JAKAVI 20 mg 56 tablet K�sa �r�n Bilgisi
{ Ruksolitinib }
1. BE�ER� TIBB� �R�N�N ADI
JAKAVI 20 mg Tablet
2. KAL�TAT�F VE KANT�TAT�F B�LE��M
Etkin madde
Ruksolitinib fosfat 26,4 mg (20 mg ruksolitinib serbest baz�na kar��l�k gelir)
Yard�mc� maddeler
Laktoz monohidrat (s���r kaynakl�) 285,8 mg
Yard�mc� maddeler i�in b�l�m 6.1'e bak�n�z.
3. FARMAS�T�K FORMU
Bir y�z�nde “NVR” ve di�er y�z�nde “L20” yazan, uzun ve e�ik kenarl�, beyaz veya beyaza
yak�n tabletler
Hidroksi�re ili�kili bacak �lseri, kontrol edilemeyen mukokutan�z belirtiler ortaya ��kmas�
4. KL�N�K �ZELL�KLER
4.1. Terap�tik endikasyonlar
Miyelofibrozis (MF):
JAKAVI, en az bir seri tedaviye yan�ts�z, DIPSS plus skoru orta/y�ksek olup, kemik ili�i nakline uygun olmayan primer miyelofibrozis, post polistemik miyelofibrozis veya esansiyel trombositemi sonras� ikincil miyelofibrozis tan�l� hastalarda splenomegaliye ba�l� semptomlar�n tedavisinde endikedir.
Polisitemi vera (PV):
En az 2 g/g�n veya hastan�n tolere edebilece�i maksimum dozda hidroksi�re kullanan polistemi vera tan�l� hastalarda en az 3 ayl�k tedaviye ra�men
Hematokriti <%45 tutmak i�in ayda 1'den fazla flebotomi ihtiyac�n�n devam etmesi
veya
4.2. Pozoloji ve uygulama �ekli
Anti-kanser ila�lar�n uygulanmas�nda deneyimli hekimler taraf�ndan tedavi yap�lmal�d�r.
JAKAVI ile tedaviye ba�lanmadan �nce beyaz k�re say�s� dahil tam kan say�m� ger�ekle�tirilmelidir.
Beyaz k�re say�s� dahil, tam kan say�mlar�, JAKAVI dozlar� stabilize olana kadar 2 ila 4 haftada bir, daha sonra klinik durumun gerektirdi�i �ekilde izlenmelidir (bkz. B�l�m 4.4).
Pozoloji:
Ba�lang�� dozu:
Ruksolitinibin MF'de �nerilen ba�lang�� dozu, trombosit say�lar�na g�re Tablo 1'de verilmektedir:
Tablo 1 Miyelofibrozis'te ba�lang�� dozu
Trombosit say�m� | Ba�lang�� dozu |
200.000/mm'ten b�y�k | G�nde iki kez a��z yoluyla 20 mg |
100.000 ile 200.000/mm aras�nda | G�nde iki kez a��z yoluyla 15 mg |
75.000 ile 100.000/mm aras�nda | G�nde iki kez a��z yoluyla 10 mg |
50.000 ile 75.000/mm aras�nda | G�nde iki kez a��z yoluyla 5 mg |
Ruksolitinibin PV'de �nerilen ba�lang�� dozu g�nde iki kez a��z yoluyla 10 mg'd�r.
Akut ve kronik Graft versus Host hastal���nda (GvHD) �nerilen JAKAVI ba�lang�� dozu, g�nde iki kez oral yolla verilen 10 mg'd�r. JAKAVI, kortikosteroidlerin ve/veya kalsin�rin inhibit�rlerinin (CNI'ler) s�rekli kullan�m�na eklenebilir.
Doz d�zenlemeleri:
Dozlar, g�venlilik ve etkililik esas al�narak titre edilebilir.
Miyelofibroz ve polistemi vera
E�er etkililik yetersiz g�r�l�rse ve kan say�m� de�erleri yeterli ise; dozlar, g�nde iki kere en
fazla 5 mg'a kadar artt�r�labilir. Maksimum doz g�nde iki kere 25 mg'� ge�memelidir.
Ba�lang�� dozu, tedavinin ilk 4 haftas�nda ve sonras�nda 2 haftada birden daha s�k aral�klarla artt�r�lmamal�d�r.
50.000/mm3'ten d���k trombosit say�mlar� ya da 500/mm3'ten d���k mutlak n�trofil say�mlar� durumlar�nda tedaviye ara verilmelidir. PV'de, hemoglobin d�zeyi 8 g/dL'nin alt�na d��t���nde tedaviye ara verilmelidir. Kan de�erleri bu d�zeylerin �zerine geri d�nd���nde tedaviye g�nde iki kez 5 mg ile tekrar ba�lanabilir ve beyaz k�re say�s� dahil, tam kan say�mlar�n�n dikkatli izlemi esas al�narak a�amal� olarak art�r�labilir.
Trombositopeni i�in doz kesintilerini �nlemek amac�yla, Tablo 2'de �zetlendi�i gibi tedavi s�ras�nda trombosit say�s� azal�rsa,dozazalt�m�d���n�lmelidir.
Tablo 2 Miyelofibroz hastalar�nda trombositopeni i�in doz �nerileri
| Trombosit azalmas� esnas�ndaki doz | ||||
| G�nde iki kez 25 mg | G�nde iki kez 20 mg | G�nde iki kez 15 mg | G�nde iki kez 10 mg | G�nde iki kez 5 mg |
Trombosit say�s� | Yeni doz | ||||
100.000 ile 125.000/ mm aras� | G�nde iki kez 20 mg | G�nde iki kez 15 mg | De�i�iklik yok | De�i�iklik yok | De�i�iklik yok |
75.000 ile 100.000/ mm aras� | G�nde iki kez 10 mg | G�nde iki kez 10 mg | G�nde iki kez 10 mg | De�i�iklik yok | De�i�iklik yok |
50.000 ile 75.000/mm aras� | G�nde iki kez 5 mg | G�nde iki kez 5 mg | G�nde iki kez 5 mg | G�nde iki kez 5 mg | De�i�iklik yok |
50.000/mm'ten az | Ara verilmeli | Ara verilmeli | Ara verilmeli | Ara verilmeli | Ara verilmeli |
PV'de hemoglobin d�zeyi 12 g/dL'nin alt�na d��t���nde de dozun azalt�lmas� d���n�lmelidir, 10 g/dL'nin alt�na d��t���nde ise dozun azalt�lmas� �nerilir.
Graft versus Host hastal���
B�y�me fakt�rleri, anti-enfektif tedaviler ve transf�zyonlar dahil olmak �zere standart destekleyici tedaviden sonra trombositopeni, n�tropeni veya total bilirubin y�ksekli�i olan GvHD hastalar�nda dozun azalt�lmas� ve tedavinin ge�ici olarak kesilmesi gerekebilir. Bir doz d�zeyinde azaltma ad�m� �nerilir (g�nde iki kez 10 mg'dan g�nde iki kez 5 mg'a veya g�nde iki kez 5 mg'dan g�nde bir kez 5 mg'a). G�nde tek doz 5 mg JAKAVI'yi tolere edemeyen hastalarda tedaviye ara verilmelidir. Ayr�nt�l� doz �nerileri Tablo 3'te verilmi�tir.
Tablo 3 Trombositopeni, n�tropeni veya total bilirubin y�ksekli�i olan GvHD hastalar� i�in ruksolitinib tedavisi s�ras�nda doz �nerileri
Laboratuvar parametresi | Doz �nerisi |
Trombosit say�s� <20.000/mm | JAKAVI'yi bir doz seviyesinde azalt�n�z. Yedi g�n i�inde trombosit say�s� ≥20.000/mm olursa, doz ba�lang�� doz d�zeyine y�kseltilebilir; aksi takdirde d���k dozdan devam ediniz. |
Trombosit say�s� <15.000/mm | Trombosit say�s� ≥20.000/mm olana kadar JAKAVI'yi durdurunuz, ard�ndan bir d���k doz seviyesinden tedaviye devam ediniz. |
Mutlak n�trofil say�s� (ANC) ≥500/mm ile <750/mm | JAKAVI'yi bir doz seviyesinde azalt�n�z.Mutlak n�trofil say�s� (ANC) >1.000/mm ise ba�lang�� doz seviyesinde devam ediniz. |
Mutlak n�trofil say�s� <500/mm | Mutlak n�trofil say�s� (ANC)>500/mm olana kadar |
| seviyesinden tedaviye devam ediniz. Mutlak n�trofil say�s� (ANC) >1.000/mm ise, dozlamaya ba�lang�� doz seviyesinde devam edilebilir. |
GvHD'nin neden olmad��� toplam bilirubin y�ksekli�i (karaci�er GvHD yoksa) | Normalin �st s�n�r�n�n (ULN) >3.0 ila 5.0 kat�: ULN'nin ≤3.0 kat� olana kadar JAKAVI'ye bir d���k doz seviyesinde devam ediniz. |
Normalin �st s�n�r�n�n (ULN)>5.0 ila 10.0 kat�: Toplam bilirubin ≤3.0 x ULN olana kadar JAKAVI'yi 14 g�ne kadar durdurunuz. Toplam bilirubin ≤3.0 x ULN ise dozlamaya mevcut dozda devam edilebilir. 14 g�n sonra ≤3.0 x ULN de�ilse, bir d���k doz seviyesinde devam ediniz. | |
Normalin �st s�n�r�n�n (ULN) >10.0 kat�: Toplam bilirubin ULN'nin ≤3.0 kat� olana kadar JAKAVI'yi durdurunuz, ard�ndan bir d���k doz seviyesinde devam ediniz. | |
GvHD'nin (karaci�er GvHD) neden oldu�u toplam bilirubin y�ksekli�i | Normalin �st s�n�r�n�n (ULN)>3.0 kat�: Toplam bilirubin ULN'nin ≤ 3.0 kat� olana kadar JAKAVI'ye bir d���k doz seviyesinde devam ediniz. |
E�zamanl� g��l� CYP3A4 inhibit�rleri veya flukonazol ile doz ayarlamas�:
Ruksolitinib, g��l� CYP3A4 inhibit�rleri veya hem CYP2C9 ve hem de CYP3A4 enzimlerinin ikili inhibit�rleri (�rn; flukonazol) ile birlikte uygulan�rken ruksolitinibin toplam g�nl�k dozu g�nde iki kez uygulamak �zere yakla��k %50 azalt�lmal�d�r (bkz. B�l�m 4.5). Ruksolitinibin g�nde 200 mg'dan fazla flukonazol dozlar�yla birlikte kullan�lmas�ndan ka��n�lmal�d�r.
G��l� bir CYP3A4 inhibit�r�ne veya CYP2C9 ve CYP3A4 enzimlerinin ikili inhibit�rlerine (�rn; flukonazol) ba�lan�rken hematolojik parametrelerin ve ruksolitinib ili�kili advers reaksiyonlar�n klinik i�aret ve semptomlar�n�n daha s�k izlemi (�rn., haftada iki kez) �nerilir.
Uygulama s�kl��� ve s�resi:
Bir doz atlan�rsa, hasta ek bir doz almamal�, re�etede belirtilen bir sonraki dozunu almal�d�r.
Uygulama �ekli:
JAKAVI, oral yolla uygulan�r, a� veya tok karn�na al�nabilir.
Tedavinin b�rak�lmas�
MF ve PV tedavisi, fayda-risk pozitif kald��� s�rece s�rd�r�lebilir. Bununla birlikte 6 aydan sonra dalak boyutunda bir azalma yoksa veya semptomlarda tedavinin ba�lang�c�ndan beri bir iyile�me g�r�lmemi�se tedavi b�rak�lmal�d�r.
Bir derece klinik iyile�me g�stermi� hastalar i�in, dalak boyutunda ba�lang�� boyutuna g�re
%40 art�� korunmu�sa (dalak hacminde kabaca %25 art��a e�de�er) ve hastal�kla ili�kili semptomlarda dikkat �ekici bir iyile�me g�r�lm�yorsa ruksolitinib tedavisinin b�rak�lmas� �nerilmektedir.
GvHD'de, yan�t veren hastalarda ve kortikosteroid tedavisi kesildikten sonra JAKAVI'nin a�amal� olarak azalt�lmas� d���n�lebilir. JAKAVI dozunun iki ayda bir %50 azalt�lmas� �nerilir. JAKAVI'nin dozunun azalt�lmas� s�ras�nda veya sonras�nda GvHD'nin belirti veya semptomlar� tekrar ortaya ��karsa, tedavi dozunun yeniden y�kseltilmesi d���n�lmelidir.
�zel pop�lasyonlara ili�kin ek bilgiler:
B�brek yetmezli�i:
Hafif ila orta derecedeki b�brek yetmezli�i olan hastalarda spesifik doz ayarlamas� gerekli
de�ildir.
�iddetli b�brek yetmezli�i olan hastalarda (kreatinin klerensi (KLkr) < 30mL/dk), trombosit say�s�na ba�l� olarak �nerilen ba�lang�� dozu MF hastalar� i�in g�nde iki kez uygulamak �zere yakla��k %50 azalt�lmal�d�r. �iddetli b�brek yetmezli�i olan PV ve GvHD hastalar�nda �nerilen ba�lang�� dozu g�nde iki kez 5 mg'd�r. Ruksolitinib tedavisi s�ras�nda hastalar etkililik ve g�venlilik a��s�ndan dikkatle izlenmelidir.
