fakt�rleri olan hastalarda, alternatif tedavi IMBRUVICA ile tedaviye ba�lamadan �nce dikkatlice yarar/risk de�erlendirilerek d���n�lebilir.
Ventrik�ler ta�iaritmi belirti ve/veya semptomlar� geli�en hastalarda, IMBRUVICA ge�ici olarak kesilmeli ve tedaviye yeniden ba�lamadan �nce kapsaml� bir klinik yarar/risk de�erlendirmesi ger�ekle�tirilmelidir.
Antikoag�lan tedavisi gerektiren, halihaz�rda atriyal fibrilasyonu olan hastalarda, IMBRUVICA'ya alternatif tedaviler d���n�lmelidir. IMBRUVICA ile tedavi s�ras�nda atriyal fibrilasyon geli�irse, hastalar tromboembolik hastal�k riski a��s�ndan incelenmelidir. Y�ksek riski olan ve IMBRUVICA alternatiflerinin uygun olmad��� hastalarda, antikoag�lanlar ile �ok yak�ndan takip ederek tedavi d���n�lmelidir.
IMBRUVICA tedavisi s�ras�nda hastalar kardiyak yetmezlik bulgu ve belirtileri a��s�ndan izlenmelidir. Bu vakalar�n baz�lar�nda, IMBRUVICA'n�n kesilmesinden veya dozun azalt�lmas�ndan sonra kalp yetmezli�i d�zelmi� veya iyile�mi�tir.
Serebrovask�ler olaylar
E�lik eden atriyal fibrilasyon ve/veya hipertansiyonu olan ve olmayan IMBRUVICA ile tedavi edilen hastalarda, serebrovask�ler olay, ge�ici iskemik atak ve �l�mleri i�eren iskemik inme vakalar� bildirilmi�tir. Bildirilen gecikmeli vakalar aras�nda, IMBRUVICA ile tedaviye ba�lanmas�ndan merkezi sinir iskemik vask�ler durumlar�n�n ba�lang�c�na kadar �o�u vakada bir ka� ay bulunmas� (%78'inde 1 aydan fazla ve vakalar�n %44'�nde 6 aydan fazla) hastalar�n d�zenli olarak izlenmesine ihtiya� oldu�unu vurgulad� (bkz. B�l�m 4.4 Kardiyak aritmi ve Hipertansiyon ve B�l�m 4.8).
T�m�r lizis sendromu
T�m�r lizis sendromu (TLS) IMBRUVICA tedavisinde bildirilmi�tir. T�m�r lizis sendromu riski ta��yan hastalar, tedavi �ncesi t�m�r y�k� y�ksek olan hastalard�r. Hastalar yak�ndan takip edilmeli ve uygun �nlemler al�nmal�d�r.
Non-melanom cilt kanseri
Havuzlanm�� randomize Faz 3 kar��la�t�rmal� �al��malarda, IMBRUVICA ile tedavi edilen hastalarda kar��la�t�rma kollar�na oranla daha s�k non-melanom cilt kanseri bildirilmi�tir. Hastalar non-melanom cilt kanseri a��s�ndan yak�ndan takip edilmelidir.
Hipertansiyon
IMBRUVICA ile tedavi edilen hastalarda hipertansiyon meydana gelmi�tir (bkz. B�l�m 4.8). IMBRUVICA ile tedavi edilen hastalarda kan bas�nc� d�zenli olarak izlenmelidir. IMBRUVICA ile tedavi boyunca uygun g�r�ld��� takdirde antihipertansif ila�lar ba�lat�lmal� veya dozu ayarlanmal�d�r.
Hemofagositik lenfohistiyositoz (HLH)
IMBRUVICA ile tedavi edilen hastalarda HLH vakalar� (�l�mc�l vakalar dahil) bildirilmi�tir. HLH, a��r� sistemik inflamasyonun klinik belirti ve semptomlar� ile karakterize, ya�am� tehdit eden bir patolojik imm�n aktivasyon sendromudur. HLH ate�, hepatosplenomegali, hipertrigliseridemi, y�ksek serum ferritin ve sitopeniler ile karakterizedir. Hastalar HLH semptomlar� hakk�nda bilgilendirilmelidir. Patolojik imm�n aktivasyonun erken belirtilerini geli�tiren hastalar derhal de�erlendirilmeli ve HLH tan�s� d���n�lmelidir.
�la�-ila� etkile�imleri
IMBRUVICA'n�n g��l� ya da orta dereceli CYP3A4 inhibit�rleri ile e� zamanl� kullan�m� ibrutinib maruziyetini art�rabilir ve dolay�s�yla toksisite i�in y�ksek risk olu�turabilir. �te yandan, CYP3A4 enzimini ind�kleyen ila�lar�n e� zamanl� kullan�m� IMBRUVICA maruziyetini azaltabilir ve dolay�s�yla etkililikte azalma riski ortaya ��kabilir. Bu sebeple, IMBRUVICA'n�n g��l� CYP3A4 inhibit�rleri ve g��l� ya da orta dereceli CYP3A4 ind�kleyicileri ile e� zamanl� kullan�m�ndan m�mk�n oldu�unca ka��n�lmal� ve e� zamanl� kullan�m sadece potansiyel yarar potansiyel zarardan �st�n oldu�unda d���n�lmelidir. CYP3A4 inhibit�r� kullan�lmas� zorunlu olan hastalar, IMBRUVICA toksisite belirtileri a��s�ndan yak�ndan izlenmelidir (bkz. B�l�m 4.2 ve 4.5). E�er bir CYP3A4 ind�kleyicisinin kullan�lmas� gerekiyorsa, hastalar IMBRUVICA etkisizlik riski belirtileri a��s�ndan izlenmelidir.
�ocuk do�urma potansiyeline sahip kad�nlar
�ocuk do�urma potansiyeline sahip kad�nlar IMBRUVICA kullan�rken y�ksek d�zeyde etkili bir do�um kontrol y�ntemi kullanmal�d�r (bkz. B�l�m 4.6).
Yard�mc� maddeler
Nadir kal�t�msal galaktoz intolerans problemi, lapp laktaz yetmezli�i ya da glukoz–galaktoz malabsorsiyonu olan hastalar�n bu ilac� kullanmamalar� gerekir.
4.5. Di�er t�bbi �r�nler ile etkile�imler ve di�er etkile�im �ekilleri
�brutinib primer olarak sitokrom P450 enzim 3A4 (CYP3A4) ile metabolize edilir. �brutinibin plazma konsantrasyonlar�n� y�kseltebilen ila�lar:
Orta g��te ya da g��l� CYP3A4 inhibit�rlerinin IMBRUVICA ile e� zamanl� kullan�m� ibrutinib maruziyetini artt�rabilir ve g��l� CYP3A4 inhibit�rlerinin kullan�mdan ka��n�lmal�d�r.
G��l� CYP3A4 inhibit�rleri
18 sa�l�kl� g�n�ll�de, �ok g��l� bir CYP3A4 inhibit�r� olan ketokonazol�n a� karn�na e� zamanl� uygulamas� ibrutinib maruziyetini (Cve EAA) s�ras�yla 29 ve 24 kat artt�rm��t�r. A� karn�na ko�ullar�n�n kullan�ld��� sim�lasyonlar g��l� bir CYP3A4 inhibit�r� olan klaritromisinin ibrutinib EAA's�n� 14 kat etkiledi�ini g�stermektedir. IMBRUVICA'y� yemek ile birlikte alan B-h�creli malignitesi olan hastalarda, g��l� CYP3A4 inhibit�r� olan
vorikonazol�n e� zamanl� uygulanmas�C'�6,7katveEA
A'y� 5,7 kat art�rm��t�r. G��l�
![]()
CYP3A4 inhibit�rlerin (�rne�in ketokonazol, indinavir, nelfinavir, ritonavir, sakuinavir,
klaritromisin, telitromisin, itrakonazol, nefazadon, kobisistat, vorikonazol ve posakonazol) kullan�m�ndan ka��n�lmal�d�r. E�er fayda riskten fazlaysa ve g��l� bir CYP3A4 inhibit�r� kullan�m� zorunluysa IMBRUVICA dozu inhibit�r kullan�m� s�resince 140 mg'a indirilir ya da IMBRUVICA ge�ici bir s�re kesilir (7 g�n ya da daha k�sa bir s�re). Hastalar toksisite a��s�ndan yak�ndan izlenmeli ve gerekti�i gibi doz modifikasyon k�lavuzlar� takip edilmelidir (bkz. B�l�m 4.2 ve 4.4).
Orta g��te CYP3A4 inhibit�rleri
IMBRUVICA'y� yemek ile birlikte alan B-h�creli malignitesi olan hastalarda CYP3A4 inhibit�r� olan eritromisinin e� zamanl� uygulanmas� C'� 3,4 kat ve EAA'y� 3 kat art�rm��t�r. E�er orta g��te bir CYP3A4 inhibit�r� (�rne�in flukonazol, eritromisin, amprenavir, aprepitant, atazanavir, siprofloksasin, krizotinib, diltiazem, fosamprenavir, imatinib, verapamil, amiodaron ve dronedaron) endike ise, IMBRUVICA dozu inhibit�r kullan�m� s�resince 280 mg'a indirilmelidir. Hastalar toksisite a��s�ndan yak�ndan izlenmeli ve gerekti�i gibi doz modifikasyon k�lavuzlar� takip edilmelidir (bkz. B�l�m 4.2 ve 4.4).
Zay�f CYP3A4 inhibit�rleri
A� karn�na ko�ullar�n�n kullan�ld��� sim�lasyonlar zay�f CYP3A4 inhibit�rleri olan azitromisin ve fluvoksaminin ibrutinib EAA's�n� 2 kattan az y�kseltebilece�ini g�stermi�tir. Zay�f g��te inhibit�rler ile kombinasyonunda herhangi bir doz ayarlamas� gerekli de�ildir. Hastalar toksisite a��s�ndan yak�ndan izlenmeli ve gerekti�i gibi doz modifikasyon k�lavuzlar� takip edilmelidir.
CYP3A4 inhibit�r� olan greyfurt suyunun e� zamanl� kullan�m�n�n de�erlendirildi�i 8 sa�l�kl� hastada ibrutinib maruziyeti (Cve EAA) s�ras�yla 4 ve 2 kat artm��t�r. Orta g��te CYP3A4 inhibit�rleri i�erdikleri i�in, IMBRUVICA tedavisi s�ras�nda greyfurt suyu ve turun�giller t�ketilmemelidir (bkz. B�l�m 4.2).
�brutinibin plazma konsantrasyonlar�n� azaltabilen ila�lar:
IMBRUVICA'n�n CYP3A4 ind�kleyicileri ile birlikte kullan�lmas� ibrutinibin plazma konsantrasyonlar�n� azaltabilir.