Diyalize giren son d�nem b�brek yetmezli�i (SDBY) olan hastalar i�in en iyi doz se�ene�ine karar verebilmek a��s�ndan s�n�rl� veri mevcuttur. Bu pop�lasyonda mevcut verilere dayal� farmakokinetik/farmakodinamik sim�lasyonlar, hemodiyalize giren SDBY'li MF hastalar�nda ba�lang�� dozunun, diyalizden sonra ve sadece hemodiyaliz g�n�nde uygulanmak �zere 15-20 mg'l�k tek doz ya da 12 saat arayla verilen 10 mg'l�k iki doz oldu�unu d���nd�rmektedir. Trombosit say�lar� 100.000/mm3 ile 200.000/mm3 aras�nda olan MF hastalar�nda 15 mg'l�k tek doz �nerilmektedir. Trombosit say�lar� >200.000/mm3 olan MF hastalar�nda 20 mg'l�k tek doz ya da 12 saat arayla verilen 10 mg'l�k iki doz �nerilmektedir. Sonraki dozlar (tek uygulama ya da 12 saat arayla verilen 10 mg'l�k iki doz) hemodiyaliz g�nlerinde her diyaliz seans�ndan sonra uygulanmal�d�r.
Diyalize giren SDBY'li PV hastalar�nda �nerilen ba�lang�� dozu 10 mg'l�k tek doz ya da 12 saat arayla verilen 5 mg'l�k iki doz olup diyaliz sonras�nda ve sadece hemodiyaliz g�n�nde uygulanmal�d�r. Bu doz �nerileri sim�lasyonlara dayanmaktad�r ve SDBY'de yap�lacak herhangi bir doz d�zenlenmesi her hastada g�venlilik ve etkililik a��s�ndan dikkatle izlenmelidir. Peritoneal diyalize ya da s�rekli venoven�z hemofiltrasyona giren hastalarda doz uygulamas�na ili�kin veri bulunmamaktad�r (bkz. B�l�m 5.2).
Son d�nem b�brek yetmezli�i (SDBY) olan GvHD hastalar� i�in veri bulunmamaktad�r.
Karaci�er yetmezli�i:
Herhangi bir d�zeyde karaci�er yetmezli�i olan MF hastalar�nda, trombosit say�m�na dayal� olan �nerilen ba�lang�� dozu g�nde iki kez uygulamak �zere yakla��k %50 azalt�lmal�d�r. Ard���k dozlar dikkatli g�venlilik ve etkililik takibi temelinde ayarlanmal�d�r. PV hastalar� i�in �nerilen ba�lang�� dozu g�nde iki kez 5 mg'd�r. Ruksolitinib kullan�rken karaci�er bozuklu�u tan�s� alan hastalarda, diferansiyel beyaz kan h�cresi say�m� dahil tam kan say�mlar� JAKAVI ile tedavi ba�lat�ld�ktan sonra ilk 6 hafta boyunca en az 1-2 haftada bir ve ard�ndan karaci�er fonksiyon testleri ve kan say�mlar� stabil seyretti�inde klinik durumun gerektirdi�i �ekilde takip edilmelidir. Ruksolitinib dozu sitopeni riskini azaltmak amac�yla titre edilebilir.
GvHD ile ili�kili olmayan hafif, orta veya �iddetli karaci�er yetmezli�i olan hastalarda, ruksolitinibin ba�lang�� dozu %50 oran�nda azalt�lmal�d�r (bkz. B�l�m 5.2).
GvHD karaci�er tutulumu olan ve toplam bilirubinin normalin �st s�n�r�n�n (ULN) >3 kat�na y�kseldi�i hastalarda, toksisite a��s�ndan kan say�mlar� daha s�k izlenmelidir ve dozun bir doz d�zeyinde azalt�lmas� �nerilir.
Pediyatrik pop�lasyon:
JAKAVI'nin MF ve PV'si olan �ocuklarda ve 18 ya�a kadar ergenlerde g�venlili�i ve etkilili�i saptanmam��t�r. Veribulunmamaktad�r (bkz. B�l�m5.1)
GvHD'li pediyatrik hastalarda (12 ya� ve �zeri) JAKAVI'nin g�venlili�i ve etkilili�i, randomize faz 3 �al��malar olan REACH2 ve REACH3'ten elde edilen kan�tlarla desteklenmektedir. 12 ya� ve �zeri GvHD'li pediyatrik hastalarda JAKAVI dozu, yeti�kinlerdeki ile ayn�d�r. JAKAVI'nin g�venlili�i ve etkilili�i 12 ya��ndan k���k hastalarda belirlenmemi�tir.
Geriyatrik pop�lasyon (≥65 ya�):
Ya�l� hastalar i�in ek olarak doz ayarlamas� gerekmemektedir.
4.3. Kontrendikasyonlar
Etkin madde
hastalarda kontrendikedir.
Gebelikte ve emziren annelerde kontrendikedir.
4.4. �zel kullan�m uyar�lar� ve �nlemleri
Miyelosupresyon
JAKAVI tedavisi, trombositopeni, anemi ve n�tropeni dahil hematolojik advers olaylara neden olabilir. JAKAVI ile tedavi ba�lat�lmadan �nce beyaz k�re say�s� dahil tam kan say�m� yap�lmal�d�r . Trombosit say�m� 50.000/mm3'ten d���k olan veya mutlak n�trofil say�m� 500/mm3'ten d���k olan MF'li hastalarda tedavi b�rak�lmal�d�r (bkz. B�l�m 4.2).
Tedavi ba�lang�c�nda trombosit say�s� d���k olan (<200.000/mm3) MF'li hastalarda tedavi
s�ras�nda trombositopeni geli�me olas�l���n�n daha y�ksek oldu�u g�zlenmi�tir.
Trombositopeni �o�u durumda geri d�n��l� olmu� ve genellikle dozun azalt�lmas� ya da JAKAVI tedavisine ge�ici olarak ara verilmesiyle kontrol edilebilmi�tir (bkz. B�l�m 4.2 ve B�l�m 4.8). Ancak, klinik gereklili�e g�re trombosit transf�zyonlar� gerekli olabilir.
Anemi geli�tiren hastalarda kan nakli gerekebilir. Anemi geli�tiren hastalarda doz d�zenlemeleri ya da tedaviye ara verilmesi de d���n�lebilir.
Hemoglobin d�zeyi tedavinin ba�lang�c�nda 10 g/dL'nin alt�nda olan hastalar, daha y�ksek ba�lang�� hemoglobin d�zeyine sahip hastalara k�yasla 8 g/ dL'lik bir hemoglobin d�zeyi geli�mesi a��s�ndan daha y�ksek risk ta��r (%30,1'e kar�� %79,3). Ba�lang�� hemoglobini 10 g/ dL olan hastalar da hematoloji parametreleri ve JAKAVI ile ili�kili advers ila� reaksiyonlar�n�n klinik belirti ve semptomlar�na ili�kin daha s�k takip �nerilmektedir.
N�tropeni (Mutlak N�trofil Say�m� (MNS) <500/mm3) genellikle geri d�n��l� olmu�tur ve JAKAVI tedavisine ge�ici olarak ara verilmesiyle kontrol edilebilmi�tir (bkz. B�l�m 4.2 ve B�l�m 4.8).
Klinik duruma g�re beyaz k�re say�s� dahil tam kan say�m� izlemi ve gerekti�i takdirde doz ayarlamas� yap�lmal�d�r (bkz. B�l�m 4.2 ve B�l�m 4.8).
Enfeksiyonlar
JAKAVI ile tedavi edilen hastalarda ciddi bakteriyel, mikobakteriyel, fungal, viral ve f�rsat�� enfeksiyonlar olu�mu�tur. Hastalar ciddi enfeksiyon geli�me riski a��s�ndan de�erlendirilmelidir. Hekimler, JAKAVI almakta olan hastalar�, enfeksiyon belirti ve semptomlar� a��s�ndan dikkatle g�zlemlemeli ve gerekti�inde tedaviyi h�zla ba�latmal�d�r. Aktif ciddi enfeksiyonlar giderilene kadar JAKAVI tedavisi ba�lanmamal�d�r.
JAKAVI alan hastalarda t�berk�loz bildirilmi�tir. Tedaviye ba�lamadan �nce hastalar aktif ve
inaktif (“latent”) t�berk�loza��s�ndanyerelgerekliliklereg�re de�erlendirilmelidir. Bu t�bbi
![]()
ve/veya interferon-gama sal�verilme analizi gibi uygun bir taramay� i�erebilir. Re�ete yazan hekimlere �zellikle a��r hasta olan veya imm�nitesi zay�flam�� hastalarda yalanc� negatif t�berk�lin deri testi bulgular� olabilece�i hat�rlat�lmal�d�r.
JAKAVI alan kronik HBV enfeksiyonu olan hastalarda alanin aminotransferaz ve aspartat aminotransferaz de�erlerinde art�� ile ili�kili olsun veya olmas�n Hepatit B viral y�k�nde (HBV-DNA titresi), art��lar bildirilmi�tir. JAKAVI ile tedaviye ba�lamadan �nce HBV taramas� yap�lmas� �nerilir. Kronik HBV enfeksiyonu olan hastalar klinik k�lavuzlara g�re tedavi edilmeli ve izlenmelidir.
Herpes Zoster
Hekimler, herpes zoster i�aret ve semptomlar� hakk�nda hastalar� e�itmeli, m�mk�n oldu�unca k�sa s�rede tedavi i�in ba�vurmalar�n� tavsiye etmelidir.
Progresif multifokal l�koensefalopati
JAKAVI tedavisinde progresif multifokal l�koensefalopati (PML) bildirilmi�tir. Hekimler, hastalar�n fark edemeyebilece�i, PML'ye i�aret eden semptomlar (�rn., kognitif, n�rolojik ya da psikiyatrik semptomlar veya belirtiler) konusunda �zellikle dikkatli olmal�d�r. Hastalar bu yeni ya da a��rla�an semptomlar ya da belirtiler i�in takip edilmeli ve bu tip semptomlar/belirtiler meydana gelirse, bir n�roloji uzman�na sevk edilmeli ve PML i�in uygun tan�sal de�erlendirmeler d���n�lmelidir. PML'den ��phelenilirse, tan� d��lan�ncaya kadar ila� verilmemelidir.
Melanom d��� deri kanseri
Ruksolitinib ile tedavi edilen hastalarda bazal h�cre, skuam�z h�cre ve Merkel h�creli karsinom dahil olmak �zere melanom d��� deri kanserleri (MDDK) bildirilmi�tir. Bu MF ve PV'li hastalar�n �o�unun �yk�s�nde hidroksi�re ile uzun s�reli tedavi ya da �nceden MDDK veya pre-malign deri lezyonlar� bulunmaktad�r. Ruksolitinib ile nedensellik ili�kisi saptanmam��t�r. Deri kanseri a��s�ndan risk alt�nda olan hastalarda periyodik deri muayenesi �nerilmektedir.
Lipid anormallikleri / y�kselmeleri
JAKAVI ile tedavi total kolesterol, y�ksek yo�unluklu lipoprotein (HDL) kolesterol, d���k yo�unluklu lipoprotein (LDL) kolesterol ve trigliserid gibi lipid parametrelerindeki art��larla ili�kilendirilmi�tir. Lipid takibi ve klinik k�lavuzlara g�re dislipidemi tedavisi �nerilmektedir.
�zel pop�lasyonlar
B�brek yetmezli�i
�iddetli b�brek yetmezli�i olan hastalarda JAKAVI'nin ba�lang�� dozu azalt�lmal�d�r. Hemodiyalize devam eden son d�nem b�brek yetmezli�i olan hastalarda, MF hastalar� i�in ba�lang�� dozunda trombosit say�lar� esas al�nmal�d�r, ancak PV hastalar� i�in �nerilen ba�lang�� dozu tek doz 10 mg'd�r (bkz. B�l�m 4.2). Sonraki dozlar (MF hastalar�nda 20 mg'l�k tek doz veya 12 saat arayla verilen 10 mg'l�k iki doz; PV hastalar�nda 10 mg'l�k tek doz veya 12 saat arayla verilen 5 mg'l�k iki doz) sadece hemodiyaliz g�nlerinde her diyaliz seans�ndan sonra uygulanmal�d�r. G�venlilik ve etkililik dikkatle izlenerek ek doz d�zenlemeleri yap�lmal�d�r (bkz. B�l�m 4.2 ve B�l�m 5.2).
Karaci�er yetmezli�i
Karaci�er yetmezli�i olan MF ve PV hastalar�nda JAKAVI'nin ba�lang�� dozu yakla��k %50 azalt�lmal�d�r. Sonraki doz d�zenlemeleri i�in ilac�n g�venlili�i ve etkilili�i esas al�nmal�d�r.
GvHD ile ili�kili olmayan karaci�er yetmezli�i olan GvHD hastalar�nda, JAKAVI'nin ba�lang�� dozu yakla��k %50 oran�nda azalt�lmal�d�r (bkz. B�l�m 4.2 ve B�l�m 5.2).
Etkile�imler
E�er JAKAVI, g��l� CYP3A4 inhibit�rleri veya hem CYP2C9 ve hem de CYP3A4 enzimlerinin inhibit�rleri (�rn; flukonazol) ile bir arada uygulanacaksa, doz g�nde iki kez uygulanmak �zere yakla��k %50 azalt�lmal�d�r (monitorizasyon s�kl��� i�in bkz. B�l�m 4.2 ve B�l�m 4.5).
Sitored�ktif tedavilerin JAKAVI ile e�zamanl� kullan�m� y�netilebilir sitopeni ile ili�kilidir. (bkz. B�l�m 4.2).
Geri �ekme etkileri:
JAKAVI'nin kesilmesini veya b�rak�lmas�n� takiben, miyelofibrozis semptomlar� yakla��k bir haftal�k bir periyotta geri d�nebilir. �zellikle akut araya giren hastal�k varl���nda, daha a��r semptomlar� olan hastalar�n JAKAVI'yi kullanmay� b�rakt��� bildirilmi�tir. JAKAVI'nin aniden b�rak�lmas�n�n bu olaylara katk�da bulunup bulunmad��� belirlenmemi�tir. Aniden b�rakma gerekli olmad�k�a, her ne kadar azaltman�n faydas� kan�tlanmam�� olsa da JAKAVI dozunun kademeli olarak azalt�lmas� d���n�lmelidir.
Yard�mc� maddeler
JAKAVI laktoz i�erir. Nadir kal�t�msal galaktoz intolerans�, Lapp laktaz eksikli�i veya glukoz-galaktoz malabsorpsiyonu olan hastalar bu t�bbi �r�n� kullanmamal�d�r.
4.5. Di�er t�bbi �r�nler ile etkile�imler ve di�er etkile�im �ekilleri
Etkile�im �al��malar� sadece eri�kinlerde y�r�t�lm��t�r.
Ruksolitinib CYP3A4 ve CYP2C9 ile katalizlenen metabolizma ile elimine olur. B�ylece bu
enzimleri inhibe eden ila�lar artm�� ruksolitinib maruziyetine yol a�abilir. Ruksolitinib dozunun azalt�lmas�n� gerektiren etkile�imler
CYP3A4 inhibit�rleri
G��l� CYP3A4 inhibit�rleri (boceprevir, klaritromisin, indinavir, itrakonazol, ketokonazol, lopinavir/ritonavir, ritonavir, mibefradil, nefazodon, nelfinavir, posakonazol, sakinavir, telaprevir, telitromisin, vorikonazol gibi, ancak bunlarla s�n�rl� olmayan) Sa�l�kl� g�n�ll�lerde ruksolitinibin (10 mg'l�k tek doz) g��l� CYP3A4 inhibit�r� ketokonazol ile bir arada uygulanmas�, tek ba��na ruksolitinib ile kar��la�t�r�ld���nda s�ras�yla %33 ve %91 daha y�ksek ruksolitinib Cve EAA de�erleri ile sonu�lanm��t�r. E�zamanl� ketokonazol uygulamas� ile yar�lanma �mr� 3,7 saatten 6 saate uzam��t�r.
Ruksolitinib, g��l� CYP3A4 inhibit�rleri ile bir arada uygulan�rken, ruksolitinibin toplam g�nl�k dozu g�nde iki kez uygulanmak �zere yakla��k %50 azalt�lmal�d�r. Hastalar, sitopeniler a��s�ndan yak�ndan izlenmelidir (haftada iki kez) ve doz, g�venlilik ve etkililik esas al�narak titre edilmelidir (bkz. B�l�m 4.2).
Hem CYP2C9 ve hem de CYP3A4 inhibit�rleri
Sa�l�kl� g�n�ll�lerde, tek ba��na ruksolitinib ile kar��la�t�r�ld���nda, ruksolitinibin (10 mg'l�k tek doz) ikili bir CYP2C9 ve CYP3A4 inhibit�r� olan flukonazol ile birlikte uygulanmas�, ruksolitinibin Cve EAA de�erlerinin s�ras�yla % 47 ve % 232 daha y�ksek ��kmas�yla sonu�lanm��t�r.
CYP2C9 ve CYP3A4 enzimlerinin inhibit�rleri olan t�bbi �r�nler (�rn., flukonazol) kullan�ld���nda dozda %50 azaltma d���n�lmelidir. Ruksolitinibin g�nde 200 mg'dan fazla flukonazol dozlar�yla birlikte kullan�lmas�ndan ka��n�lmal�d�r.
Enzim ind�kleyicileri
CYP3A4 ind�kleyicileri (avasimib, karbamazepin, fenobarbital, fenitoin, rifabutin, rifampin (rifampisin), sar� kantaron (Hypericum perforatum) gibi ancak bunlarla s�n�rl� olmayan): Hastalar yak�ndan izlenmeli ve doz g�venlilik ve etkililik temelinde titre edilmelidir (bkz. B�l�m 4.2).
Potent CYP3A4 ind�kleyicisi rifampisini (10 g�n boyunca g�nl�k 600 mg dozda) takiben 50 mg tek doz olarak ruksolitinib verilen sa�l�kl� g�n�ll�lerde, ruksolitinib EAA's� tek ba��na ruksolitinib uygulamas�ndan sonra olandan %70 daha d���k bulunmu�tur. Ruksolitinib aktif metabolitlerinin maruziyeti de�i�memi�tir. Genel olarak ruksolitinib farmakodinamik aktivitesi benzer olup, CYP3A4 ind�ksiyonunun farmakodinamik �zerinde minimum etkisinin oldu�unu d���nd�rmektedir. Bununla birlikte, bu E'a yak�n farmakodinamik etkilerle sonu�lanan y�ksek ruksolitinib dozu ile ilgili olabilir. Bir hastada g��l� bir enzim ind�kleyicisi ile tedavi ba�lat�ld���nda ruksolitinib dozunun art�r�lmas� gerekebilir.
Dikkate al�nmas� gereken di�er etkile�imler
Hafif veya orta kuvvetli CYP3A4 inhibit�rleri (siprofloksasin, eritromisin, amprenavir, atazanavir, diltiazem, simetidin gibi ancak bunlarla s�n�rl� olmayan):
Orta kuvvetli bir CYP3A4 inhibit�r� olan eritromisini d�rt g�n s�reyle g�nde iki kez 500 mg dozunda alan sa�l�kl� g�n�ll�lerde JAKAVI'nin 10 mg'l�k tek dozunun tek ba��na kullan�m�na k�yasla Cde�erinde %8, EAA de�erinde %27 art�� olmu�tur.
JAKAVI, hafif veya orta kuvvetli CYP3A4 inhibit�rleri (�rn, eritromisin) ile bir arada uygulan�rken doz ayarlamas� gerekmemektedir. Orta kuvvetli CYP3A4 inhibit�rleri ile tedavi ba�lat�l�rken hastalar sitopeniler a��s�ndan yak�ndan izlenmelidir.
Di�er ila�lar ile etkile�im:
P-glikoprotein ve di�er ta��y�c�larla ta��nan bile�ikler
Ruksolitinib ba��rsakta meme kanserine diren� proteinini (BCRP) ve P-glikoproteini inhibe edebilir. Bu inhibisyon, debigatran eteksilat, siklosporin, rosuvastatin ve muhtemelen digoksin gibi bu ta��y�c�lar�n substratlar�na sistemik maruziyeti art�rabilir. Etkilenen madde i�in terap�tik ila� takibi veya klinik takip tavsiye edilir.
Ba��rsakta meme kanserine diren� proteini ve P-gp i�in potansiyel inhibisyon, uygulamalar aras�ndaki s�re m�mk�n oldu�unca uzun tutularak en aza indirilebilir.
Sa�l�kl� g�n�ll�lerle ger�ekle�tirilen bir �al��ma ruksolitinibin oral CYP3A4 substrat� midazolam�n metabolizmas�n� inhibe etmedi�ini g�stermi�tir. Bu nedenle, ruksolitinib ile kombine edilen CYP3A4 substratlar�n�n maruziyetinde herhangi bir art�� beklenmemektedir. Sa�l�kl� g�n�ll�lerle ger�ekle�tirilen ba�ka bir �al��ma ruksolitinibin etinil estradiol ve levonorgestrel i�eren bir oral kontraseptifin farmakokineti�ini etkilemedi�ini g�stermi�tir. Dolay�s�yla, e�zamanl� ruksolitinib uygulanmas�yla bu kombinasyonun kontraseptif etkilili�inin olumsuz y�nde etkilenmesi beklenmemektedir.
�zel pop�lasyonlara ili�kin ek bilgiler
B�brek yetmezli�i:
Karaci�er yetmezli�i:
Karaci�er yetmezli�i olan hastalarla ilgili bir etkile�im �al��mas� yap�lmam��t�r. Pediyatrik pop�lasyon:
Pediyatrik hastalarla ilgili bir etkile�im �al��mas� yap�lmam��t�r.
4.6. Gebelik ve laktasyon
Gebelik kategorisi C'dir.
�ocuk do�urma potansiyeli bulunan kad�nlar/Do�um kontrol� (Kontrasepsiyon)
�ocuk do�urma potansiyeli bulunan kad�nlar, JAKAVI ile tedavi s�ras�nda etkili do�um kontrol y�ntemi kullanmal�d�r.
JAKAVI ile tedavi s�ras�nda gebelik olu�ursa, hasta baz�nda fayda-risk de�erlendirmeleri yap�lmal� ve fet�s �zerine potansiyel riskler ile ilgili olarak hasta bilgilendirilmelidir (bkz. B�l�m 5.3).
Gebelik d�nemi
Gebe kad�nlarda JAKAVI ile yeterli �al��ma bulunmamaktad�r.
Hayvanlar �zerinde yap�lan �al��malar ruksolitinibin embriyotoksik ve fetotoksik oldu�unu g�stermi�tir. S��anlarda veya tav�anlarda teratojenisite g�zlenmemi�tir. Bununla birlikte, maruz kalma marjlar� en y�ksek klinik doza k�yasla d���kt�r ve dolay�s�yla sonu�lar�n insanlarla ili�kisi s�n�rl�d�r (bkz. B�l�m 5.3).
�nsanlar i�in potansiyel risk bilinmemektedir. JAKAV� gebelikte kontrendikedir. Gebelik s�resince JAKAVI kullan�lmamal�d�r (bkz. B�l�m 4.3).
Laktasyon d�nemi
Emzirme esnas�nda JAKAVI kullan�lmamal�d�r (bkz. B�l�m 4.3), bu nedenle tedavi ba�lad���nda emzirmeye ara verilmelidir. Ruksolitinib ve/veya metabolitlerinin anne s�t� ile at�l�p at�lmad��� bilinmemektedir. Emzirilen �ocuk i�in risk g�z ard� edilemez. Hayvanlardan elde edilen farmakodinamik/toksikolojik veriler ruksolitinib ve/veya metabolitlerinin anne s�t�ne ge�ti�ini g�stermi�tir.
�reme yetene�i/Fertilite
Ruksolitinibin fertilite �zerindeki etkisine dair insanda veri bulunmamaktad�r. Hayvan �al��malar�nda, fertilite �zerinde herhangi bir etki g�zlenmemi�tir.
4.7. Ara� ve makine kullan�m� �zerindeki etkiler
JAKAVI'nin sedatif etkisi yoktur, ya da g�z ard� edilebilecek kadar azd�r. Bununla birlikte, JAKAVI ald�ktan sonra ba� d�nmesi ya�ayan hastalar, ara� veya makine kullanmaktan ka��nmal�d�r.
4.8. �stenmeyen etkiler
G�venlilik profilinin �zeti MiyelofibrozisEk s�k bildirilen advers ila� reaksiyonlar� trombositopeni ve anemidir.
Hematolojik advers reaksiyonlar (t�m Advers Olaylar i�in Ortak Terminoloji Kriterleri [CTCAE] dereceleri) anemi (% 83,8), trombositopeni (% 80,5) ve n�tropeniyi (% 20,8) i�ermektedir.
Anemi, trombositopeni ve n�tropeniyi doz ili�kili yan etkilerdir.
Hematolojik olmayan en s�k �� advers reaksiyon morarma (% 33,3), di�er kanamalar (burun kanamas�, prosed�r sonras� kanama ve hemat�ri dahil) (% 24,3) ve ba� d�nmesidir (% 21,9). Hematolojik olmayan en s�k �� laboratuvar anomalisi alanin transaminaz (% 40,7), y�ksek aspartat aminotransferaz (% 31,5) ve hipertrigliseridemidir (% 25,2). Faz 3 MF klinik �al��malarda CTCAE derece 3 veya 4 hipertrigliseridemi veya aspartat aminotransferaz y�kselmesi ya da CTCAE derece 4 alanin aminotransferaz y�kselmesi veya hiperkolesterolemi g�zlenmemi�tir.
Nedensellikten ba��ms�z olarak advers olaylar nedeniyle tedavinin kesilmesi hastalar�n % 30,0'unda g�r�lm��t�r.
Polisitemi vera
En s�k bildirilen advers ila� reaksiyonlar� anemi ve alanin aminotransferaz art��� olmu�tur.
Hematolojik advers reaksiyonlar (herhangi bir CTCAE derecesi) anemi (% 61,8) ve trombositopeni (% 25,0) ve n�tropeniyi (% 5,3) i�ermektedir. CTCAE derecesi 3 ve 4 anemi ve trombositopeni s�ras�yla % 2,9 ve % 2,6 oran�nda bildirilmi�tir.