18 sa�l�kl� g�n�ll�de, g��l� bir CYP3A4 ind�kleyicisi olan rifampisinin a� karn�na e� zamanl� uygulamas� ibrutinib maruziyetini (Cve EAA) s�ras�yla %92 ve %90 azaltm��t�r. G��l� veya orta g��te CYP3A4 ind�kleyicilerinin (�rne�in karbamazepin, rifampisin, fenitoin) e� zamanl� kullan�m�ndan ka��n�lmal�d�r. St. John's Wort (Sar� kantaron) i�eren preparatlar ile IMBRUVICA'n�n e� zamanl� kullan�m� etkililik azalabilece�inden kontrendikedir. Daha d���k bir CYP3A4 ind�ksiyonu sa�layan alternatif ila�lar d���n�lmelidir. E�er fayda riskten fazlaysa ve g��l� veya orta g��te bir CYP3A4 ind�kleyicisinin kullan�m� zorunluysa hastalar etkisizlik a��s�ndan yak�ndan takip edilmelidir (bkz. B�l�m 4.3 ve 4.4). Zay�f ind�kleyiciler IMBRUVICA ile e� zamanl� kullan�labilir ama yine de hastalar etkisizlik a��s�ndan yak�ndan takip edilmelidir.
�brutinib pH'a ba�l� ��z�n�rl�k sergiler ve daha y�ksek pH de�erinde daha d���k ��z�n�rl��e sahiptir. 5 g�n s�reyle g�nde bir kere 40 mg omeprazol ald�ktan sonra a� karn�na 560 mg'l�k tek bir ibrutinib dozu uygulanan sa�l�kl� g�n�ll�lerde daha d���k bir Cde�eri g�zlenmi�tir (bkz. B�l�m 5.2). Daha d���k bir Cde�erinin klinik anlam� oldu�una dair hi�bir bulgu mevcut de�ildir ve midenin pH de�erini art�ran t�bbi �r�nler (�rne�in proton pompas� inhibit�rleri) pivot klinik �al��malarda hi�bir k�s�tlama olmaks�z�n kullan�lm��t�r.
Plazma konsantrasyonlar� ibrutinib ile de�i�en ila�lar:
�brutinib in vitro bir P-gp ve meme kanseri rezistan proteini (BCRP) inhibit�r�d�r. Bu etkile�imi kan�tlayan bir klinik �al��ma olmad���ndan, ibrutinibin terap�tik bir dozda intestinal P-gp'yi ve BCRP'yi inhibe edebilece�i de olas�l�k d��� b�rak�lamaz. Gastrointestinal kanalda bir etkile�im potansiyelini en aza indirmek �zere, digoksin veya metotreksat gibi oral dar bir terap�tik aral���na sahip P-gp veya BCRP substratlar� IMBRUVICA'dan en az 6 saat �nce ya da sonra kullan�lmal�d�r. �brutinib ayn� zamanda karaci�erde BCRP'yi inhibe edebilir ve BCRP-arac�l� hepatik at�l�ma u�rayan t�bbi �r�nlerin, rosuvastatin gibi, maruziyetini art�r�r.
Venetoklaks (400 mg) ile kombinasyon halinde ibrutinib (420 mg) �al��malar�ndaki KLL hastalar�nda, venetoklaks i�in monoterapi verilerine k�yasla venetoklaks maruziyetinde bir art�� (EAA baz�nda yakla��k 1,8 kat) g�zlenmi�tir.
B-h�creli maligniteleri olan hastalarda yap�lan bir ila� etkile�imi �al��mas�nda, tek seferlik 560 mg ibrutinib dozunun, CYP3A4 substrat� midazolam�n maruziyeti �zerinde klinik olarak anlaml� bir etkisi olmam��t�r. Ayn� �al��mada, g�nde 560 mg ibrutinib ile 2 haftal�k tedavinin oral kontraseptiflerin (etinilestradiol ve levonorgestrel), CYP3A4 substrat� midazolam�n veya CYP2B6 substrat� bupropionun farmakokineti�i �zerinde klinik olarak anlaml� bir etkisi olmam��t�r.
�zel pop�lasyonlara ili�kin ek bilgiler
Herhangi bir etkile�im �al��mas� yap�lmam��t�r.
Pediyatrik pop�lasyon:
Herhangi bir etkile�im �al��mas� yap�lmam��t�r.
4.6. Gebelik ve laktasyon
Genel tavsiyeGebelik kategorisi: D
�ocuk do�urma potansiyeli bulunan kad�nlar/Do�um kontrol� (Kontrasepsiyon) Hayvanlardan elde edilen bulgulara dayal� olarak, IMBRUVICA gebe kad�nlara uyguland���nda fetusa zarar verebilir. IMBRUVICA kullan�rken ve tedavinin tamamlanmas�n� takiben 3 ay s�resince kad�nlar gebe kalmaktan ka��nmal�d�r. Bu sebeple �ocuk do�urma potansiyeline sahip kad�nlar IMBRUVICA kullan�rken ve tedavinin tamamlanmas�n� takiben 3 ay s�resince y�ksek d�zeyde etkili bir do�um kontrol y�ntemi kullanmal�d�r. �brutinibin hormonal do�um kontrol y�ntemlerini etkileyip etkilemedi�i hen�z bilinmemektedir. Bu sebeple, hormonal do�um kontrol y�ntemi kullanan hastalar ikinci bir bariyer y�ntemi de ilave etmelidir.
Gebelik d�nemi
IMBRUVICA gebelik s�ras�nda kullan�lmamal�d�r. IMBRUVICA'n�n gebe kad�nlarda kullan�m�na ili�kin veri mevcut de�ildir. Hayvan �al��malar� �reme toksisitesi ortaya koymu�tur (bkz. B�l�m 5.3).
Laktasyon d�nemi
�brutinibin veya metabolitlerinin anne s�t�yle at�l�p at�lmad��� bilinmemektedir. Emzirilen bebe�e olan riskler g�z ard� edilemez. IMBRUVICA tedavisi s�ras�nda emzirme kesilmelidir.
Fertilite
Erkek ya da di�i s��anlarda test edilen maksimum doza kadar (100 mg/kg/g�n – insana e�de�er doz 16 mg/kg/g�n) fertilite ya da �reme kapasitesine bir etki g�r�lmemi�tir (bkz. B�l�m 5.3). �brutinibin insan fertilitesine etkileri �zerine veri bulunmamaktad�r.
4.7. Ara� ve makine kullan�m� �zerindeki etkiler
IMBRUVICA'n�n ara� ve makine kullanma yetene�i �zerinde min�r bir etkisi bulunmaktad�r.
IMBRUVICA kullanan baz� hastalarda yorgunluk, ba� d�nmesi ve asteni bildirilmi� olup, bir hastan�n ta��t ya da makine kullanma kapasitesini de�erlendirirken bu dikkate al�nmal�d�r.
4.8. �stenmeyen etkiler
G�venlilik profili �zeti
G�venlilik profili, d�rt Faz 2 klinik �al��mada ve sekiz randomize Faz 3 �al��mada IMBRUVICA ile tedavi edilen 1981 hastadan ve pazarlama sonras� deneyimlerden birle�tirilen verilere dayanmaktad�r. Klinik �al��malarda MHL ile tedavi edilen hastalar g�nde bir kez 560 mg IMBRUVICA alm�� ve klinik �al��malarda KLL ya da Waldenstr�m makroglobulinemisi (WM) i�in tedavi edilen hastalar da g�nde bir kez 420 mg IMBRUVICA alm��t�r. Hastalara IMBRUVICA ile venetoklaks�n kombinasyon halinde sabit s�reli verildi�i �al��malar (CLL3011 ve PCYC-1142-CA �al��malar�) haricinde klinik �al��malardaki t�m hastalar IMBRUVICA'y� hastal�k progresyonuna ya da art�k tolere edilemez hale gelene kadar alm��t�r. Birle�tirilmi� veri seti genelinde IMBRUVICA tedavisinin medyan s�resi 14,7 ayd�. KLL/SLL i�in medyan tedavi s�resi 14,7 ayd� (52 aya kadar); MHL i�in medyan tedavi s�resi 11,7 ayd� (28 aya kadar); WM i�in medyan tedavi s�resi 21,6 ayd� (37 aya kadar).
En yayg�n g�r�len advers ila� reaksiyonlar� (≥ %20) diyare, n�tropeni, kas-iskelet a�r�s�, d�k�nt�, hemoraji (�rne�in morarma), d�k�nt�, bulant�, trombositopeni, artralji ve �st solunum yolu enfeksiyonu olmu�tur. En yayg�n Derece 3 veya 4 advers ila� reaksiyonlar� (≥
%5) aras�nda n�tropeni, lenfositoz, trombositopeni, hipertansiyon ve pn�moni yer alm��t�r.
Tablola�t�r�lm�� advers ila� reaksiyonlar�
B-h�creli maligniteleri olan hastalar�n tedavilerinde advers ila� reaksiyonlar� ve pazarlama sonras� advers reaksiyonlar sistem organ s�n�f�na ve s�kl�k derecelerine g�re a�a��da listelenmi�tir. S�kl�k dereceleri ��yledir: �ok yayg�n (≥1/10); yayg�n (≥1/100 ila <1/10); yayg�n olmayan (≥1/1.000 ila <1/100); seyrek (≥1/10.000 ila <1/1.000); �ok seyrek (<1/10.000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). �stenmeyen etkiler her s�kl�k gruplamas�nda ciddiyette azalma s�ras�na g�re sunulmaktad�r.
Tablo 1: B-h�creli Maligniteleri Olan Hastalarda Klinik �al��malarda ve Pazarlama Sonras� Raporlanan Advers Reaksiyonlar
Sistem Organ S�n�f� |
S�kl�k (t�m dereceler) |
Advers reaksiyon | T�m Dereceler (%) | Derece ≥ 3 (%) | |
Enfeksiyonlar | �ok yayg�n | Pn�moni* |
| 12 | 7 |
ve |
| �st solunum | yolu | 21 | 1 |
enfestasyonlar |
| enfeksiyonu |
| 15 | 2 |
|
| Deri enfeksiyonu* |
|
|
|
Sistem Organ S�n�f� |
S�kl�k (t�m dereceler) |
Advers reaksiyon | T�m Dereceler (%) | Derece ≥ 3 (%) |
| Yayg�n | Sepsis* �riner sistem enfeksiyonu Sin�zit* | 3 9 9 | 3 1 1 |
Yayg�n olmayan | Cryptococcal enfeksiyonlar* Pneumocystis enfeksiyonlar* Aspergillus enfeksiyonlar�* Hepatit B reaktivasyonu | <1 <1 <1 <1 | 0 <1 <1 <1 | |
�yi huylu ve k�t� huylu neoplazmalar (kist ve polipler de dahil olmak �zere) | Yayg�n | Non-melanom cilt kanseri* Bazal h�creli karsinom Skuam�z h�creli karsinom | 5 3 1 | 1 <1 <1 |
Kan ve lenf sistemi hastal�klar� | �ok yayg�n | N�tropeni* Trombositopeni* Lenfositoz* | 39 29 15 | 31 8 11 |
Yayg�n | Febril n�tropeni L�kositoz | 4 4 | 4 4 | |
Seyrek | L�kostaz sendromu | <1 | <1 | |
Ba����kl�k sistemi hastal�klar� | Yayg�n | �nterstisyel akci�er hastal���* | 2 | <1 |
Metabolizma ve beslenme hastal�klar� | Yayg�n | Hiper�risemi | 9 | 1 |
Yayg�n olmayan | T�m�r lizis sendromu | 1 | 1 | |
Sinir sistemi hastal�klar� | �ok yayg�n | Ba� d�nmesi Ba� a�r�s� | 12 19 | <1 1 |
Yayg�n | Periferal n�ropati* | 7 | <1 | |
Yayg�n olmayan | Serebrovask�ler olaylar Ge�ici iskemik atak �skemik inme | <1 <1 <1 | <1 <1 <1 | |
G�z hastal�klar� | Yayg�n | G�rme bulan�kl��� | 6 | 0 |
Yayg�n olmayan | G�z kanamas� | <1 | 0 | |
Kardiyak hastal�klar | Yayg�n | Kardiyak yetmezlik* Atriyal fibrilasyon | 2 8 | 1 4 |
Yayg�n olmayan | Ventrik�ler ta�iaritmi* Kardiyak arrest | 1 <1 | <1 <1 | |
Vask�ler hastal�klar | �ok yayg�n | Kanama* Morarma* Hipertansiyon* | 35 27 18 | 1 <1 8 |
Yayg�n | Burun kanamas� Pete�i | 9 7 | <1 0 | |
Yayg�n olmayan | Subdural hematom | 1 | <1 |
Sistem Organ S�n�f� |
S�kl�k (t�m dereceler) |
Advers reaksiyon | T�m Dereceler (%) | Derece ≥ 3 (%) |
Gastrointestinal hastal�klar | �ok yayg�n | Diyare Kusma Stomatit* Bulant� Kab�zl�k Dispepsi | 47 15 17 31 16 11 | 4 1 1 1 <1 <1 |
Hepato-biliyer hastal�klar | Yayg�n olmayan | Karaci�er yetmezli�i* | <1 | <1 |
Deri ve deri alt� doku hastal�klar� | �ok yayg�n | D�k�nt�* | 34 | 3 |
Yayg�n | �rtiker Eritem T�rnak k�r�lmas� | 1 3 4 | <1 <1 0 | |
Yayg�n olmayan | Anjiyo�dem Pannik�lit* N�trofilik dermatozlar* | <1 <1 <1 | <1 <1 <1 | |
�ok seyrek | Stevens Johnson sendromu | <1 | <1 | |
Kas-iskelet bozukluklar�, ba� doku ve kemik hastal�klar� | �ok yayg�n | Artralji Kas spazmlar� Kas-iskelet a�r�s�* | 24 15 36 | 2 <1 3 |
Genel hastal�klar ve uygulama b�lgesine ili�kin hastal�klar | �ok yayg�n | Pireksi Periferik �dem | 19 16 | 1 1 |
Ara�t�rmalar | �ok yayg�n | Kan kreatinin art��� | 10 | <1 |
* �oklu advers reaksiyon �artlar�n� i�erir.
Se�ilmi� advers reaksiyonlar�n tan�m�
Advers reaksiyonlara ba�l� tedavi sonland�rmas� ve doz azalt�m�
B-h�creli maligniteler i�in IMBRUVICA ile tedavi olan 1981 hastadan %6's� temelde advers ila� reaksiyonlar�na ba�l� olarak tedaviyi kesmi�tir. Bu advers ila� reaksiyonlar�n�n ba��nda pn�moni, atriyal fibrilasyon, n�tropeni, d�k�nt�, trombositopeni ve kanama gelir. Hastalar�n yakla��k %8'inde dozun azalt�lmas�na neden olan advers ila� reaksiyonlar� g�r�lm��t�r.
Geriatrik pop�lasyon
IMBRUVICA ile tedavi g�ren 1981 hastan�n %50'si 65 ya� veya daha �zerinde idi. IMBRUVICA ile tedavi edilen ya�l� hastalarda Derece 3 veya daha y�ksek pn�moni (65 ya� ve �zeri hastalar�n %11'ine kar��l�k 65 ya� alt�ndaki hastalar�n %4'�nde) ve trombositopeni (65 ya� ve �zeri hastalar�n %11'ine kar��l�k 65 ya� alt�ndakilerin %5'i) daha s�k meydana gelmi�tir.
Uzun d�nem g�venlilik verileri
IMBRUVICA ile tedavi edilen 1284 hastada (tedavi g�rmemi� KLL/SLL n=162, n�ks/diren�li KLL/SLL n=646, n�ks/diren�li MHL n=370 ve WM n=106) 5 y�ll�k bir s�rede elde edilen uzun s�reli g�venlilik verileri analiz edilmi�tir. KLL/SLL i�in medyan tedavi s�resi 51 ayd�r (aral�k; 0,2 ila 98 ay) ve hastalar�n %70'i 2 y�l ve %52'si ise 4 y�ldan uzun bir s�reyle tedavi g�rm��t�r. MHL i�in medyan tedavi s�resi 11 ayd�r (aral�k; 0 ila 87 ay) ve hastalar�n %31'i 2 y�l ve %17'si ise 4 y�ldan uzun s�reyle tedavi g�rm��t�r. WM i�in medyan tedavi s�resi 47 ayd�r (aral�k; 0,3 ila 61 ay) ve hastalar�n %78'i 2 y�l ve %46'si ise 4 y�ldan uzun s�reyle tedavi g�rm��t�r. IMBRUVICA'ya maruz kalan hastalar�n bilinen genel g�venlik profili tutarl� kal�rken hipertansiyon prevalans�ndaki bir art�� d���nda hi�bir yeni g�venlilik endi�esi belirlenmemi�tir. Derece 3 veya �zeri hipertansiyon prevalans� %4 (0-1 y�l), %7 (1-2 y�l), %9 (2-3 y�l), %9 (3-4 y�l) ve %9 (4-5 y�l) ve 5 y�ll�k d�nemdeki insidans�
%11 olmu�tur.
Pediatrik pop�lasyon
G�venlik de�erlendirmesi, rituksimab, ifosfamid, karboplatin, etoposid ve deksametazon (RICE) rejimi ve ya rituksimab, vinkristin, ifosfamid, karboplatin, idarubisin ve deksametazon (RVICI) rejimi ile kombinasyon halinde arka plan tedavisi olarak ya da n�ks eden ve ya refrakter mat�r B h�creli non-Hodgkin lenfomal� pediyatrik ve gen� eri�kin hastalarda (3 ila 19 ya� aras�) tek ba��na arka plan tedavisi olarak IMBRUVICA'n�n bir Faz 3 �al��mas�ndan elde edilen verilere dayanmaktad�r (bkz. b�l�m 5.1). Bu �al��mada hi�bir yeni advers reaksiyon g�zlenmemi�tir.
��pheli advers reaksiyonlar�n raporlanmas�
Ruhsatland�rma sonras� ��pheli ila� advers reaksiyonlar�n�n raporlanmas� b�y�k �nem ta��maktad�r. Raporlama yap�lmas�, ilac�n yarar/risk dengesinin s�rekli olarak izlenmesine olanak sa�lar. Sa�l�k mesle�i mensuplar�n�n herhangi bir ��pheli advers reaksiyonu T�rkiye Farmakovijilans Merkezi (T�FAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e- posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks: 0 312 218 35 99).
4.9. Doz a��m� ve tedavisi
IMBRUVICA doz a��m�n�n etkilerine ili�kin veriler s�n�rl�d�r. Hastalar�n 12,5 mg/kg/g�n'e kadar dozlar kulland��� (1.400 mg/g�n) Faz 1 �al��mas�nda maksimum tolere edilen bir doza ula��lmam��t�r. Ba�ka bir �al��mada, 1.680 mg dozunda ila� kullanan sa�l�kl� bir birey Derece
4 geri d�n���ml� hepatik enzim art��� [aspartat aminotransferaz (AST) ve alanin aminotransferaz (ALT)] deneyimlemi�tir. IMBRUVICA'n�n spesifik bir antidotu yoktur. �nerilen dozdan fazlas�n� alan hastalar yak�ndan izlenmeli ve uygun destekleyici tedavi almal�d�r.
5. FARMAKOLOJ�K �ZELL�KLER
5.1. Farmakodinamik �zellikler
Farmakoterap�tik grup: Antineoplastik ajanlar, protein kinaz inhibit�rleri, ATC kodu: L01EL01.
Etki mekanizmas�
�brutinib k���k molek�ll�, g��l� bir BTK (Bruton tirozin kinaz) inhibit�r�d�r. �brutinib, BTK aktif b�lgesindeki bir sistein molek�l�yle (Cys-481) kovalent ba� olu�turarak, BTK enzimatik aktivitesinin s�rekli inhibisyonuna yol a�ar. Tec kinaz ailesinin bir �yesi olan BTK,
![]()
molek�l�d�r. BCR yola��, Mantle h�creli lenfoma (MHL), yayg�n b�y�k B h�creli lenfoma
(DLBCL), folik�ler lenfoma ve KLL dahil, �e�itli B-h�creli malignitelerin patogenezinde rol oynamaktad�r. BTK'nin B h�cresi y�zey resept�rleri arac�l� ger�ekle�en sinyal iletimindeki rol�, B-h�cre trafi�i, kemotaksis ve adhezyon i�in gerekli yolaklar�n aktivasyonuyla sonu�lan�r. Preklinik �al��malar, ibrutinibin in-vivo malign B-h�cre �o�almas� ve sa� kal�m�n�n yan� s�ra, in-vitro h�cre g��� ve substrat adezyonunu engelledi�ini g�stermektedir. Klinik �ncesi t�m�r modellerinde, ibrutinib ve venetoklaks kombinasyonu, her iki ajan�n tek ba��na kullan�lmas�na k�yasla artm�� h�cresel apoptoz ve anti-t�m�r aktivite ile sonu�lanm��t�r. �brutinib taraf�ndan ger�ekle�tirilen BTK inhibisyonu, KLL h�crelerinin bir h�cre sa�kal�m yolu olan BCL-2'ye ba��ml�l���n� artt�r�rken, venetoklaks da BCL-2'yi inhibe ederek apoptoza �nc�l�k eder.
Lenfositoz
Tedaviye ba�lanmas�yla birlikte, IMBRUVICA ile tedavi edilen KLL hastalar�n�n yakla��k d�rtte ���nde s�kl�kla lenfadenopatide k���lme ile ili�kili, geri d�n���ml� bir lenfosit art��� (yani ba�lang�ca g�re %50 ve �zerinde bir art�� ve mutlak say�s� 5.000/mcL'nin �zerinde) g�zlenmi�tir. Bu etki ayr�ca IMBRUVICA ile tedavi g�ren relaps ya da refrakter MHL hastalar�n�n da ��te birinde g�r�lm��t�r. G�zlenen bu lenfositoz farmakodinamik bir etkidir ve di�er klinik bulgular�n yoklu�unda progresif hastal�k olarak de�erlendirilmemelidir. Her iki hastal�k tipinde de lenfositoz tipik olarak IMBRUVICA tedavisinin ilk ay�nda meydana gelir ve tipik olarak MHL hastalar�nda medyan 8 hafta ve KLL hastalar�nda medyan 14 haftada d�zelir. Baz� hastalarda dola��mdaki lenfosit say�s�nda b�y�k art��lar g�r�lm��t�r (�rne�in >400.000/mcL).
IMBRUVICA ile tedavisi g�ren WM'li hastalarda lenfositoz g�zlenmemi�tir.
In vitro platelet agregasyonu
Bir in vitro �al��mada, ibrutinib kolajen etkisi ile olu�an platelet agregasyon inhibisyonu g�stermi�tir. �brutinib, platelet agregasyonunun di�er agonistlerini kullanarak anlaml� bir platelet agregasyon inhibisyonu g�stermemi�tir.