Hematolojik olmayan en s�k �� advers reaksiyon kilo art��� (% 20,3), ba� d�nmesi (% 19,4) ve
ba� a�r�s� (% 17,9) olmu�tur.
Advers reaksiyonlar olarak tan�mlanan en s�k g�r�len �� hematoloji d��� laboratuvar anormalli�i (herhangi bir CTCAE derecesi) alanin aminotransferaz d�zeyinde y�kselme (% 45,3), aspartat aminotransferaz d�zeyinde y�kselme (% 42,6) ve hiperkolesterolemi (% 34,7) olmu�tur. CTCAE derece 4 alanin aminotransferaz y�kselmesi veya hiperkolesterolemi g�zlenmemi� ve bir adet CTCAE derece 4 artm�� aspartat aminotransferaz g�zlenmi�tir.
Hastalar�n % 19,4'�nde nedensellikten ba��ms�z olarak advers reaksiyonlar nedeniyle
tedavinin kesilmesi g�zlenmi�tir.
Akut GvHD
En s�k bildirilen genel advers ila� reaksiyonlar� trombositopeni, anemi ve n�tropenidir.
Advers ila� reaksiyonlar� olarak tan�mlanan hematolojik laboratuvar anormallikleri trombositopeni (%85,2), anemi (%75,0) ve n�tropeniyi (%65,1) i�ermi�tir. Hastalar�n
%47,7'sinde derece 3 anemi rapor edilmi�tir (CTCAE v4.03'e g�re derece 4 uygulanamaz).
Hastalar�n s�ras�yla %31,3 ve %47,7'sinde derece 3 ve 4 trombositopeni bildirilmi�tir.
En s�k g�r�len hematolojik olmayan advers ila� reaksiyonlar� sitomegalovir�s (CMV) enfeksiyonu (%32,3), sepsis (%25,4) ve idrar yolu enfeksiyonlar�d�r (%17,9).
Advers ila� reaksiyonlar� olarak tan�mlanan en s�k g�r�len hematolojik olmayan laboratuvar anormallikleri, alanin aminotransferaz art��� (%54,9), aspartat aminotransferaz art��� (%52,3) ve hiperkolesterolemi (%49,2) olmu�tur. �o�unlu�u derece 1 ve 2'dir.
Nedenselli�e bak�lmaks�z�n advers olaylar nedeniyle tedavinin kesilmesi hastalar�n
%29,4'�nde g�zlemlenmi�tir.
![]()
En s�k bildirilen genel advers ila� reaksiyonlar� anemi, hiperkolesterolemi ve aspartat aminotransferaz art���d�r.
Advers ila� reaksiyonlar� olarak tan�mlanan hematolojik laboratuvar anormallikleri anemi (%68,6), trombositopeni (%34,4) ve n�tropeniyi (%36,2) i�ermi�tir. Derece 3 anemi hastalar�n %14,8'inde rapor edilmi�tir (CTCAE v4.03'e g�re derece 4 uygulanamaz). Hastalar�n s�ras�yla %9,5 ve %6,7'sinde derece 3 ve 4 n�tropeni bildirilmi�tir.
Hematolojik olmayan en s�k g�r�len �� advers ila� reaksiyonu hipertansiyon (%15,0), ba�
a�r�s� (%10,2) ve idrar yolu enfeksiyonlar�d�r (%9,3).
Advers ila� reaksiyonlar� olarak tan�mlanan en s�k g�r�len hematolojik olmayan laboratuvar anormallikleri hiperkolesterolemi (%52,3), aspartat aminotransferaz art��� (%52,2) ve alanin aminotransferaz art���d�r (%43,1). �o�unlu�u derece 1 ve 2'dir.
Nedenselli�e bak�lmaks�z�n advers olaylar nedeniyle tedavinin kesilmesi hastalar�n
%18,1'inde g�zlemlenmi�tir.
Klinik �al��malardan bildirilen advers ila� reaksiyonlar�n�n tablo halinde listesi
MF hastalar�nda g�venlilik, ba�lang��ta ruksolitinibe (n=301) randomize edilen ve kontrol tedavilerinden �apraz ge�i�ten sonra ruksolitinib alan hastalardan (n=156) elde edilen verileri i�eren iki faz 3 �al��madan (COMFORT-I ve COMFORT-II) elde edilen uzun vadeli takip verileri kullan�larak de�erlendirilmi�tir. MF hastalar� i�in advers ila� reaksiyonu s�kl�k kategorilerinin dayand��� medyan maruziyet 30.5 ayd�r (aral�k 0,3 ila 68,1 ay).
PV hastalar�nda g�venlilik, ba�lang��ta ruksolitinibe (n=184) randomize edilmi� hastalardan ve kontrol tedavilerinden �apraz ge�i�ten sonra ruksolitinib alan hastalardan elde edilen verileri i�eren iki faz 3 �al��madan (RESPONSE, RESPONSE 2) uzun vadeli takip verileri kullan�larak de�erlendirilmi�tir. (n=156). PV hastalar� i�in ADR s�kl�k kategorilerinin dayand��� ortanca maruziyet 41,7 ayd�r (aral�k 0,03 ila 59,7 ay).
JAKAVI'nin akut GvHD hastalar�nda g�venlili�i, faz 3 REACH2 �al��mas�nda de�erlendirilmi� olup bu de�erlendirme de ba�lang��ta JAKAVI'ye randomize edilen hastalardan (n=152) ve mevcut en iyi tedaviden ge�i� yapt�ktan sonra JAKAVI alan hastalardan (n=49) elde edilen verileri i�ermi�tir. Advers ila� reaksiyonu s�kl�k kategorilerinin dayand��� medyan maruziyet 8,9 haftad�r (aral�k 0,3 ila 66,1 hafta).
JAKAVI'nin kronik GvHD hastalar�nda g�venlili�i, faz 3 REACH3 �al��mas�nda de�erlendirilmi� olup bu de�erlendirme de ba�lang��ta JAKAVI'ye randomize edilen hastalardan (n=165) ve mevcut en iyi tedaviden ge�i� yapt�ktan sonra JAKAVI alan hastalardan (n=61) elde edilen verileri i�ermi�tir. Advers ila� reaksiyonu s�kl�k kategorilerinin dayand��� medyan maruziyet 41,4 haftad�r (aral�k 0,7 ila 127,3 hafta).
Klinik �al��ma program�nda advers ila� reaksiyonlar�n�n �iddeti, derece 1=hafif, derece 2=orta, derece 3=�iddetli, derece 4=hayat� tehdit eden veya sakat b�rakan, derece 5=�l�m olarak tan�mlanan CTCAE'ye dayal� olarak de�erlendirilmi�tir.
MF ve PV'de (Tablo 4) ve akut ve kronik GvHD'deki (Tablo 5) klinik �al��malardan elde edilen advers ila� reaksiyonlar� MedDRA sistem organ s�n�f�na g�re listelenmi�tir. Klinik �al��malar program�nda advers ila� reaksiyonlar�n�n �iddeti, CTCAE esas al�narak de�erlendirilmi�tir: derece 1=hafif,derece2=orta,derece3= �iddetli ve derece 4= ya�am�
MF ve PV'de (Tablo 4) ve akut ve kronik GvHD'deki (Tablo 5) klinik �al��malardan elde edilen advers ila� reaksiyonlar� MedDRA sistem organ s�n�f�na g�re listelenmektedir. Her bir organ s�n�f� i�inde advers ila� reaksiyonlar� s�kl��a g�re s�ralanmakta, en s�k reaksiyonlar ilk s�rada g�sterilmektedir. Ayr�ca, her advers ila� reaksiyonu i�in kar��l�k gelen s�kl�k kategorisi, a�a��daki sisteme g�redir (CIOMS III): �ok yayg�n (≥1/10); yayg�n (≥1/100 ila <1/10); yayg�n olmayan (≥1/1,000 ila <1/100); seyrek (≥1/10,000 ila <1/1,000); �ok seyrek (<1/10,000); s�kl��� bilinmiyor.
Tablo 4 MF ve PV'deki Faz 3 �al��malarda bildirilen advers ila� reaksiyonlar�n�n s�kl�k
kategorisi
Advers ila� reaksiyonu | MF hastalar� i�in s�kl�k kategorisi | PV hastalar� kategorisi | i�in | s�kl�k |
Enfeksiyonlar ve enfestasyonlar |
|
| ||
�drar yolu enfeksiyonlar� | �ok yayg�n | �ok yayg�n | ||
Herpes zoster | �ok yayg�n | �ok yayg�n | ||
Pn�moni | �ok yayg�n | Yayg�n | ||
Sepsis | Yayg�n | Yayg�n olmayan | ||
T�berk�loz | Yayg�n olmayan | Bilinmiyor | ||
HBV reaktivasyonu | Bilinmiyor | Yayg�n olmayan | ||
Kan ve hastal�klar� | lenf | sistemi |
|
|
Anemi |
|
| ||
CTCAE derece 4 (<6,5 g/ ) | �ok yayg�n | Yayg�n olmayan | ||
CTCAE derece 3 (<8,0 – 6,5g/ ) | �ok yayg�n | Yayg�n | ||
Herhangi derecesi | bir | CTCAE | �ok yayg�n | �ok yayg�n |
Trombositopeni |
|
| ||
CTCAE derece 4 (<25.000/mm) | Yayg�n | Yayg�n olmayan | ||
CTCAE derece 3 (50.000 – 25.000/mm) | �ok yayg�n | Yayg�n | ||
Herhangi derecesi | bir | CTCAE | �ok yayg�n | �ok yayg�n |
N�tropeni |
|
| ||
CTCAE derece 4 (<500/mm) | Yayg�n | Yayg�n olmayan | ||
CTCAE derece 3 (<1.000 – 500/mm) | Yayg�n | Yayg�n olmayan | ||
Herhangi derecesi | bir | CTCAE | �ok yayg�n | Yayg�n |
Pansitopeni | Yayg�n | Yayg�n | ||
Kanama (intrakraniyal ve gastrointestinal kanama, morarma ve di�er kanamamalar dahil herhangi bir kanama) | �ok yayg�n | �ok yayg�n | ||
Morarma | �ok yayg�n | �ok yayg�n | ||
Gastrointestinal kanama | �ok yayg�n | Yayg�n | ||
�ntrakraniyal kanama | Yayg�n | Yayg�n olmayan | ||
Di�er kanama (burun kanamas�, prosed�r sonras� kanama, hemorajive hemat�ri dahil) | �ok yayg�n | �ok yayg�n | ||
Metabolizma ve hastal�klar� | beslenme |
|
| |
Hiperkolesterolemi Herhangi bir derecesi |
CTCAE | �ok yayg�n | �ok yayg�n | |
Hipertrigliseridemi Herhangi bir derecesi |
CTCAE | �ok yayg�n | �ok yayg�n | |
Kilo art��� | �ok yayg�n | �ok yayg�n | ||
Sinir sistemi hastal�klar� |
|
| ||
Sersemlik hali | �ok yayg�n | �ok yayg�n | ||
Ba� a�r�s� | �ok yayg�n | �ok yayg�n | ||
Gastrointestinal hastal�klar |
|
| ||
Y�ksek lipaz seviyesi Herhangi bir CTCAE derecesi | �ok yayg�n | �ok yayg�n | ||
Konstipasyon | �ok yayg�n | �ok yayg�n | ||
Flatulans | Yayg�n | Yayg�n | ||
Hepato-biliyer hastal�klar |
|
| ||
Alanin aminotransferaz y�kselmesi |
|
| ||
CTCAE derece 3 (> 5x – 20 x ULN) | Yayg�n | Yayg�n | ||
Herhangi bir derecesi | CTCAE | �ok yayg�n | �ok yayg�n | |
Aspartat y�kselmesi | aminotransferaz |
|
| |
Herhangi derecesi | bir | CTCAE | �ok yayg�n | �ok yayg�n |
Vask�ler hastal�klar |
|
| ||
Hipertansiyon | �ok yayg�n | �ok yayg�n | ||
<100 g/l, trombosit say�s� <100x10/l ve n�trofil say�s� <1.5x10/l (veya n�trofil say�s� eksikse, derece 2 d���k beyaz kan h�cresi say�s�) olarak tan�mlan�r. tehlike olu�turan | ||||
Tedavi kesildikten sonra MF hastalar�nda yorgunluk, kemik a�r�s�, y�ksek ate�, prurit, gece terlemeleri, semptomatik splenomegali ve kilo kayb� gibi MF semptomlar�nda d�n�� olabilir. Klinik �al��malarda MF semptomlar� i�in toplam semptom skoru, doz uygulamas� kesildikten sonraki 7 g�n i�inde a�amal� olarak ba�lang�� de�erlerine d�nm��t�r (bkz. B�l�m 4.4).