Kardiyak elektrofizyoloji ve QT/QTc aral��� �zerindeki etki
�brutinibin QTc aral��� �zerindeki etkisi, plasebo ve pozitif kontrollerle randomize, �ift k�r ayr�nt�l� bir QT �al��mas�nda 20 sa�l�kl� erkek ve kad�n g�n�ll�de de�erlendirilmi�tir. �brutinib 1.680 mg'lik supraterap�tik bir dozda QTc aral���n� klinik olarak ilgili bir derecede uzatmam��t�r. �brutinib ile plasebo aras�ndaki ba�lang��taki d�zeltilmi� ortalama bak�m�ndan farkl�l�klar i�in 2 yanl� %90 GA'n�n en b�y�k �st s�n�r� 10 ms'nin alt�nda olmu�tur. Ayn� �al��mada, QTc aral���nda konsantrasyona ba�l� bir k�salma g�zlenmi�tir (1.680 mg'l�k supraterap�tik dozu takiben 719 ng/mL Cde�erinde -5,3 ms [%90 GA: -9,4, -1,1]).
Klinik etkililik ve g�venlilik MHL
Relaps ya da refrakter MHL hastalar�nda IMBRUVICA'n�n g�venlili�i ve etkilili�i, 111 hastay� kapsayan a��k-etiketli, �ok-merkezli bir Faz 2 �al��mada de�erlendirilmi�tir (PCYC- 1104-CA �al��mas�). Medyan ya� 68 y�l idi (aral�k: 40 ila 84 y�l), hastalar�n %77'si erkek ve
%92'si beyaz �rktand�. ECOG performans durumu 3 ya da �zerinde olan hastalar �al��maya al�nmam��t�r. Tan�dan itibaren ge�en medyan s�re 42 ayd�; �nceki tedavilerin medyan say�s� 3 olup (aral�k: 1 ila 5 tedavi), hastalar�n %35'ine daha �nceden y�ksek doz kemoterapi,
%43'�ne bortezomib, %24'�ne lenalidomid ve %11'ine de daha �nce otolog veya allojenik k�k h�cre nakli uygulanm��t�r. Ba�lang��ta, taramada hastalar�n %39'unda kitlesel (bulky) hastal�k (≥5 cm), %49'unda Basitle�tirilmi� MHL Uluslararas� Prognostik �ndeks (MIPI) ile
�l��len y�ksek riskli skor ve%72'sindedeilerlem i� hastal�k mevcuttu (ekstranodal tutulum
ve/veya kemik ili�i tutulumu).
IMBRUVICA hastal�k progresyonu veya kabul edilemez toksisite meydana gelene kadar oral olarak g�nde bir kez 560 mg dozda uyguland�. T�m�r yan�t�, non-Hodgkin lenfoma (NHL) i�in revize edilmi� Uluslararas� �al��ma Grubu (IWG) kriterlerine g�re de�erlendirildi. Bu �al��madaki birincil sonlan�m noktas�, ara�t�rmac� taraf�ndan de�erlendirilen genel yan�t oran�yd� (ORR). IMBRUVICA'ya verilen yan�tlar Tablo 2'de g�sterilmektedir.
Tablo 2: Relapsl� ya da Refrakter MHL Hastalar�nda ORR ve DOR (PCYC-1104-CA �al��mas�)
| Toplam N = 111 |
ORR (%) | 67,6 |
%95 GA (%) | (58,0;76,1) |
CR (%) | 20,7 |
PR (%) | 46,8 |
Medyan DOR (CR+PR) (ay) | 17,5 (15,8, NR) |
�lk yan�ta kadar ge�en medyan s�re, ay (aral�k) | 1,9 (1,4-13,7) |
CR'ye kadar ge�en medyan s�re, ay (aral�k) | 5,5 (1,7-11,5) |
GA = g�ven aral���; CR = tam yan�t; DOR = yan�t s�resi; ORR = genel yan�t oran�; PR = k�smi yan�t; NR = ula��lmam��t�r
Bir Ba��ms�z �nceleme Komitesi (IRC) etkililik verilerini ilave olarak de�erlendirmi� ve
%69'luk bir ORR, %21'lik bir CR ve %48'lik bir PR oran� ortaya koymu�tur. IRC DOR'yi 19,6 ay olarak saptanm��t�r.
IMBRUVICA'ya verilen genel yan�t bortezomib ve lenalidomidi kapsayan ge�mi� tedaviden ya da temelde yatan risk/prognoz fakt�rlerinden, kitlesel (bulky) hastal�k, cinsiyet ya da ya�tan ba��ms�z olarak g�zlenmi�tir.
IMBRUVICA'n�n g�venlili�i ve etkilili�i, daha �nce en az bir tedavi g�rm�� 280 MHL hastas�n� kapsayan randomize, a��k etiketli, �ok merkezli Faz 3 �al��mada ortaya konmu�tur (�al��ma MCL3001). Hastalar, 21 g�n s�reyle g�nde bir kez oral yoldan 560 mg IMBRUVICA ya da ilk k�r�n 1, 8 ve 15. g�nlerinde 175 mg ve onu takiben her 21 g�nl�k k�r�n 1, 8 ve 15. g�nlerinde intraven�z yoldan 75 mg temsirolimus alacak �ekilde 1:1 oran�nda randomize edilmi�tir. Her iki kolda da tedaviye, hastal�k progresyonuna veya kabul edilemez toksisiteye kadar devam edilmi�tir. Medyan hasta ya�� 68 idi (aral�k: 34 ila 88); hastalar�n %74'� erkek ve %87'si beyaz �rktand�. Tan�dan itibaren ge�en medyan s�re 43 ayd� ve �nceki tedavilerin medyan say�s� 2 olup (aral�k: 1 ila 9 tedavi), hastalar�n %51'ine daha �nceden y�ksek doz kemoterapi, %18'ine bortezomib, %5'ine lenalidomid ve %24'�ne de daha �nce k�k h�cre nakli uygulanm��t�r. Ba�lang��ta, taramada hastalar�n %53'�nde kitlesel (bulky) hastal�k (≥5 cm), %21'inde Basitle�tirilmi� MIPI ile �l��len y�ksek riskli skor,
%60'�nda ekstranodal hastal�k ve %54'�nde de kemik ili�i tutulumu mevcuttu.
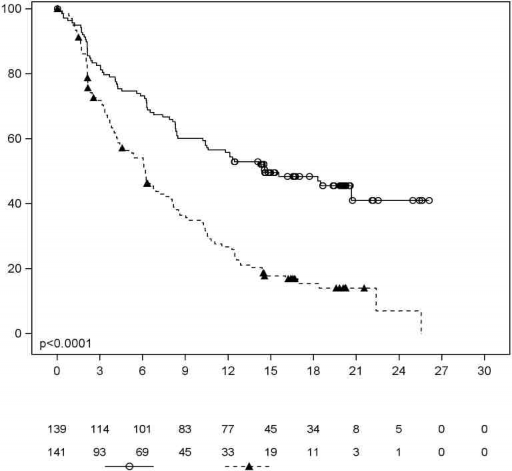
Progresyonsuz sa�kal�m (PFS), IRC taraf�ndan non-Hodgkin lenfoma (NHL) i�in revize edilmi� Uluslararas� �al��ma Grubu (IWG) kriterlerine g�re de�erlendirildi. MCL3001 �al��mas�na y�nelik etkililik sonu�lar� Tablo 3'te ve PFS i�in Kaplan-Meier e�risi �ekil 1'de g�sterilmektedir.
Tablo 3: Relaps veya Refrakter MHL Hastalar�nda Etkililik Sonu�lar� (�al��ma MCL3001)
Sonlan�m Noktas� | IMBRUVICA N = 139 | Temsirolimus N = 141 |
PFS | ||
Medyan PFS (%95 GA), (ay) | 14,6 (10,4, NE) | 6,2 (4,2, 7,9) |
| HR = 0,43 [%95 GA: 0,32, 0,58] | |
ORR (%) | 71,9 | 40,4 |
p de�eri | p < 0,0001 | |
NE = tahmin edilemez; HR = tehlike oran�; GA = g�ven aral���; ORR = genel yan�t oran�;
PFS = progresyonsuz sa�kal�m
�brutinib ile tedavi edilen daha az hasta temsirolimusa kar�� lenfoma semptomlar�nda klinik olarak anlaml� k�t�le�me ya�am�� (%27 kar��s�nda %52) ve semptomlar�n k�t�le�mesine kadar ge�en s�re ibrutinib ile temsirolimus kar��s�nda daha yava� olmu�tur (HR 0,27, p
<0,0001).
�ekil 1: MCL3001 �al��mas�na Ait Kaplan-Meier PFS (ITT Pop�lasyonu) E�risi
Progresyonsuz hayatta kalan hasta %
Imbruvica
Temsirolimus
Risk alt�nda olan g�n�ll�ler Imbruvica
Temsirolimus
Ay
Imbruvica Temsirolimus
KLL
KLL i�in daha �nce tedavi almam�� hastalar Tek ajan
Daha �nce tedavi almam�� 65 ya� ve �zeri KLL hastalar�nda IMBRUVICA'n�n klorambusil ile kar��la�t�r�ld��� randomize, �ok merkezli, a��k etiketli bir Faz 3 �al��mas� (PCYC-1115- CA) ger�ekle�tirilmi�tir. 65-70 ya� aras�ndaki hastalarda fludarabin, siklofosfamid ve rituksimab ile birinci basamak kemoimm�noterapi kullan�m�na engel olan en az bir komorbidite olmas� ko�ulu konmu�tur. Hastalar (n=269) hastal�k progresyonu veya kabul edilemez toksisiteye kadar g�nde 420 mg IMBRUVICA veya her bir 28 g�nl�k k�r�n 1. ve
15. g�nlerinde 0,5 mg/kg ba�lang�� dozunda klorambusil almak ve 0,8 mg/kg'l�k doz art��lar� tolere edildi�i s�rece maksimum 12 k�r i�in izin verilir �ekilde 1:1 oranda kollara randomize edildiler. Hastal�k progresyonunun do�rulanmas� ard�ndan klorambusil grubundaki hastalar ibrutinib grubuna ge�i� yapabilmi�tir.
Medyan hasta ya�� 73 idi (aral�k 65 ila 90); hastalar�n %63'� erkek ve %91'i beyaz �rktand�. Hastalar�n %91'inin ba�lang��taki ECOG performans durumu 0 ya da 1 ve %9'unun ECOG performans durum 2'ydi. �al��maya 269 KLL hastas� kaydedilmi�tir. Ba�lang��ta, hastalar�n
%45'inde ileri klinik evre (Rai Evre III veya IV), %35'inde ≥ 5 cm'lik en az bir t�m�r,
%39'unda ba�lang��ta anemi, %23'�nde ba�lang��ta trombositopeni, %65'inde > 3500 mcg/L β2 mikroglobulin y�ksekli�i, %47'sinde < 60 ml/dak CrCL, %20'sinde del11q, %6's�nda del 17p/t�m�r protein 53 (TP53) mutasyonu ve %44'�nde mutasyonsuz imm�noglobulin a��r zincir de�i�ken b�lgesi (IGHV) mevcuttu.