Tablo 5 GvHD'deki faz 3 �al��malar�nda bildirilen advers ila� reaksiyonlar�n�n
s�kl�k kategorisi
| Akut GvHD (REACH2) | Kronik GvHD (REACH3) |
Advers ila� reaksiyonu | S�kl�k kategorisi | S�kl�k kategorisi |
Enfeksiyonlar ve enfestasyonlar |
|
|
CMV enfeksiyonlar� | �ok yayg�n | Yayg�n |
CTCAE derece ≥ 3 | �ok yayg�n | Yayg�n |
Sepsis | �ok yayg�n | |
CTCAE derece ≥ 3 | �ok yayg�n | - |
�drar yolu enfeksiyonlar� | �ok yayg�n | Yayg�n |
CTCAE derece ≥ 3 | Yayg�n | Yayg�n |
BK vir�s� enfeksiyonlar� | - | Yayg�n |
CTCAE derece ≥ 3 | - | Yayg�n olmayan |
Kan ve lenf sistemi hastal�klar� |
|
|
Trombositopeni | �ok yayg�n | �ok yayg�n |
CTCAE derece 3 | �ok yayg�n | Yayg�n |
CTCAE derece 4 | �ok yayg�n | �ok yayg�n |
Anemi | �ok yayg�n | �ok yayg�n |
CTCAE derece 3 | �ok yayg�n | �ok yayg�n |
N�tropeni | �ok yayg�n | �ok yayg�n |
CTCAE derece 3 | �ok yayg�n | Yayg�n |
CTCAE derece 4 | �ok yayg�n | Yayg�n |
Pansitopeni | �ok yayg�n | - |
Metabolizma ve beslenme hastal�klar� |
|
|
Hiperkolesterolemi | �ok yayg�n | �ok yayg�n |
CTCAE derece 3 | Yayg�n | Yayg�n |
CTCAE derece 4 | Yayg�n | Yayg�n olmayan |
Kilo alma | - | Yayg�n |
CTCAE derece ≥3 | - | Uygulanamaz |
Sinir sistemi hastal�klar� |
|
|
Ba� a�r�s� | Yayg�n | �ok yayg�n |
CTCAE derece ≥3 | Yayg�n olmayan | Yayg�n |
Vask�ler hastal�klar |
|
|
Hipertansiyon | �ok yayg�n | �ok yayg�n |
CTCAE derece ≥3 | Yayg�n | Yayg�n |
Gastrointestinal hastal�klar |
|
|
Lipaz art��� | �ok yayg�n | |
CTCAE derece 3 | - | Yayg�n |
CTCAE derece 4 | - | Yayg�n olmayan |
Amilaz art��� | �ok yayg�n | |
CTCAE derece 3 | Yayg�n | |
CTCAE derece 4 | Yayg�n |
Bulant� | �ok yayg�n | - |
CTCAE derece ≥3 | Yayg�n olmayan | - |
Kab�zl�k | Yayg�n | |
CTCAE derece >3 | - | Uygulanamaz |
Hepato-biliyer hastal�klar |
|
|
Alanin aminotransferaz art��� | �ok yayg�n | �ok yayg�n |
CTCAE derece 3 | �ok yayg�n | Yayg�n |
CTCAE derece 4 | Yayg�n | Yayg�n olmayan |
Aspartat aminotransferaz art��� | �ok yayg�n | �ok yayg�n |
CTCAE derece 3 | Yayg�n | Yayg�n |
CTCAE derece 4 | Uygulanamaz | Yayg�n olmayan |
Kas-iskelet sistemi ve ba� dokusu hastal�klar� |
|
|
Kan kreatin fosfokinaz art��� | - | �ok yayg�n |
CTCAE derece 3 | - | Yayg�n |
CTCAE derece 4 | - | Yayg�n |
Renal ve �riner hastal�klar |
|
|
Kan kreatinin art��� | �ok yayg�n | |
CTCAE derece 3 | Yayg�n | |
CTCAE derece 4 | - | Uygulanamaz |
<100 g/l, trombosit say�s� <100x10/l ve n�trofil say�s� <1.5x10/l (veya n�trofil say�s� eksikse, derece 2 d���k beyaz kan h�cresi say�s�) olarak tan�mlan�r. | ||
Se�ilen advers ila� reaksiyonlar�n�n tan�m�
Anemi
MF'teki Faz 3 klinik �al��malarda, ilk CTCAE derece 2 veya daha y�ksek dereceli aneminin ba�lamas�na kadar ge�en medyan s�re 1,5 ay olmu�tur. Bir hasta (%0,3) anemi nedeniyle tedaviden ayr�lm��t�r.
Ruksolitinib alan hastalarda ortalama hemoglobin d����leri, tedavinin 8 ila 12 haftas�ndan sonra ba�lang�c�n yakla��k 10 g/L alt�nda en d���k de�erlere ula�m��t�r ve ard�ndan a�amal� olarak d�zelerek, ba�lang�c�n yakla��k 5 g/L alt�nda yeni bir kararl� duruma ula�m��t�r. Hastalarda bu patern, tedavi s�ras�nda transf�zyon alm�� olup olmamalar�ndan ba��ms�z bir �ekilde g�zlenmi�tir.
Randomize, plasebo kontroll� �al��mada (COMFORT-I), JAKAVI tedavisindeki hastalar�n
%60,6's� ve plasebo uygulanan hastalar�n %37,7'si randomize tedavi s�ras�nda eritrosit transf�zyonu alm��t�r. COMFORT-II �al��mas�nda eritrosit transf�zyonu oran� JAKAVI kolunda %53,4 iken en iyi mevcut tedavi (BAT) kolunda %41,1 olmu�tur.
Pivot �al��malar�n randomize periyodunda anemi, PV hastalar�nda MF hastalar� ile kar��la�t�r�ld���nda daha d���k s�kl�kta g�r�lm��t�r (%40,8 kar��s�nda % 82,4). PV pop�lasyonunda, CTCAE derecesi 3 ve 4 olaylar %2,7 oran�nda bildirilirken MF hastalar�nda s�kl�k %42,56 olmu�tur.
Faz 3 akut ve kronik GvHD �al��malar�nda, hastalar�n s�ras�yla %47,7'sinde ve %14,8'inde CTCAE derece 3 anemi bildirilmi�tir.
Trombositopeni
MF'teki Faz 3 klinik �al��malarda, derece 3 veya 4 trombositopeni geli�en hastalarda, trombositopeninin ba�lang�c�na kadar ge�en medyan s�re 8 hafta olmu�tur. Trombositopeni genellikle dozun azalt�lmas� ya da ara verilmesiyle geri d�n���ml� olmu�tur. 50.000/mm3 �zerindeki trombosit say�lar�na d�n�� i�in medyan s�re 14 g�n olmu�tur. Randomize periyod boyunca trombosit transf�zyonlar�, ruksolitinib alan hastalar�n %4,7'sine ve kontrol rejimleri alan hastalar�n %4,0'�ne uygulanm��t�r. Ruksolitinib tedavisindeki hastalar�n %0,7'si ve kontrol rejimleri alan hastalar�n %0,9'u trombositopeni nedeniyle �al��madan ayr�lm��t�r. �al��malar�n randomize periyodu boyunca ruksolitinibe ba�lamadan �nce trombosit say�s� 100.000/mm3 ila 200.000/mm3 olan hastalarda derece 3 ya da 4 trombositopeni s�kl���, trombosit say�s� >200.000/mm3 olan hastalar ile kar��la�t�r�ld���nda daha y�ksek olmu�tur (%64,2 kar��s�nda %38,5).
Pivot �al��malar�n randomize periyodunda, trombositopeni ya�ayan hastalar�n oran� MF (%69,8) hastalar� ile kar��la�t�r�ld���nda PV (%16,8) hastalar�nda daha d���k olmu�tur. �iddetli (CTCAE derecesi 3 ve 4) trombositopeni s�kl��� PV hastalar�nda (%2,7), MF hastalar�na (%11,6) k�yasla daha d���k bulunmu�tur.
Faz 3 akut GvHD �al��mas�nda, hastalar�n s�ras�yla %31,3 ve %47,7'sinde derece 3 ve 4 trombositopeni g�zlemlenmi�tir. Faz 3 kronik GvHD �al��mas�nda, derece 3 ve 4 trombositopeni, akut GvHD'dekine k�yasla daha d���k olmu�tur (%5,9 ve %10,7).
N�tropeni
MF'teki Faz 3 klinik �al��malarda, derece 3 veya 4 n�tropeni geli�tiren hastalarda, n�tropenin ba�lang�c�na kadar ge�en medyan s�re 12 hafta olmu�tur. N�tropeni nedeniyle doz uygulamalar�na ara verilmesi ya da dozun azalt�lmas�, hastalar�n %1.0'�nda s�z konusu olmu�tur ve hastalar�n %0,3'� n�tropeni nedeniyle �al��madan ayr�lm��t�r.
PV hastalar�nda yap�lan Faz 3 �al��malar�n�n randomize periyodunda, n�tropeni referans tedavilerde %7'ye k�yasla ruksolitinibe maruz kalan hastalar�n % 1,6's�nda bildirilmi�tir. Ruksolitinib kolunda bir hastada CTCAE derece 4 n�tropeni geli�mi�tir. Ruksolitinib ile tedavi edilen hastalar�n uzun s�reli takibi, CTCAE derece 4 n�tropeni bildiren 2 hasta g�stermi�tir.
Faz 3 akut GvHD �al��mas�nda, hastalar�n s�ras�yla %17,9'unda ve %20,6's�nda derece 3 ve 4 n�tropeni g�zlemlenmi�tir. Faz 3 kronik GvHD �al��mas�nda derece 3 ve 4 n�tropeni, akut GvHD'dekine k�yasla daha d���k bulunmu�tur (%9,5 ve %6,7).
Kanama
Faz 3 pivotal �al��malarda MF kanama olaylar� (intrakranial ve gastrointestinal, morarma ve di�er kanama olaylar� dahil) ruksolitinibe maruz kalan hastalar�n %32,6's� ve referans tedavileri (plasebo veya en iyi mevcut tedavi) kullananlar�n %23,2'sinde bildirilmi�tir. Derece 3-4 olaylar�n s�kl��� ruksolitinib veya referans tedavilerle tedavi edilen hastalar i�in benzerdir (%3,1'e kar�� %4,7). Tedavi s�ras�nda kanama olaylar� g�r�len hastalar�n �o�u morarma bildirmi�tir (%65,3). Morarma olaylar� referans tedavilere k�yasla ruksolitinib kullanan hastalarda daha s�k bildirilmi�tir (%11,6'ya kar�� %21,3). �ntrakranial kanama ruksolitinibe maruz kalan hastalar�n %1'inde ve referans tedavilere maruz kalanlar�n %0,9'unda bildirilmi�tir. Gastrointestinal kanama referans tedavilere maruz kalanlar�n %3,1'ine k�yasla ruksolitinibe maruz kalan hastalar�n%5'indebildirilmi�tir.Di�er kanama olaylar� (burun
kanamas�, prosed�r sonras� hemoraji ve hemat�ri gibi olaylar dahil) ruksolitinib ile tedavi
edilen hastalar�n %13,3 ve referans tedavilerle tedavi edilenlerin %10,3'�nde bildirilmi�tir.
MF'de Faz 3 klinik �al��malar�n uzun s�reli takibi s�ras�nda, kanama olaylar�n�n k�m�latif s�kl���, takip s�resindeki art��la orant�l� olarak artm��t�r. Morarma olaylar� en s�k bildirilen kanama olaylar� olmu�tur (% 33,3). �ntrakraniyal ve gastrointestinal kanama olaylar� s�ras�yla hastalar�n % 1,3 ve % 10,1'inde bildirilmi�tir.
PV hastalar�ndaki faz 3 �al��malar�n kar��la�t�rmal� periyodunda kanama olaylar� (intrakranial ve gastrointestinal kanama, morarma ve di�er kanama olaylar�n� i�erir) ruksolitinib ile tedavi edilen hastalar�n %16,8'inde, RESPONSE �al��mas�nda en iyi mevcut tedavi alan hastalar�n
%15,3'�nde ve RESPONSE 2 �al��mas�nda en iyi mevcut tedavi alan hastalar�n %12'sinde bildirilmi�tir. Morarma, ruksolitinib ile tedavi edilen hastalar�n %10,3'�nde, RESPONSE �al��mas�nda en iyi mevcut tedavi alan hastalar�n %8,1'inde ve RESPONSE 2 �al��mas�nda en iyi mevcut tedavi alan hastalar�n %2,7'sinde bildirilmi�tir. Ruksolitinib uygulanan hastalarda herhangi bir intrakranial kanama ya da gastrointestinal hemoraji olay� bildirilmemi�tir. Ruksolitinib ile tedavi edilen bir hasta bir adet derece 3 kanama olay� ya�am��t�r (prosed�r sonras� kanama); hi�bir derece 4 kanama bildirilmemi�tir. Di�er kanama olaylar� (burun kanamas�, prosed�r sonras� hemoraji, di�eti kanamas� dahil), ruksolitinib ile tedavi edilen hastalar�n %8,7'sinde, RESPONSE �al��mas�nda en iyi mevcut tedavi alan hastalar�n %6,3'�nde ve RESPONSE 2 �al��mas�nda en iyi mevcut tedavi alan hastalar�n
%6,7'sinde bildirilmi�tir.
PV'deki Faz-3 �al��malar�n�n uzun s�reli takibi s�ras�nda, kanama olaylar�n�n k�m�latif s�kl��� takip s�resindeki art��la orant�l� olarak artm��t�r. Morarma olaylar�, en �ok s�k bildirilen kanama olaylar�d�r (% 17,4). Kafa i�i ve gastrointestinal kanama olaylar� s�ras�yla hastalar�n
% 0,3 ve % 3,5'inde rapor edilmi�tir.
Faz 3 akut GvHD �al��mas�n�n kar��la�t�rmal� d�neminde, ruksolitinib ve mevcut en iyi tedavi kollar�ndaki hastalar�n s�ras�yla %25,0 ve %22,0'sinde kanama olaylar� rapor edilmi�tir. Kanama olaylar�n�n alt gruplar� genellikle tedavi kollar� aras�nda benzer olmu�tur: morarma olaylar� (ruksolitinibte %5,9'a kar�� mevcut en iyi tedavi kolunda %6,7), gastrointestinal olaylar (%9,2'ye kar�� %6,7) ve di�er kanama olaylar� (%13,2'ye kar�� %10,7). �ntrakraniyal kanama olaylar� mevcut en iyi tedavi kolundaki hastalar�n %0.7'sinde rapor edilirken ve ruksolitinib kolunda hi�bir hastada rapor edilmemi�tir.
Faz 3 kronik GvHD �al��mas�n�n kar��la�t�rmal� d�neminde, ruksolitinib ve mevcut en iyi tedavi kollar�ndaki hastalar�n s�ras�yla %11,5 ve %14,6's�nda kanama olaylar� rapor edilmi�tir. Kanama olaylar�n�n alt gruplar� genellikle tedavi kollar� aras�nda benzer olmu�tur: morarma olaylar� (ruksolitinibde %4,2'ye kar�� mevcut en iyi tedavi kolunda %2,5), gastrointestinal olaylar (%1,2'ye kar�� %3,2) ve di�er kanama olaylar� (%6,7'ye kar�� %10,1). Her iki tedavi kolunda da intrakraniyal kanama olay� bildirilmemi�tir.
Enfeksiyonlar
Faz 3 pivotal �al��malarda hastalar�n %1'inde derece 3 veya 4 idrar yolu enfeksiyonu,
%4,3'�nde herpes zoster ve %1'inde t�berk�loz bildirilmi�tir. Faz 3 klinik �al��malarda sepsis hastalar�n %3'�nde bildirilmi�tir. Ruksolitinib ile tedavi edilen hastalar�n uzun s�reli takibi, zaman i�inde sepsis oran�nda art�� y�n�nde bir e�ilim olmad���n� g�stermi�tir.
PV hastalar�nda yap�lan Faz 3 �al��malar�n�n randomize periyodunda, bir (% 0,5) CTCAE derece 3 idrar yolu enfeksiyonu bildirilmi�, herhangi bir derece 4 idrar yolu enfeksiyonu bildirilmemi�tir. Herpes zosteroran�PV(%4,3)hastalar�ile MF (% 4,0) hastalar� aras�nda
rapor s�z konusu olmu�tur. Referans tedavilerde hastalar�n %1.6's�na k�yasla ruksolitinib ile tedavi edilen hastalar�n % 0,5'inde pn�moni bildirilmi�tir. Ruksolitinib kolundaki hi�bir hasta sepsis veya t�berk�loz bildirmemi�tir.
PV'de Faz 3 �al��malar�n�n uzun s�reli takibi s�ras�nda s�kl�kla bildirilen enfeksiyonlar idrar
yolu enfeksiyonlar� (% 11,8), herpes zoster (% 14,7) ve pn�moni (% 7,1) olmu�tur. Hastalar�n
% 0,6's�nda sepsis bildirilmi�tir. Uzun s�reli takipte hi�bir hasta t�berk�loz bildirmemi�tir.
Faz 3 akut GvHD �al��mas�nda, kar��la�t�rmal� d�nemde, mevcut en iyi tedavi kolundaki hastalar�n %10,7'sine (derece ≥3, %6,0) k�yasla ruksolitinib kolundaki hastalar�n %9,9'unda (derece ≥3, %3,3) idrar yolu enfeksiyonlar� bildirilmi�tir. Mevcut en iyi tedavi kolunda %24,0 (derece ≥3, %10,0) ile kar��la�t�r�ld���nda, ruksolitinib kolundaki hastalar�n %28,3'�nde (derece ≥3, %9,3) CMV enfeksiyonlar� bildirilmi�tir. Mevcut en iyi tedavi kolundaki hastalar�n %8,7'sine (derece ≥3, %6,0) k�yasla ruksolitinib kolundaki hastalar�n %12,5'inde (derece ≥3, %11,1) sepsis olaylar� bildirilmi�tir. BK vir�s� enfeksiyonu sadece ruksolitinib kolunda, bir derece 3 olay� olan 3 hastada rapor edilmi�tir. Ruksolitinib ile tedavi edilen hastalar�n uzun s�reli takipleri s�ras�nda, hastalar�n %17,9'unda (derece ≥3, %6,5) idrar yolu enfeksiyonlar� ve hastalar�n %32,3'�nde (derece ≥3, %11,4) CMV enfeksiyonlar� bildirilmi�tir. Organ tutulumlu CMV enfeksiyonu �ok az hastada g�r�lm��t�r; S�ras�yla d�rt, iki ve bir hastada CMV koliti, CMV enteriti ve herhangi bir derecedeki CMV gastrointestinal enfeksiyonu bildirilmi�tir. Herhangi bir derecedeki septik �ok dahil sepsis olaylar� hastalar�n
%25,4'�nde (derece ≥3, %21,9) rapor edilmi�tir.
Faz 3 kronik GvHD �al��mas�nda, kar��la�t�rmal� d�nemde, ruksolitinib kolundaki hastalar�n
%8,5'inde (derece ≥3, %1,2) idrar yolu enfeksiyonlar� rapor edilirken, bu oran mevcut en iyi tedavi kolunda %6,3't�r (derece ≥3, %1,3). BK vir�s� enfeksiyonu mevcut en iyi tedavi kolunda %1,3'e k�yasla ruksolitinib kolundaki hastalar�n %5,5'inde (derece ≥3, %0,6) bildirilmi�tir. CMV enfeksiyonlar� ruksolitinib kolundaki hastalar�n %9,1'inde (derece ≥3,
%1,8), BAT kolundaki hastalar�n ise %10,8'inde (derece ≥3, %1,9) bildirilmi�tir. Mevcut en iyi tedavi kolundaki hastalar�n %6,3'�ne (derece ≥3, %5,7) k�yasla ruksolitinib kolundaki hastalar�n %2,4'�nde (derece ≥3, %2,4) sepsis olaylar� bildirilmi�tir. Ruksolitinib ile tedavi edilen hastalar�n uzun s�reli takipleri s�ras�nda, hastalar�n s�ras�yla %9,3'�nde (derece ≥3,
%1,3) ve %4,9'unda (derece ≥3, %0,4) idrar yolu enfeksiyonlar� ve BK vir�s� enfeksiyonlar� bildirilmi�tir. Hastalar�n s�ras�yla %8,8'inde (derece ≥3, %1,3) ve %3,5'inde (derece ≥3,
%3,5) CMV enfeksiyonlar� ve sepsis olaylar� bildirilmi�tir.
Lipaz y�kselmesi
RESPONSE �al��mas�n�n randomize periyodunda, lipaz de�erlerinde k�t�le�me, kontrol koluna k�yasla ruksolitinib kolunda daha y�ksek olup bunun ana nedeni 1. derece y�kselmeler aras�ndaki farklar olmu�tur (% 18,2'ye kar�� % 8,1). Derece ≥ 2 y�kselmeler tedavi kollar� aras�nda benzer bulunmu�tur. RESPONSE 2'de, s�kl�klar ruksolitinib ve kontrol kolu aras�nda kar��la�t�r�labilir olmu�tur (% 10,8'e kar�� % 8). Faz 3 PV �al��malar�n�n uzun s�reli takibi s�ras�nda hastalar�n % 7,4 ve % 0,9'u derece 3 ve derece 4 lipaz de�erlerinde y�kselme bildirmi�tir. Bu hastalarda y�ksek lipaz de�erleri ile e�zamanl� pankreatit belirti ve semptomlar� bildirilmemi�tir.
MF'de yap�lan faz 3 �al��malarda, COMFORT-I ve COMFORT-II �al��malar�nda kontrol kollar�nda s�ras�yla % 16,6 ve % 14,0 ile kar��la�t�r�ld���nda ruksolitinib kollar�ndaki hastalar�n % 18,7 ve % 19,3'�nde y�ksek lipaz de�erleri bildirilmi�tir. Lipaz de�erleri y�kselmi� hastalarda, e� zamanl� pankreatit belirti ve semptomlar� bildirilmemi�tir.
k�t�le�mi� lipaz de�erleri rapor edilmi�tir; kar��l�k gelen derece 3 (%3,1'e kar�� %5,1) ve derece 4 (%0'a kar�� %0,8) art��lar benzerdir. Ruksolitinib ile tedavi edilen hastalar�n uzun s�reli takipleri s�ras�nda, hastalar�n %32,2'sinde artm�� lipaz de�erleri bildirilmi�tir; Derece 3 ve 4, hastalar�n s�ras�yla %8,7 ve %2,2'sinde bildirilmi�tir.
Faz 3 kronik GvHD �al��mas�n�n kar��la�t�rmal� d�neminde, mevcut en iyi tedavi kolundaki hastalar�n %23,5'ine k�yasla ruksolitinib kolundaki hastalar�n %32,1'inde yeni veya k�t�le�mi� lipaz de�erleri rapor edilmi�tir; kar��l�k gelen derece 3 (%10,6'ya kar�� %6,2) ve derece 4 (%0,6'ya kar�� %0) art��lar benzerdir. Ruksolitinib ile tedavi edilen hastalar�n uzun s�reli takipleri s�ras�nda, hastalar�n %35,9'unda lipaz de�erlerinde art�� bildirilmi�tir; Derece 3 ve 4, hastalar�n s�ras�yla %9,5 ve %0,4'�nde g�zlemlenmi�tir.
Artm�� sistolik kan bas�nc�
Faz 3 pivotal klinik �al��malarda, en az bir vizitte kontrol ile tedavi edilen hastalar�n
%19,5'ine k�yasla hastalar�n %31,5'inde sistolik kan bas�nc�nda ba�lang�ca g�re 20 mmHg veya daha fazla bir art�� kaydedilmi�tir. COMFORT-I �al��mas�nda, sistolik kan bas�nc�nda ba�lang�ca g�re ortalama art�� ruksolitinib ile 0-2 mmHg iken, plasebo kolunda 2-5 mmHg azalma g�r�lm��t�r. COMFORT-II �al��mas�nda, ortalama de�erler ruksolitinib ile tedavi edilen ve kontrol ile tedavi edilen hastalar aras�nda k���k bir fark g�stermi�tir.
PV hastalar�ndaki pivot �al��malar�n randomize periyodunda, ortalama sistolik kan bas�nc�
ruksolitinib kolunda 0,65 mmHg y�kselirken BAT kolunda 2 mmHg d���� olmu�tur.
Pediyatrik hastalar
GvHD'li 12 ila <18 ya�lar� aras�ndaki toplam 20 hasta g�venlilik a��s�ndan analiz edilmi�tir: REACH2 �al��mas�nda 9 hasta (ruksolitinib kolunda 5 ve mevcut en iyi tedavi kolunda 4) ve REACH3 �al��mas�nda 11 hasta (ruksolitinib kolunda 4 ve mevcut en iyi tedavi kolunda 7 hasta). Ad�lesanlarda ve yeti�kinlerde g�zlemlenen benzer maruziyete dayanarak, g�nde iki kez 10 mg dozunda �nerilen ruksolitinibin g�venlili�i, s�kl�k ve �iddet a��s�ndan benzerdir.
Geriyatrik pop�lasyon
65 ya� �st� olup ruksolitinib ile tedavi edilen, REACH2 �al��mas�nda toplam 29 hasta ve REACH3'te 25 hasta g�venlilik a��s�ndan analiz edilmi�tir. Genel olarak, yeni g�venlilik endi�eleri tan�mlanmam��t�r ve 65 ya� �st� hastalardaki g�venlilik profili genellikle 18-65 ya��ndaki hastalar�nkiyle uyumludur.
��pheli advers reaksiyonlar�n raporlanmas�
Ruhsatland�rma sonras� ��pheli ila� advers reaksiyonlar�n�n raporlanmas� b�y�k �nem ta��maktad�r. Raporlama yap�lmas�, ilac�n yarar / risk dengesinin s�rekli olarak izlenmesine olanak sa�lar. Sa�l�k mesle�i mensuplar�n�n herhangi bir ��pheli advers reaksiyonu T�rkiye Farmakovijilans Merkezi (T�FAM)'ne bildirmeleri gerekmektedir. (www.titck.gov.tr; e- posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks: 0 312 218 35 99)
4.9. Doz a��m� ve tedavisi
5. FARMAKOLOJ�K �ZELL�KLER
5.1. Farmakodinamik �zellikler
Farmakoterap�tik grubu: Antineoplastik ila�lar, Protein kinaz inhibit�rleri, Janus-�li�kili Kinaz inhibit�r�
ATC kodu: L01EJ01
Etki mekanizmas�
Ruksolitinib, Janus �li�kili Kinazlar (JAKs) JAK1 ve JAK2'in selektif bir inhibit�r�d�r (ICde�erleri JAK1 ve JAK2 enzimleri i�in s�ras�yla 3,3 nM ve 2,8 nM). Bunlar, hematopoez ve imm�n fonksiyon a��s�ndan �nemli olan bir dizi sitokininin ve b�y�me fakt�r�n�n sinyallemesine arac�l�k ederler.