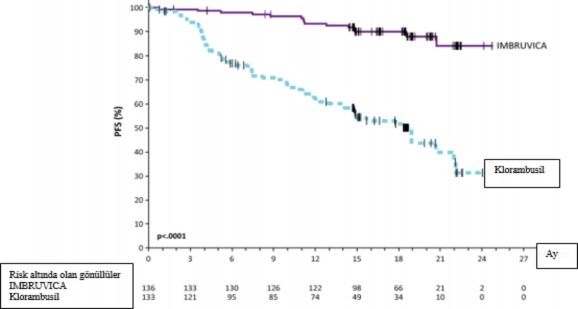
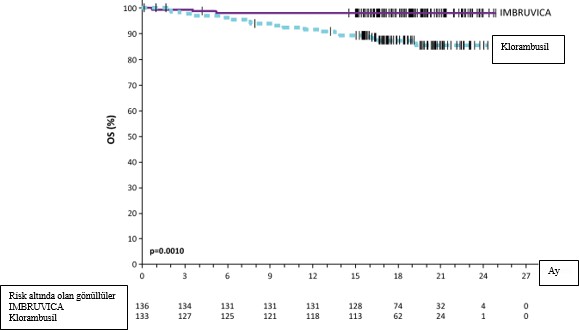
Uluslararas� KLL �al��ma Grubu (IWCLL) kriterlerine g�re Ba��ms�z De�erlendirme Kurulu (IRC) taraf�ndan de�erlendirilen progresyonsuz sa�kal�m (PFS), IMBRUVICA kolunda �l�m ya da progresyon riskinde %84 oran�nda istatistiksel olarak anlaml� bir azalma g�stermi�tir. PCYC-1115-CA �al��mas�na ait etkililik sonu�lar� Tablo 4'te ve PFS ve OS i�in Kaplan- Meier e�rileri s�ras�yla �ekil 2 ve 3'te g�sterilmektedir.
ITT pop�lasyonunda klorambusil kar��s�nda ibrutinib lehine istatistiksel olarak anlaml� bir s�rekli trombosit veya hemoglobin d�zelmesi g�zlenmi�tir. Ba�lang��ta sitopenileri olan hastalarda s�rekli hematolojik d�zelme a�a��daki gibi olmu�tur: ibrutinib ve kloramb�sil i�in s�ras�yla trombosit i�in %77,1'e %42,9, hemoglobin i�in %84,3'e %45,5'tir.
Tablo 4: PCYC-1115-CA �al��mas�na Ait Etkililik Sonu�lar�
Sonlan�m noktas� | IMBRUVICA N = 136 | Kloramb�sil N = 133 |
PFS | ||
Olay say�s� (%) | 15 (11,0) | 64 (48,1) |
Medyan (%95 GA), ay | Ula��lmad� | 18,9 (14,1, 22,0) |
HR (%95 GA) | 0,161 (0,091, 0,283) | |
ORR(CR + PR) | % 82,4 | % 35,3 |
P-de�eri | <0,0001 | |
OS |
| |
�l�mlerin say�s� (%) | 3 (2,2) | 17 (12,8) |
HR (%95 GA) | 0,163 (0,048, 0,558) | |
GA = g�ven aral���; HR = tehlike oran�; CR = tam yan�t; ORR = genel yan�t oran�; OS = genel sa�kal�m; PFS = progresyonsuz sa�kal�m; PR = k�smi yan�t
IRC taraf�ndan de�erlendirilenmedyan18,4ayl�ktakip.
Medyan OS'ye iki tedavi kolunda da ula��lmam��t�r.OSi�inp<0,005
�ekil 2: PCYC-1115-CA �al��mas�nda Kaplan-Meier PFS E�risi (ITT Pop�lasyonu)
�ekil 3: PCYC-1115-CA �al��mas�nda Kaplan-Meier OS E�risi (ITT Pop�lasyonu)
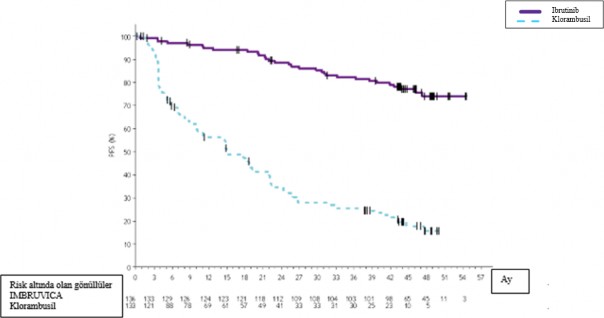
48 ayl�k takip
PCYC-1115-CA ve ona ait uzatma �al��mas�nda 48 ayl�k medyan takip s�resi ile IMBRUVICA kolundaki hastalar i�in, ara�t�rmac� de�erlendirmesine g�re �l�m veya progresyon riskinde %86 oran�nda bir azalma g�zlenmi�tir. Ara�t�rmac� taraf�ndan de�erlendirilen medyan PFS'ye IMBRUVICA kolunda ula��lmazken, klorambusil kolunda 15 ay [%95 GA (10,22, 19,35)] olarak bulunmu�tur (HR=0,14 [%95 GA (0,09, 0,21)]).
olarak bulunmu�tur. G�ncellenmi� Kaplan-Meier PFS e�risi �ekil 4'te g�sterilmektedir.
Ara�t�rmac� taraf�ndan de�erlendirilen ORR, IMBRUVICA kolunda %91,2 ve klorambusil kolunda %36,8 olmu�tur. IWCLL kriterlerine g�re CR oran�, IMBRUVICA kolunda %16,2 ve klorambusil kolunda %3 olmu�tur. Uzun s�reli takip s�ras�nda, ilk ba�ta klorambusil koluna randomize edilen toplam 73 hasta (%54,9) daha sonra �apraz ge�i� tedavisi olarak ibrutinib alm��t�r. K�rk sekizinci aydaki Kaplan-Meier OS d�n�m noktas� tahmini IMBRUVICA kolunda %85,5 olmu�tur.
PCYC-1115-CA �al��mas�nda ibrutinib ile g�r�len tedavi etkisi, del 17p/TP53 mutasyonu, del 11q ve/veya mutasyonsuz IGHV olan y�ksek riskli hastalar genelinde tutarl�l�k g�stermi�tir.
�ekil 4: PCYC-1115-CA �al��mas�nda 48 Ayl�k Takip ile Kaplan-Meier PFS E�risi (ITT Pop�lasyonu)
Kombinasyon tedavisi
IMBRUVICA'n�n daha �nce tedavi g�rmemi� KLL/SLL hastalar�ndaki g�venlili�i ve etkilili�i, obinutuzumabla kombinasyon halinde IMBRUVICA'n�n obinutuzumabla kombinasyon halinde klorambusille kar��la�t�r�ld��� randomize, �ok merkezli, a��k etiketli, Faz 3 �al��mas�nda (PCYC-1130-CA) daha ayr�nt�l� bi�imde incelenmi�tir. �al��maya 65 ya��nda veya daha b�y�k ya da <65 ya��nda olan ve mevcut hastal�klar olan, kreatinin klirensine g�re b�brek i�levinde d���� olan (<70 mL/dk.) veya del 17p/TP53 mutasyonu olan hastalar kaydedilmi�tir. Hastalar (n=229) 6 d�ng� boyunca 28 g�nl�k her bir d�ng�n�n 1. ve
15. g�nlerinde hastal�k ilerlemesine veya kabul edilemez toksisiteye kadar g�nl�k IMBRUVICA 420 mg'a ya da 0,5 mg/kg klorambusil dozuna 1:1 oran�nda randomize edilmi�tir. Her iki kolda da hastalara birinci d�ng�n�n 1., 8. ve 15. g�n�nde 1000 mg obinutuzumab verilmi�, daha sonra izleyen 5 d�ng�n�n birinci g�n�nde tedavi uygulanm��t�r (toplam 6 d�ng�, her biri 28 g�nl�k). �lk obinutuzumab dozu 1. g�n (100 mg) ve 2. g�n (900 mg) aras�nda b�l�nm��t�r.
Medyan ya� 71'dir (aral�k, 40 ila 87) ve hastalar�n %64'� erkek, %96's� beyazd�r. T�m hastalar�n bazal ECOG performansstat�s�0(%48)ve1-2'dir (%52). Ba�lang��ta hastalar�n
d�r
%44'�nde bazal anemi, %22'sinde bazal trombositopeni, %28'sinde CrCL <60 mL/dk var
ve medyan Ya�l�larda K�m�latif Hastal�k De�erlendirme �l�e�i (CIRS-G) 4't�r (aral�k, 0 ila 12). Ba�lang��ta, hastalar�n %65'inde y�ksek risk fakt�rleriyle KLL/SLL g�r�lm��t�r (del17p/TP53 mutasyonu [%18], del11q [%15] veya mutasyona u�ramam�� IGHV [%54]).
�lerlemesiz sa�kal�m (PFS), IWCLL kriterlerine g�re IRC taraf�ndan incelenmi�tir ve IMBRUVICA kolunda �l�m ve ilerleme riskinde istatistik a��dan anlaml� %77 oran�nda bir d���� oldu�una i�aret etmi�tir. �al��madaki 31 ayl�k medyan izlem s�resinde, IMBRUVICA
+ obinutuzumab kolunda medyan PFS'ye ula��lmam��t�r ve klorambusil + obinutuzumab kolunda 19 ayd�r. PCYC-1130-CA i�in etkililik sonu�lar� Tablo 5'te g�sterilmektedir ve PFS i�in Kaplan-Meier e�risi �ekil 5'te g�sterilmektedir.
Tablo 5: PCYC-1130-CA �al��mas�ndaki Etkililik Sonu�lar�
Biti� noktas� | IMBRUVICA + Obinutuzumab N=113 | Klorambusil + Obinutuzumab N=116 |
�lerlemesiz sa�kal�m |
|
|
Olay say�s� (%) | 24 (21,2) | 74 (63,8) |
Medyan (%95 GA), ay | Ula��lmam��t�r | 19 (15,1; 22,1) |
HR (%95 GA) | 0,23 (0,15; 0,37) | |
Toplam Yan�t Oran� | 88,5 | 73,3 |
CR | 19,5 | 7,8 |
PR | 69 | 65,5 |
GA=G�ven Aral���; HR = tehlike oran�; CR = tam yan�t; PR = k�smi yan�t
a IRC taraf�ndan de�erlendirilmi�tir.
b IMBRUVICA + obinutuzumab kolunda tam yan�t veren ve tamamlanamam�� kemik ili�i iyile�mesi ile tam yan�t (CRi) olan 1 hasta dahil.
c PR = PR + nPR
�ekil 5: PCYC-1130-CA �al��mas�nda PFS ��in Kaplan-Meier E�risi (ITT Pop�lasyonu)
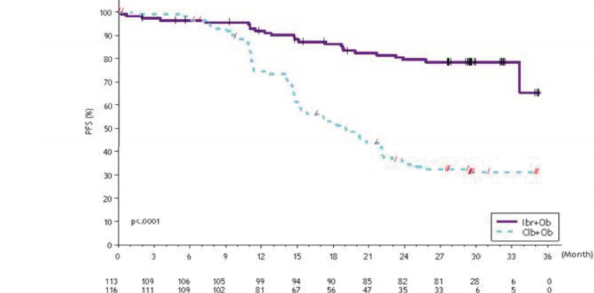
�brutinibin tedavi etkisi y�ksekriskiKLL/SLLpop�lasyonda tutarl�d�r (del 17p/TP53
l�k
PFS HR 0,15'tir [%95 GA (0,09; 0,27)]. Y�ksek riskli KLL/SLL pop�lasyonu i�in 2 y�l
PFS oran� tahminleri IMBRUVICA + obinutuzumab ve klorambusil + obinutuzumab kollar�nda s�ras�yla %78,8 [%95 GA (67,3; 86,7)] ve %15,5'tir [%95 GA (8,1; 25,2)].