MF ve PV, disreg�le JAK1 ve JAK2 sinyallemesi ile ili�kili oldu�u bilinen miyeloproliferatif bir neoplazmad�r (MPN). Bu disreg�lasyonun temelinde, JAK-STAT yola��n� aktive eden dola��mdaki sitokinlerin artm�� d�zeyinin, JAK2V617F gibi fonksiyon kazand�r�c� mutasyonlar�n ve negatif reg�lat�r mekanizmalar�n etkisizle�mesinin yatt���na inan�lmaktad�r. MF hastalar� JAK2V617F mutasyon durumu fark etmeksizin disreg�le JAK sinyallemesi g�sterirler. PV hastalar�n�n >%95'inde JAK2'de (V617F veya ekson 12) aktive edici mutasyonlar bulunmu�tur.
Ruksolitinib, JAK-STAT sinyallemesini ve hematolojik malignitelerin sitokine ba��ml� h�cre modellerinde h�cre proliferasyonunu inhibe eder. Ayr�ca JAK2V617F mutasyonu eksprese etti�i i�in sitokinden ba��ms�z hale gelen Ba/F3 h�crelerinin proliferasyonunu, 80-320 nM aral���ndaki ICde�erleri ile inhibe eder.
JAK-STAT sinyal yolaklar�, GvHD patogenezi i�in �nemli olan �e�itli imm�n h�cre tiplerinin geli�imi, proliferasyonu ve aktivasyonunun d�zenlenmesinde rol oynar.
Farmakodinamik etkiler
Ruksolitinib, sa�l�kl� g�n�ll�lerin, MF ve PV hastalar�n�n tam kanlar�nda sitokinin ind�kledi�i STAT3 fosforilasyonunu inhibe eder. Ruksolitinib, doz uyguland�ktan 2 saat sonra STAT3 fosforilasyonunda maksimal inhibisyon sa�lam��, gerek sa�l�kl� g�n�ll�lerde gerekse miyelofibrozis hastalar�nda bu de�er 8 saatte ba�lang�� d�zeylerine yak�n de�erlere d�nerek ana bile�ik ya da aktif metabolitlerde birikme olmad���na i�aret etmi�tir.
Miyelofibrozisli hastalarda TNF, IL-6, ve CRP gibi, konstit�syonel semptomlarla ili�kili enflamatuvar belirte�lerde ba�lang��taki y�kselmeler, ruksolitinib ile tedavi sonras�nda d��m��t�r. Miyelofibrozisli hastalar zaman i�inde ruksolitinib tedavisinin farmakodinamik etkilerine refrakter hale gelmemi�tir. Benzer �ekilde, PV hastalar� da ba�lang��ta enflamatuvar belirte�lerde y�kselmeler ile ba�vurmu�lard�r ve bu belirte�ler ruksolitinib ile tedavi sonras�nda azalm��t�r.
Sa�l�kl� g�n�ll�lerde kapsaml� bir QT �al��mas�nda, 200 mg supraterap�tik dozlara kadarki tek dozlarda ruksolitinib verilmesinin QT/QTc uzat�c� etkisine dair bir belirti bulunamam��t�r. Bu da ruksolitinibin kardiyak repolarizasyon �zerinde bir etkisi olmad���na i�aret etmi�tir.
Klinik etkililik ve g�venlilik
Miyelofibrozis
MF (Primer MF, Polisitemi Vera Sonras� MF ya da Esansiyel Trombositemi Sonras� MF) hastalar�nda iki randomize faz 3 �al��ma ger�ekle�tirilmi�tir (COMFORT-I ve COMFORT- II). �ki �al��mada da hastalarda kot kavsi alt�nda en az 5 cm ele gelen splenomegali ve Uluslararas� �al��ma Grubu (IWG) Konsens�s Kriterlerine g�re orta-2 veya daha y�ksek risk kategorisi s�z konusudur.JAKAVI ba�lang�� dozundatrombosit say�s� esas al�nm��t�r.
Trombosit say�lar� ≤100.000/mm3 olan hastalar COMFORT �al��malar�na kay�t i�in uygun de�ildir. Ancak ba�lang�� trombosit say�lar� ≥50.000 ve <100.000/mm3 olan 69 hasta, MF hastalar�nda (Primer MF, Polisitemi Vera sonras� MF ya da Esansiyel Trombositemi sonras� MF) bir Faz Ib a��k etiketli, doz belirleme �al��mas� olan EXPAND �al��mas�na dahil edilmi�tir.
COMFORT-I, mevcut tedaviye diren�li ya da bu tedaviler i�in aday olamayan 309 hastan�n dahil edildi�i, �ift k�r, randomize, plasebo kontroll� bir �al��mad�r. Hastalara JAKAVI ya da plasebo uygulanm��t�r. Birincil etkililik sonlan�m noktas�, 24. haftada manyetik rezonans g�r�nt�leme (MR) ya da bilgisayarl� tomografi (BT) ile g�r�nt�lemede dalak hacminde ba�lang�ca g�re ≥%35 k���lme elde eden hastalar�n oran�d�r.
�kincil sonlan�m noktalar� dalak hacminde ba�lang�ca g�re ≥%35 azalman�n korundu�u s�re, toplam semptom skorunda ≥%50 azalma olan hastalar�n oran�, ba�lang��tan 24. haftaya kadar toplam semptom skorlar�nda de�i�iklikler (modifiye MF Semptom De�erlendirme Formu [MFSAF] v2.0 g�nl��� ile �l��len) ve genel sa�kal�m� i�ermi�tir.
COMFORT-II, 219 hasta ile y�r�t�len a��k etiketli, randomize bir �al��mad�r. Hastalar 2:1 oran�nda ruksolitinib ya da mevcut en iyi tedaviye randomize edilmi�tir. Mevcut en iyi tedavi ara�t�rmac� taraf�ndan hasta baz�nda se�ilmi�tir. Mevcut en iyi tedavi kolunda hastalar�n
%47'si hidroksi�re ve %16's� glukokortikoidler alm��t�r. Birincil etkililik sonlan�m noktas�,
48. haftada MR ya da BT ile g�r�nt�lemede dalak hacminde ba�lang�ca g�re ≥%35 k���lme
elde eden hastalar�n oran� olmu�tur.
�kincil sonlan�m noktalar� 24. haftada dalak hacminde ba�lang�ca g�re ≥%35 azalmaya ula�an hastalar�n oran� ve dalak hacminde ba�lang�ca g�re ≥%35 azalman�n korundu�u s�reyi i�ermi�tir.
COMFORT-I ve COMFORT-II �al��malar�nda hasta ba�lang�� demografik �zellikleri ve hasta karakteristikleri tedavi kollar� aras�nda benzerdir.
Tablo 6 COMFORT-I'de 24. haftada ve COMFORT-II'de 48 haftada (ITT) dalak boyutunda ba�lang�ca g�re ≥%35 k���lme elde eden hastalar�n y�zdeleri
| COMFORT-I | COMFORT-II | ||
| JAKAVI (N=155) | Plasebo (N=153) | JAKAVI (N=144) | Mevcut En �yi Tedavi (N=72) |
Zaman noktalar� | 24. hafta | 48. hafta | ||
Dalak boyutu ≥%35 k���len hasta say�s� (%) | 65 (41,9) | 1 (0,7) | 41 (28,5) | 0 |
%95 g�ven aral�klar� | 34,1-50,1 | 0-3,6 | 21,3-36,6 | 0-5 |
P de�eri | < 0,0001 | < 0,0001 | ||
JAK2V617F mutasyonunun veya hastal�k alt tipinin (primer MF, polisitemi vera sonras� MF, esansiyel trombositemi sonras� MF) varl���na veya yoklu�una bak�lmaks�z�n, JAKAVI grubundaki hastalar�n anlaml� derecede daha b�y�k bir oran�, dalak hacminde ba�lang�ca g�re
≥%35 k���lme elde etmi�tir.
Tablo 7 JAK mutasyon durumuna g�re dalak hacminde ba�lang�ca g�re ≥%35 azalma ya�ayan hasta y�zdesi (g�venlilik seti)
| COMFORT-I | COMFORT-II | ||||||
| JAKAVI | Plasebo | JAKAVI | En iyi mevcut tedavi | ||||
JAK mutasyon durumu | Pozitif (N=113) n (%) | Negatif (N=40) n (%) | Pozitif (N=121) n (%) | Negatif (N=27) n (%) | Pozitif (N=110) n (%) | Negatif (N=35) n (%) | Pozitif (N=49) n (%) | Negatif (N=20) n (%) |
Dalak hacmi ≥%35 azalm�� g�n�ll� say�s� (%) | 54 (47,8) | 11 (27,5) | 1 (0,8) | 0 | 36 (32,7) | 5 (14.3) | 0 | 0 |
Zaman noktas� | 24 hafta sonra | 48 hafta sonra | ||||||
JAKAVI tedavisinde en az 24 hafta s�reyle dalak yan�t�n� (≥%35 azalma) s�rd�rme olas�l��� COMFORT-I �al��mas�nda %89 ve COMFORT-II �al��mas�nda %87 olmu�tur; COMFORT- II �al��mas�nda hastalar�n %52'si dalak yan�tlar�n� en az 48 hafta s�reyle korumu�tur.
COMFORT-I �al��mas�nda JAKAVI grubundaki hastalar�n %45,9'unda toplam semptom skorunda (MFSAF g�nl��� v2.0 kullan�larak �l��len) 24. haftada ba�lang�ca g�re ≥%50 d�zelme elde edilirken bu oran plasebo grubunda %5,3 bulunmu�tur (ki-kare testi kullan�larak p<0,0001). 24. haftada Avrupa Kanser Ara�t�rma ve Tedavi Organizasyonu (EORTC) QLQ C30 ile �l��len global sa�l�k durumundaki ortalama de�i�iklik JAKAVI i�in +12,3 iken plasebo i�in -3,4 olmu�tur (p<0,0001).
COMFORT-I �al��mas�nda 34,3 ayl�k medyan takip sonras�nda �l�m oran�, ruksolitinib koluna randomize edilen hastalarda %27,1 iken plaseboya randomize edilen hastalarda %35.1 bulunmu�tur; HR 0,687; %95 GA 0,459-1,029; p=0,0668.
COMFORT-I �al��mas�nda 61,7 ayl�k medyan takip sonras�nda �l�m oran�, ruksolitinib koluna randomize edilen hastalarda %44,5 (155 hastan�n 69'u) iken plaseboya randomize edilen hastalarda %53,2 (154 hastan�n 82'si) bulunmu�tur. Ruxolitinib kolunda plaseboya g�re �l�m riskinde % 31'lik bir azalma olmu�tur (HR 0,69; % 95 GA 0,50-0,96; p = 0,025).
COMFORT-II �al��mas�nda 34,7 ayl�k medyan takip sonras�nda �l�m oran�, ruksolitinib koluna randomize edilen hastalarda %19,9 iken mevcut en iyi tedaviye (BAT) randomize edilen hastalarda %30,1 olmu�tur; HR 0,48; %95 GA 0,28-0,85; p=0,009. Her iki �al��mada da ruksolitinib kolunda belirlenen daha d���k �l��m oranlar�n�n a��rl�kl� sebebi, post- polisitemi vera ve post-esansiyel trombositemi alt gruplar�nda elde edilen sonu�lar olmu�tur.
COMFORT-II �al��mas�nda 55,9 ayl�k medyan takip sonras�nda �l�m oran�, ruksolitinib koluna randomize edilen hastalarda %40,4 (146 hastan�n 59'u) iken mevcut en iyi tedaviye (BAT) randomize edilen hastalarda %47,9 (73 hastan�n 35'i) olmu�tur; Ruksolitinib kolunda plaseboya g�re �l�m riskinde % 33'lik bir azalma olmu�tur (HR 0,67; % 95 GA 0,44-1,02; p
= 0,062).
Polisitemi vera
Avrupa LeukemiaNet (ELN) uluslararas� �al��ma grubunun yay�mlam�� oldu�u kriterlere g�re tan�mlanan, hidroksi�re tedavisine diren�li veya yan�t vermeyen 222 PV hastas� ile randomize, a��k etiketli, aktif kontroll� bir faz 3 �al��ma (RESPONSE) ger�ekle�tirilmi�tir. 110 hasta ruksolitinib koluna ve 112 hasta BAT koluna randomize edilmi�tir. JAKAVI'nin ba�lang�� dozu g�nde iki kez 10 mg olmu�tur. Dozlar daha sonra tolerabilite ve etkililik esas al�narak, maksimum doz g�nde iki kez 25 mg olacak �ekilde hasta baz�nda d�zenlenmi�tir. BAT ara�t�rmac� taraf�ndan hasta baz�nda se�ilmi�tir ve �unlar� i�ermi�tir: hidroksi�re (%59,5), interferon/pegile interferon (%11,7), anagrelid (%7,2), pipobroman (%1,8) ve
g�zlem (%15,3).