Tablo 6: PFS Alt Grup Analizi (PCYC-1130-CA �al��mas�)
| N | Tehlike Oran� | %95 GA |
T�m hastalar | 229 | 0,231 | 0,145; 0,367 |
Y�ksek risk (del17p/TP53/del11q/mutasyona u�ramam�� IGHV) | |||
Evet | 148 | 0,154 | 0,087; 0,27 |
Hay�r | 81 | 0,521 | 0,221; 1,231 |
Del17p/TP53 | |||
Evet | 41 | 0,109 | 0,031; 0,38 |
Hay�r | 188 | 0,275 | 0,166; 0,455 |
FISH | |||
Del17p | 32 | 0,141 | 0,039; 0,506 |
Del11q | 35 | 0,131 | 0,03; 0,573 |
Di�erleri | 162 | 0,302 | 0,176; 0,52 |
Mutasyona u�ramam�� IHGV | |||
Evet | 123 | 0,15 | 0,084; 0,269 |
Hay�r | 91 | 0,3 | 0,12; 0,749 |
Ya� | |||
<65 | 46 | 0,293 | 0,122; 0,705 |
≥65 | 183 | 0,215 | 0,125; 0,372 |
Kitlesel hastal�k | |||
<5 cm | 154 | 0,289 | 0,161; 0,521 |
≥5 cm | 74 | 0,184 | 0,085; 0,398 |
Rai evresi | |||
0/I/II | 110 | 0,221 | 0,115; 0,424 |
III/IV | 119 | 0,246 | 0,127; 0,477 |
CRF'ye g�re ECOG | |||
0 | 110 | 0,226 | 0,11; 0,464 |
1-2 | 119 | 0,239 | 0,13; 0,438 |
Katmanla�t�r�lmam�� analize g�re tehlike oran�
IMBRUVICA + obinutuzumabla tedavi edilen hastalar�n %25'inde, klorambusil ve obinutuzumabla tedavi edilen hastalar�n %58'inde t�m derecelerde inf�zyon ili�kili reaksiyon g�zlemlenmi�tir. IMBRUVICA + obinutuzumabla tedavi edilen hastalar�n %3'�nde, klorambusil ve obinutuzumabla tedavi edilen hastalar�n %9'unda Derece 3 veya daha ciddi inf�zyon ili�kili reaksiyon g�zlemlenmi�tir.
IMBRUVICA'n�n daha �nce tedavi edilmemi� KLL veya SLL hastalar�ndaki g�venlili�i ve etkilili�i randomize, �ok merkezli, a��k etiketli bir Faz 3 �al��mas�nda (E1912), rituksimabla kombinasyon halinde IMBRUVICA (IR) ile standart fludarabin, siklofosfamid ve rituksimab (FCR) kemoimm�noterapisi kar��la�t�r�larak incelenmi�tir. �al��maya daha �nce tedavi edilmemi� ve 70 ya��nda veya daha k���k KLL veya SLL hastalar� kaydedilmi�tir. Hastalar (n=529) 2:1 oran�nda IR veya FCR'ye atanm��t�r. IMBRUVICA, hastal�k ilerlemesi veya kabul edilemez toksisite olana kadar 420 mg/g�n dozunda uygulanm��t�r. Fludarabin 25 mg/m dozunda ve siklofosfamid 250 mg/m dozunda, her ikisi de 1.-6. D�ng�lerin 1., 2. ve
3. g�nlerinde uygulanm��t�r.RituksimabIRkolundatoplam6 d�ng� boyunca, 2. d�ng�de,
![]()
birinci d�ng�n�n 2. g�n�nde 325 mg/m dozunda, izleyen 5 d�ng�n�n 1. g�n�nde 500 mg/m dozunda uygulanm��t�r. Her bir d�ng� 28 g�nd�r.
Medyan ya� 58'dir (aral�k 28 ila 70) ve hastalar�n %67'si erkek, %90'� beyazd�r. T�m hastalar�n bazal ECOG performans stat�s� 0 veya 1 (%98) veya 2'dir (%2). Bazalda, hastalar�n %43'�nde Rai evresinin 3. veya 4. evrede oldu�u g�r�lm��t�r ve hastalar�n
%59'unda y�ksek risk fakt�rl� KLL/SLL g�r�lm��t�r (TP53 mutasyonu [%6], del11q [%22] veya mutasyonsuz IGHV [%53]).
37 ayl�k medyan takip s�resi olan E1912 �al��mas�n�n etkililik sonu�lar� Tablo 7'de g�sterilmektedir. PFS (IWCLL kriterlerine g�re de�erlendirilmi�tir) ve OS i�in Kaplan-Meier e�rileri s�ras�yla �ekil 6 ve �ekil 7'de g�sterilmektedir.
Tablo 7: E1912 �al��mas�ndaki Etkililik Sonu�lar�
Biti� noktas� | �brutinib+ rituksimab (IR) N=354 | Fludarabin, Siklofosfamid ve Rituksimab (FCR) N=175 |
�lerlemesiz sa�kal�m | ||
Olay say�s� (%) | 41 (12) | 44 (25) |
Hastal�k ilerlemesi | 39 | 38 |
�l�m olaylar� | 2 | 6 |
Medyan (%95 GA), ay | NE (49,4; NE) | NE (47,1; NE) |
HR (%95 GA) | 0,34 (0,22; 0,52) | |
P-de�eri | <0,0001 | |
Toplam sa�kal�m | ||
�l�m say�s� (%) | 4 (1) | 10 (6) |
HR (%95 GA) | 0,17 (0,05; 0,54) | |
P-de�eri | 0,0007 | |
Toplam Yan�t Oran�(%) | 96,9 | 85,7 |
a P de�eri katmanla�t�r�lmam�� log-s�ra testine g�redir.
b Ara�t�rmac� de�erlendirmeli.
HR = tehlike oran�; NE = de�erlendirilemez
![]()
�ekil 6: E1912 �al��mas�nda PFS'nin Kaplan-Meier E�risi (ITT Pop�lasyonu)
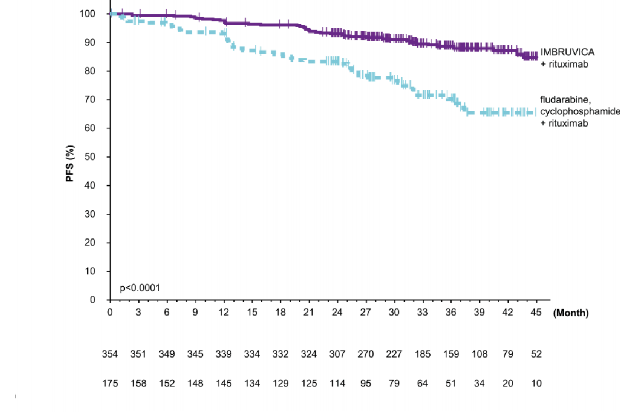
�brutinibin tedavi etkisi y�ksek riskli KLL/SLL pop�lasyonunda (TP53 mutasyonu, del11q veya mutasyonsuz IGHV) tutarl�d�r; PFS HR de�eri 0,23 [%95 GA (0,13; 0,40)], p <0,0001, Tablo 8'de g�sterilmektedir. Y�ksek riskli KLL/SLL pop�lasyonu i�in 3 y�ll�k PFS oran� tahminleri IR ve FCR kollar�nda s�ras�yla %90,4 [%95 GA (85,4; 93,7)] ve %60,3't�r [%95
GA (46,2; 71,8)].
Tablo 8: PFS Alt Grup Analizi (E1912 �al��mas�)
![]()
| N | Tehlike | %95 GA |
T�m hastalar | 529 | 0,340 | 0,222; 0,522 |
Y�ksek riskli (TP53/del11q/mutasyonsuz IGHV) | |||
Evet | 313 | 0,231 | 0,132; 0,404 |
Hay�r | 216 | 0,568 | 0,292; 1,105 |
del11q | |||
Evet | 117 | 0,199 | 0,088; 0,453 |
Hay�r | 410 | 0,433 | 0,260; 0,722 |
Mutasyonsuz IGHV | |||
Evet | 281 | 0,233 | 0,129; 0,421 |
Hay�r | 112 | 0,741 | 0,276; 1,993 |
Kitlesel hastal�k | |||
<5 cm | 316 | 0,393 | 0,217; 0,711 |
≥5 cm | 194 | 0,257 | 0,134; 0,494 |
Rai evresi | |||
0/I/II | 301 | 0,398 | 0,224; 0,708 |
III/IV | 0,148; 0,534 | ||
0 | 335 | 0,242 | 0,138; 0,422 |
1-2 | 194 | 0,551 | 0,271; 1,118 |
Tehlike oran� katmanla�t�r�lmam�� analize dayanmaktad�r.
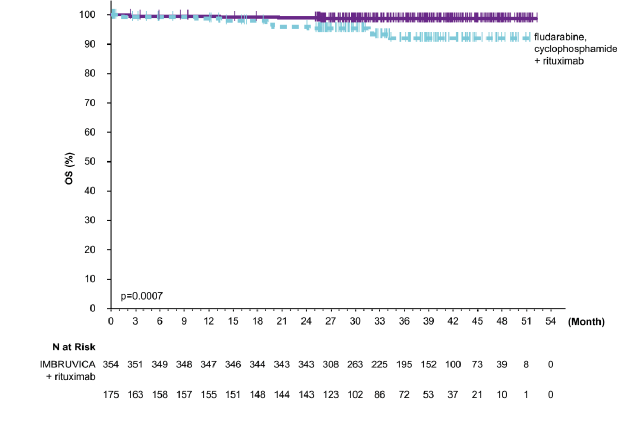
�ekil 7: E1912 �al��mas�nda OS'nin Kaplan-Meier E�risi (ITT Pop�lasyonu)
Sabit s�reli kombinasyon tedavisi
�nceden tedavi edilmemi� KLL hastalar�nda IMBRUVICA'n�n venetoklaks ile kombinasyon halinde sabit s�reli tedavisinin g�venlili�i ve etkilili�i, randomize, a��k etiketli bir faz 3 �al��mas�nda (CLL3011) de�erlendirilmi�tir. �al��maya, daha �nce tedavi edilmemi�, CIRS skoru >6 veya CrCL ≥30 ila <70 mL/dak olan <65 ya� KLL hastalar� ve 65 ya� ve �st� yeti�kin hastalar dahil edilmi�tir. Del 17p veya bilinen TP53 mutasyonlar� olan hastalar �al��ma d��� b�rak�lm��t�r. Hastalar (n=211), venetoklaks ile kombinasyon halinde IMBRUVICA veya obinutuzumab ile kombinasyon halinde klorambusil almak �zere 1:1 oran�nda randomize edilmi�tir. IMBRUVICA + venetoklaks kolundaki hastalar, 3 k�r boyunca tek ajan IMBRUVICA ve ard�ndan 12 k�r (5 haftal�k doz titrasyon program� dahil) venetoklaks ile kombinasyon halinde IMBRUVICA alm��t�r. Her k�r 28 g�nd�r. IMBRUVICA g�nde 420 mg dozda uygulanm��t�r. Venetoklaks, 1 hafta boyunca 20 mg ile ba�lanarak, ard�ndan 1 hafta boyunca her bir 50 mg, 100 mg ve 200 mg doz seviyesinde, ard�ndan �nerilen g�nl�k doz olan 400 mg olarak uygulanm��t�r. Klorambusil + obinutuzumab koluna randomize edilen hastalar, 6 k�r boyunca tedavi g�rm��t�r. Obinutuzumab, 1. k�r 1., 8. ve 15. g�nlerinde 1.000 mg dozunda uygulanm��t�r. 2 ila 6.
k�rlerde, 1. g�nde 1.000 mg obinutuzumab verilmi�tir. Klorambusil, 1 ila 6. k�rlerin 1. ve 15. g�nlerinde 0.5 mg/kg v�cut a��rl��� dozunda uygulanm��t�r. Sabit s�reli rejimlerden herhangi birinin tamamlanmas�n�n ard�ndan IWCLL kriterlerine g�re progrese oldu�u tespit edilen hastalar, tek ajan IMBRUVICA ile tedavi edilebilir.
ta,
hastalar�n %18'i del 11q'lu KLL ve %52'si mutasyona u�ramam�� IGHV ile ba�vurmu�tur. T�m�r lizis sendromu riski i�in temel de�erlendirmede, hastalar�n %25'inde y�ksek t�m�r y�k� vard�r. 3 k�r tek ajan IMBRUVICA �nc� tedavisinden sonra, hastalar�n %2'sinde y�ksek t�m�r y�k� g�r�lm��t�r. Y�ksek t�m�r y�k�, herhangi bir lenf nodu ≥10 cm veya herhangi bir lenf nodu ≥5 cm ve mutlak lenfosit say�s� ≥25×10/L olarak tan�mlanm��t�r. 28 ayl�k medyan takip s�resinde, IWCLL kriterlerine g�re CLL3011 �al��mas� i�in IRC taraf�ndan de�erlendirilen etkililik sonu�lar� Tablo 9'da, PFS i�in Kaplan-Meier e�risi �ekil 8'de ve minimal kal�nt� hastal�k oranlar� (MRD) negatifli�i oranlar� Tablo 10'da sunulmaktad�r.
Tablo 9: CLL3011 �al��mas�ndaki Etkililik Sonu�lar�
Biti� noktas� | IMBRUVICA + Venetoklaks N=106 | Klorambusil + Obinutuzumab N=105 |
�lerlemesiz Sa�kal�m |
|
|
Olay say�s� (%) | 22 (20,8) | 67 (63,8) |
Medyan (%95 GA), ay | Ula��lmam��t�r (31,2, Ula��lmam��t�r) | 21 (16,6, 24,7) |
HR (%95 GA) | 0,22 (0,13, 0,36) | |
P-de�eri | <0,0001 | |
Tam Yan�t Oran� (%) | 38,7 | 11,4 |
%95 GA | (29,4, 48) | (5,3, 17,5) |
P-de�eri | <0,0001 | |
Toplam Yan�t Oran� (%) | 86,8 | 84,8 |
%95 GA | (80,3, 93,2) | (77,9, 91,6) |
CR = tam yan�t; CRi = tamamlanamam�� kemik ili�i iyile�mesi ile tam yan�t; HR = tehlike oran�; NE = ula��lamam��t�r; nPR = nod�ler k�smi yan�t; PR = k�smi yan�t
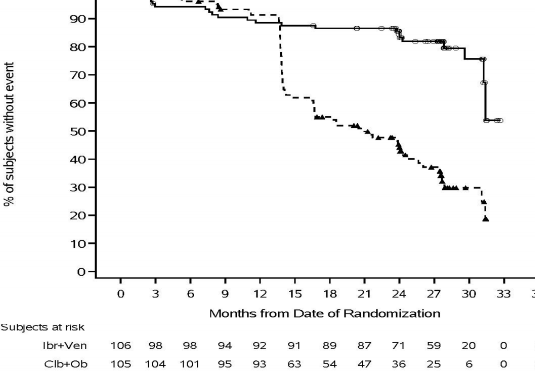
�ekil 8: CLL3011 �al��mas� KLL Hastalar�na ait Kaplan-Meier PFS (ITT pop�lasyonu) E�risi
IMBRUVICA + venetoklaks�n tedavi etkisi, 0,23 PFS HR [%95 GA (0,13, 0,41)] ile y�ksek riskli KLL pop�lasyonunda (TP53 mutasyonu, del 11q veya mutasyona u�ramam�� IGHV) ile tutarl�d�r.
Genel sa�kal�m verileri hen�z olgun de�ildir. 28 ayl�k medyan takipte, toplam 23 �l�mle tedavi kollar� aras�nda anlaml� fark yoktur: IMBRUVICA + venetoklaks kolunda 11 (%10,4) ve klorambusil + obinutuzumab kolunda 12 (%11,4) OS HR 1,048 [%95 GA (0,454, 2,419)].
6 ayl�k ek takibin ard�ndan, IMBRUVICA + venetoklaks kolunda ve klorambusil + obinutuzumab kolunda s�ras�yla 11 (%10,4) ve 16 (%15,2) �l�m rapor edilmi�tir ve tahmini
OS HR 0,76'd�r [%95 GA (0,352, 1,642].
Tablo 10: CLL3011 �al��mas� MRD Negatiflik Oranlar�
| NGS Testi | Ak�� sitometrisi | ||
| IMBRUVICA + Venetoklaks N=106 | Klorambusil + Obinutuzumab N=105 | IMBRUVICA + Venetoklaks N=106 | Klorambusil + Obinutuzumab N=105 |
MRD Negatiflik oran� | ||||
Kemik ili�i, n (%) | 59 (55,7) | 22 (21) | 72 (67,9) | 24 (22,9) |
%95 GA | (46,2, 65,1) | (13,2, 28,7) | (59, 76,8) | (14,8, 30,9) |
Periferik kan, n (%) | 63 (59,4) | 42 (40) | 85 (80,2) | 49 (46,7) |
%95 GA | (50,1, 68,8) | (30,6, 49,4) | (72,6, 87,8) | (37,1, 56,2) |
Tedavinin Tamamlanmas�ndan Sonraki �� Aydaki MRD Negatiflik Oran� | ||||
Kemik ili�i, n (%) | 55 (51,9) | 18 (17,1) | 60 (56,6) | 17 (16,2) |
%95 GA | (42,4, 61,4) | (9,9, 24,4) | (47,2, 66) | (9,1, 23,2) |
Periferik kan, n (%) | 58 (54,7) | 41 (39) | 65 (61,3) | 43 (41) |
%95 GA | (45,2, 64,2) | (29,7, 48,4) | (52, 70,6) | (31,5, 50,4) |
P de�erleri Cochran-Mantel-Haenszel ki-kare testinden al�nm��t�r. NGS ile kemik ili�inde MRD negatiflik oran� i�in P de�eri birincil MRD analizidir.
Yeni nesil s�ralama testi (clonoSEQ) kullan�larak 10-4 e�i�ine dayal�d�r
5.2. Farmakokinetik �zellikler
Genel �zellikler Emilim:
�brutinib oral uygulama sonras�nda 1 ila 2 saatlik medyan Tde�eri ile h�zla emilir. A� ko�ullarda (n=8) mutlak biyoyararlan�m %2,9'dur (%90 GA = 2,1 – 3,9) ve bir ���n ile birlikte kullan�ld���nda iki kat�na ��kar. �brutinibin farmakokineti�i farkl� B h�creli maligniteleri olan hastalarda anlaml� bir farkl�l�k g�stermez. �brutinib maruziyeti 840 mg'a kadar olan dozlarda artar. Dozun 560 mg olarak uyguland��� hastalarda g�zlenen kararl� durum EAA de�eri (ortalama ± standart sapma) 953 ± 705 ng sa/mL'dir. �brutinibin a� karn�na al�nmas� ile y�ksek oranda ya� i�eren bir kahvalt�dan 30 dakika �nce, 30 dakika sonra (tok ko�ullar) ya da y�ksek ya�l� bir kahvalt�dan 2 saat sonras�na k�yasla maruziyetin yakla��k
%60'�na (EAA) neden olmu�tur.
�brutinib pH'a ba�l� ��z�n�rl�k sergiler ve daha y�ksek pH de�erinde daha d���k ��z�n�rl��e sahiptir. 5 g�n s�reyle g�nde bir kere 40 mg omeprazol ald�ktan sonra a� karn�na 560 mg'l�k tek bir ibrutinib uygulanan sa�l�kl� g�n�ll�lerde, geometrik ortalama oranlar� (%90 GA) EAAi�in %83 (%68-102), EAAi�in %92 (%78-110) ve Ci�in %38
(%26-53) olmu�tur.
Da��l�m:
�brutinibin insan plazma proteinlerine in vitro geri d�n���ml� ba�lanmas� %97,3 olup, 50 ila
1.000 ng/mL aral���nda hi�bir konsantrasyon ba��ml�l��� yoktur. Kararl� durumdaki g�r�n�r da��l�m hacmi (V/F) yakla��k 10.000 L'dir.
Biyotransformasyon:
�brutinib primer olarak CYP3A4 ile metabolize olur ve BTK'y� inhibe edici aktivitesi ibrutinibden yakla��k 15 kat daha d���k olan bir dihidrodiol metaboliti olu�ur. CYP2D6 ibrutinib metabolizmas�nda minimal bir rol oynar.
Bu nedenle, farkl� CYP2D6 genotiplerine sahip hastalarda �nlem al�nmas� gerekli de�ildir. Eliminasyon:
G�r�n�r klerens (CL/F) yakla��k 1000 L/saat'tir. �brutinibin yar�lanma �mr� 4 ila 13 saattir. Sa�l�kl� deneklerde radyoaktif olarak etiketlenmi� tek doz oral [C]-ibrutinib uygulamas�ndan sonra radyoaktivitenin yakla��k %90'� 168 saat i�erisinde at�lm�� olup, bunun b�y�k b�l�m� (%80) fe�este ve %10'undan az� idrarda at�lm��t�r. De�i�memi� ibrutinib radyoaktif olarak etiketlenmi� bo�alt�m �r�n�n�n fe�este yakla��k %1'ini olu�turur ve idrarda g�r�lmez.
![]()
Do�rusall�k/do�rusal olmayan durum:
�brutinib maruziyeti doz ile orant�l� olarak artar.
Hastalardaki karakteristik �zellikler
Geriyatik pop�lasyon:
Pop�lasyon farmakokineti�i ya��n, dola��mdan ibrutinib klerensini anlaml� d�zeyde etkilemedi�ini g�stermi�tir.
Pediyatrik pop�lasyon:
Farmakokinetik veriler, n�kseden veya refrakter mat�r B-h�creli non-Hodgkin lenfomal� �ocuklarda, 12 ya� ve �zeri �ocuklarda g�nl�k 329 mg/m doz ve 3 ya� ila 12 ya� alt� �ocuklarda g�nl�k 440 mg/m doz uygulanan ibrutinib maruziyetlerinin genellikle g�nl�k 560 mg doz uygulanan yeti�kin hastalarda g�zlenen maruziyet aral��� i�inde oldu�unu g�stermektedir.
Cinsiyet:
Pop�lasyon farmakokineti�i cinsiyetin, dola��mdan ibrutinib klerensini anlaml� d�zeyde etkilemedi�ini g�stermi�tir.
Irk:
Irk�n ibrutinib farmakokineti�i �zerindeki potansiyel etkisini kar��la�t�rmak i�in yeterli bir veri yoktur.
V�cut a��rl���:
Pop�lasyon farmakokineti�i verileri v�cut a��rl���n�n (aral�k: 41-146 kg; ortalama [standart sapma]: 83 [19 kg]) ibrutinib klerensi �zerinde ihmal edilebilir bir etkiye sahip oldu�unu g�stermi�tir.
B�brek yetmezli�i:
�brutinib minimal bir renal klerense sahiptir; metabolitlerin �riner yolla at�lan miktar� dozun
<%10'udur. Bug�ne kadar b�brek fonksiyon bozuklu�u olan hastalarda spesifik �al��malar yap�lmam��t�r. A��r b�brek yetmezli�i olan hastalara veya diyaliz hastalar�na ait hi�bir veri mevcut de�ildir (bkz. B�l�m 4.2).
Karaci�er yetmezli�i:
�brutinib karaci�erde metabolize olur. Kanserli olmayan hastalarda, a�l�k ko�ullar�nda 140 mg IMBRUVICA'n�n verildi�i bir karaci�er yetmezli�i �al��mas� y�r�t�lm��t�r. Hafif (n=6, Child Pugh S�n�f A), orta (n=10, Child Pugh S�n�f B) ve �iddetli (n=8, Child Pugh S�n�f C) karaci�er yetmezli�i olan hastalarda bozulmu� karaci�er fonksiyonu bireyler aras�nda �nemli �l��de de�i�mekte olup, ortalama olarak ibrutinib maruziyetinde (EAA) s�ras�yla 2,7-, 8,2- ve 9,8-kat bir art�� g�zlenmi�tir. Ayr�ca ibrutinibin serbest fraksiyonu da yetmezli�in derecesi artt�k�a y�kselmi�tir; bu �al��mada benzer �zelliklere sahip sa�l�kl� kontrollerde plazmadaki
%3,3 oran�na k�yasla hafif, orta ve �iddetli karaci�er yetmezli�i olan g�n�ll�lerde s�ras�yla
%3, 3,8 ve 4,8 oranlar� g�r�lm��t�r. Ba�l� olmayan ibrutinib maruziyetindeki (EAA) ilgili art���n hafif, orta ve �iddetli karaci�er yetmezli�i olan g�n�ll�lerde s�ras�yla 4,1-, 9,8- ve 13-kat oldu�u hesaplanmaktad�r (bkz. B�l�m 4.2).
CYP substratlar� ile e� zamanl� kullan�m:
In vitro �al��malar ibrutinibin CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 ve intestinal (ancak hepatik olmayan) CYP3A4'�n zay�f bir reversibl inhibit�r� oldu�unu ve CYP1A2,
CYP2B6, CYP2C8, CYP2C9,CYP2C19veCYP2D6'n�nzam
ana ba�l� klinik olarak anlaml�
![]()
inhibisyonunu sergilemedi�ini g�stermi�tir. �brutinibin dihidrodiol metaboliti CYP2B6,
CYP2C8, CYP2C9 ve CYP2D6'n�n zay�f bir inhibit�r�d�r. Dihidrodiol metaboliti in vitro ortamda en zay�f CYP450 izoenzim ind�kleyicidir. Ibrutinib'in hassas bir CYP3A4 substrat� olmas� sebebiyle, kendi maruziyeti �zerinde klinik olarak ili�kili bir etkisi bulunmamaktad�r.
Transport substratlar�/inhibit�rleri ile e� zamanl� kullan�m:
In vitro �al��malar ibrutinibin ne P glikoproteinin ne de OCT2 hari� di�er maj�r ta��y�c�lar�n bir substrat� olmad���n� g�stermi�tir. Dihidrodiol metaboliti ve di�er metabolitler P glikoprotein substrat�d�r. �brutinib, P glikoproteinin ve BCRP'nin in vitro inhibit�r�d�r (bkz. B�l�m 4.5).
5.3. Klinik �ncesi g�venlilik verileri
A�a��daki advers etkiler s��anlar ve k�peklerde yap�lan 13 haftal�k �al��malarda g�r�lm��t�r. �brutinibin her iki t�rde de 30 mg/kg/g�n'l�k bir Yan Etki G�zlenmeyen Seviye (No Observed Adverse Effect Level; NOAEL) dozunda s��anlarda ve k�peklerde gastrointestinal etkileri (yumu�ak fe�es/diyare ve/veya enflamasyon) ve lenfoid t�kenmesini ind�kledi�i saptanm��t�r. 560 mg/g�n klinik dozundaki ortalama maruziyete dayal� olarak, EAA oranlar� NOAEL'de erkek ve di�i s��anlarda s�ras�yla 2,6 ve 21 ile erkek ve di�i k�peklerde s�ras�yla 0,4 ve 1,8 saptanm��t�r. G�zlenen En D���k Etki Seviyesi (Lowest Observed Effect Level; LOEL) (60 mg/kg/g�n) marjinleri k�peklerde 3,6 (erkekler) ve 2,3 mislidir (di�i). S��anlarda, orta dereceli pankreatik asiner h�cre atrofisi (advers kabul edilir) erkek s��anlarda 100 mg/kg ve �zerindeki dozlarda (EAA maruziyet marjini: 2,6 kat) g�zlenirken, 300 mg/kg/g�n'e kadar dozlarda di�i s��anlarda g�r�lmemi�tir (EAA maruziyet marjini: 21,3 kat). > 100 mg/kg/g�n (EAA maruziyet marjini: 20,3 kat) verilen di�i s��anlarda hafif d�zeyde azalm�� trabek�ler ve kortikal kemik g�r�lm��t�r. T�m gastrointestinal, lenfoid ve kemik bulgular� 6 ila 13 haftal�k s�re�ler sonunda iyile�mi�tir. Pankreatik bulgular benzer geri d�n���m s�re�lerinde k�smen d�zelmi�tir.
Juvenil toksisite �al��malar� yap�lmam��t�r.
Karsinojenisite/genotoksisite
Transjenik farelerde (Tg.rasH2) 2000 mg/kg/g�n'e kadar oral dozlarla y�r�t�len 6 ayl�k bir �al��mada, ibrutinib karsinojenisite g�stermemi�tir. Bu, insanlarda 560 mg/g�n olarak uygulanan doz ile kar��la�t�r�ld���nda, EAA bak�m�ndan erkeklerde yakla��k 23 kat, kad�nlarda ise yakla��k 37 kat ibrutinib maruziyetine kar��l�k gelmektedir.
�brutinib bakterilerde, memeli h�crelerinde ya da farelerde test edildi�inde genotoksik �zellikler sergilememi�tir.
�reme toksisitesi
�brutinib gebe s��anlara 80 mg/kg/g�n dozunda verildi�inde, g�nde 560 mg doz uygulanan hastalardaki maruziyetin (EAA) yakla��k 14 kat� bir maruziyet marjini ile visseral malformasyonlar (kalp ve ana damarlar) ve iskeletsel de�i�imler ile artm�� bir post- implantasyon kayb�yla ili�kilendirilmi�tir. Hayvanlarda uygulanan ≥40 mg/kg/g�n dozunda, ibrutinib (g�nde 560 mg doz uygulanan hastalara k�yasla, EAA oran� ≥5,6) azalm�� fetus a��rl�klar�na neden olmu�tur. Sonu�ta, f�tal NOAEL 10 mg/kg/g�n olarak belirlenmi�tir (g�nde 560 mg dozuna k�yasla yakla��k 1,3 kat� EAA) (bkz. B�l�m 4.6).
![]()
KLL ve WM hastalar�nda maruziyetin (EAA) yakla��k 2,8 kat�) uygulanan tav�anlarda
malformasyonlara sebep olmu�tur. Sonu�ta, f�tal NOAEL 5 mg/kg/g�n olarak belirlenmi�tir (g�nde 560 mg dozuna k�yasla yakla��k 0,7 kat� EAA) (bkz. B�l�m 4.6).
Fertilite
Erkek ya da di�i s��anlarda test edilen maksimum doza kadar (100 mg/kg/g�n – insana e�de�er doz 16 mg/kg/g�n) fertilite ya da �reme kapasitesine bir etki g�r�lmemi�tir.
6. FARMAS�T�K �ZELL�KLER
6.1. Yard�mc� maddelerin listesi
Tablet �ekirde�i Kolloidal susuz silika Kroskarmelloz sodyum
Laktoz monohidrat (s���r s�t� kaynakl�) Magnezyum stearat
Mikrokristalin sel�loz Povidon
Sodyum lauril s�lfat (E487)
Film kaplama Makrogol Polivinil alkol
Titanyum dioksit (E171) Siyah demir oksit (E172) Sar� demir oksit (E172) Talk
6.2. Ge�imsizlikler
Ge�erli de�il.
6.3. Raf �mr�
24 ay.
6.4. Saklamaya y�nelik �zel tedbirler
30 °C alt�ndaki oda s�cakl���nda saklay�n�z.
6.5. Ambalaj�n niteli�i ve i�eri�i
Her bir karton c�zdanda poliklorotrifloroetilen (PCTFE) ile lamine edilmi� polivinil klor�r (PVC)/al�minyum blister ile 10 film kapl� tablet i�erir. Her kartonda (30 film kapl� tablet) 3 c�zdan bulunur.
6.6. Be�eri t�bbi �r�nden arta kalan maddelerin imhas� ve di�er �zel �nlemler
Kullan�lmam�� olan �r�nler ya da at�k materyaller “T�bbi At�klar�n Kontrol� Y�netmeli�i” ve “Ambalaj At�klar�n�n Kontrol� Y�netmelik”lerine uygun olarak imha edilmelidir.
 Grip, So�uk Alg�nl��� ve �ks�r�k
Grip ve so�uk alg�nl��� (nezle) semptomlar� aras�ndaki fark� bilmek �nemlidir. So�uk alg�nl��� gripten daha hafif belirtiler g�steren bir solunum yolu hastal���d�r.
Grip, So�uk Alg�nl��� ve �ks�r�k
Grip ve so�uk alg�nl��� (nezle) semptomlar� aras�ndaki fark� bilmek �nemlidir. So�uk alg�nl��� gripten daha hafif belirtiler g�steren bir solunum yolu hastal���d�r. |
 Deri Kanseri
Deri kanseri �ok rastlanan bir hastal�kt�r. �� ana t�r� bulunur ;genelde kemirici �lser olarak bilinen bazal h�creli karsinom, yass� h�creli karsinom ve k�t� huylu t�m�r.
Deri Kanseri
Deri kanseri �ok rastlanan bir hastal�kt�r. �� ana t�r� bulunur ;genelde kemirici �lser olarak bilinen bazal h�creli karsinom, yass� h�creli karsinom ve k�t� huylu t�m�r. |
 |
Mide Kanseri Mide kanseri genellikle mideyi t�m�yle kaplayan ve mukus �retmekle g�revli h�crelerde ba�lar. Bu kanser tipine adenokarsinom denir. |
 |
Parkinson Hastal��� Hastal�k ilk kez 1817 de �ngiliz doktor James Parkinson taraf�ndan tan�mlanm�� ve Dr. Parkinson hastal��� “sallay�c� fel�” olarak kaleme alm��. |
 |
Depresyonu Anlamak Depresyon farkl� ki�ileri farkl� bi�imlerde etkiler. Duygusal veya fiziksel olmak �zere geni� alanda belirtilere sebep olabilir.Depresyona neler sebep olur? |