Ba�lang��taki demografik �zellikler ve hastal�k karakteristikleri iki tedavi kolu aras�nda kar��la�t�r�labilir niteliktedir. Medyan ya� 60't�r (aral�k 33-90 ya�). Ruksolitinib kolundaki hastalarda medyan 8,2 y�l PV tan�s� mevcuttur ve bu hastalar �nceden medyan olarak yakla��k 3 y�l s�reyle hidroksi�re tedavisi alm��t�r. �o�u hastaya (>%80) tarama �ncesindeki son 24 haftada en az iki flebotomi uygulanm��t�r. Uzun s�reli sa�kal�m ve hastal�k komplikasyonlar�n�n insidans�na y�nelik kar��la�t�rmal� veri mevcut de�ildir.
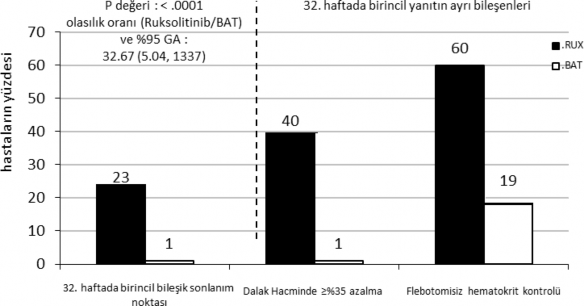
Birincil bile�ik sonlan�m noktas�n� hem flebotomiye uygunlu�un (HCT kontrol�) olmay��� hem de 32. haftada dalak b�y�kl���nde ba�lang�ca g�re ≥%35 azalma durumuna ula�an hastalar�n oran� olu�turmu�tur. Flebotomiye uygunluk, do�rulanan HCT >%45 �eklinde, yani ba�lang��ta elde edilen HCT'ye k�yasla en az 3 y�zde puan� fazla ya da do�rulanan HCT
>%48 �eklinde (hangisinin daha d���k oldu�una ba�l� olarak) tan�mlanm��t�r. Ba�l�ca ikincil sonlan�m noktalar� aras�nda birincil sonlan�m noktas�na ula�an ve 48. haftada progresyonsuz kalan hastalar�n yan� s�ra 32. haftada tam hematolojik remisyona ula�an hastalar�n oran� yer alm��t�r.
�al��ma birincil amac�n� kar��lam��t�r ve JAKAVI grubundaki hastalar�n daha y�ksek bir oran� birincil bile�ik sonlan�m noktas�na ve bile�enlerinin her birine ula�m��t�r. BAT (%0,9) ile kar��la�t�r�ld���nda, JAKAVI ile tedavi edilen hastalar�n anlaml� derecede daha �o�u (%23) bir birincil yan�ta ula�m��t�r (p<0,0001). Hematokrit kontrol� JAKAVI kolundaki hastalar�n
%60'�nda elde edilirken bu oran BAT kolunda %18,8 bulunmu�tur ve dalak hacminde ≥%35 azalma, JAKAVI kolundaki hastalar�n %40'�nda, BAT kolundaki hastalar�n ise %0,9'unda elde edilmi�tir (�ekil 1).
Ayr�ca her iki ikincil sonlan�m noktas� da kar��lanm��t�r. Tam hematolojik remisyona ula�an
hastalar�n oran� JAKAVI kolunda %23,6 iken BAT kolunda %8,0 bulunmu�tur (p=0,0013) ve
48. haftada kal�c� birincil yan�ta ula�an hastalar�n oran� JAKAVI ile %20 ve BAT ile %0,9 olmu�tur (p<0,0001).
�ekil 1 32. haftada birincil sonlan�m noktas�na ve birincil sonlan�m noktas�n�n bile�enlerine ula�an hastalar
Semptom y�k�, 14 sorudan olu�an MPN-SAF toplam semptom skoru (TSS) elektronik hasta g�nl��� kullan�larak de�erlendirilmi�tir. 32. haftada ruksolitinib ile tedavi edilen hastalar�n s�ras�yla %49'unda ve %64'�nde TSS-14 ve TSS-5'te ≥%50 azalma elde edilirken BAT tedavisindeki hastalarda bu oranlar sadece %5 ve %11 olmu�tur.
Tedavi faydas� alg�s�, Hasta Global De�i�iklik �zlenimi (PGIC) anketi kullan�larak �l��lm��t�r. BAT ile tedavi edilen hastalar�n %19'u kar��s�nda ruksolitinib ile tedavi edilen hastalar�n %66's�, tedavi ba�lad�ktan sonra hen�z 4 hafta gibi k�sa bir s�rede bir d�zelme bildirmi�tir. Tedavi faydas� alg�s�nda d�zelme 32. haftada da ruksolitinib ile tedavi edilen hastalarda daha y�ksek olmu�tur (%78 kar��s�nda %33).
RESPONSE �al��mas�nda, randomizasyonun ard�ndan 80. haftada ve 256. haftada yan�t�n kal�c�l���na y�nelik ek analizler ger�ekle�tirilmi�tir. 32. haftada birincil yan�t elde edilen 25 hastadan 3 hasta 80. hafta itibariyle, 6 hasta 256. hafta itibariyle progrese olmu�tur. Yan�t�n
32. haftadan sonra 80. hafta ve 256. haftaya kadar devam etme olas�l��� s�ras�yla % 92 ve % 74 olmu�tur (bkz. Tablo 4).
Tablo 8 RESPONSE �al��mas�nda birincil yan�t�n kal�c�l���
| Hafta 32 | Hafta 80 | Hafta 256 |
32. haftada ula��lan birincil yan�t * n/N (%) | 25/110 (% 23) | n/a | n/a |
Birincil yan�t� s�rd�ren hastalar | n/a | 22/25 | 19/25 |
Birincil yan�t� s�rd�rme olas�l��� | n/a | % 92 | % 74 |
* Birincil yan�t bile�ik sonlan�m noktas� kriterlerine g�re: flebotomi uygunlu�unun olmamas� (HCT kontrol�) ve dalak hacminde ba�lang�ca g�re ≥ % 35 azalma.
n/a : uygulanamaz
Hidroksi�reye diren�li olan ya da hidroksi�reyi tolere edemeyen fakat palpabl splenomegalisi olmayan 149 PV hastas�nda ikinci bir randomize, a��k etiketli, aktif kontroll�, faz 3b �al��ma (RESPONSE 2) ger�ekle�tirilmi�tir. 28. haftada HCT kontrol� (flebotomiye uygunlu�un olmamas�) elde edilen hastalar�n oran� �eklinde tan�ml� birincil sonlan�m noktas� kar��lanm��t�r (JAKAVI kolunda %62,2 kar��s�nda BAT kolunda %18,7). 28. haftada tam hematolojik remisyona ula�an hastalar�n oran� �eklinde tan�ml� ana ikincil sonlan�m noktas�na da ula��lm��t�r (JAKAVI kolunda %23 kar��s�nda BAT kolunda %5,3).
Graft-versus-host hastal���
�ki randomize faz 3, a��k etiketli, �ok merkezli �al��mada allojenik hematopoietik k�k h�cre transplantasyonundan (alloSCT) sonra akut GvHD (REACH2) ve kronik GvHD'si (REACH3) olan ve kortikosteroidlere/veya di�er sistemik tedavilere yetersiz yan�t veren 12 ya� ve �zerindeki hastalarda JAKAVI incelenmi�tir. JAKAVI'nin ba�lang�� dozu g�nde iki kez 10 mg olmu�tur.
Akut graft-versus-host hastal���
REACH2'de, kortikosteroidlere derece II ila IV diren�li, akut GvHD'li 309 hasta, JAKAV� veya mevcut en iyi tedaviye 1:1 oran�nda randomize edilmi�tir. Hastalar, randomizasyon s�ras�nda akut GvHD'nin ciddiyetine g�re katmanland�r�lm��t�r. Kortikosteroid refrakterli�i, hastalarda en az 3 g�n sonra progresyon oldu�unda, 7 g�n sonra yan�t al�namad���nda veya kortikosteroid azalt�m�n�n ba�ar�s�z oldu�u durumlar olarak belirlenmi�tir.
Mevcut en iyi tedavi(BAT), ara�t�rmac� taraf�ndan hasta baz�nda se�ilmi�tir ve anti-timosit globulin (ATG), ekstrakorporeal fotoferez (ECP), mezenkimal stromal h�creler (MSC), d���k doz metotreksat (MTX), mikofenolat mofetil (MMF), mTOR inhibit�rleri (everolimus veya sirolimus), etanersept veya infliksimabi i�ermi�tir.
JAKAVI veya mevcut en iyi tedaviye ek olarak, hastalar anti-enfektif t�bbi �r�nler ve transf�zyon deste�i dahil olmak �zere standart allojenik k�k h�cre nakli destekleyici bak�m alm�� olabilir. Ruksolitinib, enstit�lerin k�lavuzlar�na g�re kortikosteroidlerin ve/veya siklosporin veya takrolimus gibi kalsin�rin inhibit�rlerinin (CNI'ler) ve/veya topikal veya inhale kortikosteroid tedavilerinin s�rekli kullan�m�na eklenmi�tir.
Akut GvHD i�in kortikosteroidler ve kalsin�rin inhibit�rlerinin (CNI) d���nda �nceden bir sistemik tedavi alm�� hastalar �al��maya dahil edilmeye uygun bulunmu�tur. Kortikosteroidler ve kalsin�rin inhibit�rlerine (CNI) ek olarak, akut GvHD i�in �nceki sistemik t�bbi �r�n�n, yayg�n t�bbi uygulamaya g�re yaln�zca akut GvHD profilaksisi i�in kullan�lmas� durumunda (yani akut GvHD te�hisinden �nce ba�lam��sa), devam�na izin verilmi�tir.
Mevcut en iyi tedavi alan hastalar, a�a��daki kriterleri kar��lamalar� halinde 28. g�nden sonra ruksolitinibe ge�ebilmi�lerdir:
28. g�nde birincil sonlan�m noktas� yan�t tan�m� (tam yan�t [CR] veya k�smi yan�t [PR]) kar��lanamam��sa; VEYA
5.2. Farmakokinetik �zellikler
Genel �zelliklerEmilim
Biyofarmas�tik S�n�fland�rma Sistemine g�re ruksolitinib y�ksek permeabilite, y�ksek ��z�n�rl�k ve h�zl� da��l�m karakteristiklerine sahip bir S�n�f 1 molek�ld�r. Klinik �al��malarda, ruksolitinib, oral uygulamadan sonra h�zla emilmekte, maksimal plazma konsantrasyonuna (C) dozdan yakla��k 1 saat sonra ula��lmaktad�r. �nsanda ruksolitinibin oral emilimi ≥ %95'tir. Ortalama ruksolitinib Cve toplam maruziyet (EAA) de�erleri, 5 ila 200 mg tek doz aral���nda orant�l� olarak artm��t�r. Y�ksek oranda ya� i�eren bir ���nle birlikte uyguland���nda ruksolitinibin farmakokineti�inde klinik olarak anlaml� bir de�i�iklik olmam��t�r. Y�ksek oranda ya� i�eren ���nle verildi�inde Corta d�zeyde azalma g�stermi� (%24), ortalama EAA ise neredeyse hi� de�i�memi�tir (%4 art��).
Da��l�m
MF ve PV hastalar�nda kararl� durumda g�r�n�r da��l�m hacmi yakla��k 75 litredir. Klinik olarak anlaml� ruksolitinib konsantrasyonlar�nda, in vitro ortamda plazma proteinlerine ba�lanma yakla��k %97 olup a��rl�kl� olarak alb�mine ba�lan�r. S��anlarda tam v�cut otoradyografi �al��mas�, ruksolitinibinkan-beyinbariyerinige�medi�ini g�stermi�tir.
Biyotransformasyon
Ruksolitinib CYP2C9'un ilave katk�s� ile ba�l�ca CYP3A4 ile metabolize edilir (>%50). Ana bile�ik, insanlarda bask�n olup dola��mdaki ila�la ili�kili maddelerin yakla��k %60'�n� olu�turmaktad�r. Plazmada ana EAA'n�n %25 ve %11'ini temsil eden iki ana ve aktif metabolit mevcuttur. Bu metabolitler, ana bile�i�in JAK ili�kili farmakolojik aktivitesinin yar�s� ila be�te birine sahiptir. T�m aktif metabolitlerin toplam�, ruksolitinibin toplam farmakodinamik etkisine %18 katk� yapmaktad�r. �n vitro �al��malara g�re, ruksolitinib, klinik olarak anlaml� konsantrasyonlarda CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 ya da CYP3A4'� inhibe etmez ve CYP1A2, CYP2B6 ya da hepatik CYP3A4'�n kuvvetli ind�kleyicisi de�ildir. In vitro �al��malara g�re ruksolitinib P-gp ve meme kanserine diren� proteini inhibe edebilir.
Eliminasyon:
Ruksolitinib, b�y�k oranda metabolizma yoluyla elimine edilir. Ruksolitinibin ortalama eliminasyon yar�lanma �mr� yakla��k 3 saattir. Sa�l�kl� eri�kin g�n�ll�lerde [14C] i�aretli ruksolitinibin tek oral dozundan sonra eliminasyon ba�l�ca metabolizma yoluyla olmu�, radyoaktivitenin %74'� idrarda ve %22'si fe�este tespit edilmi�tir. De�i�memi� ila�, at�lan toplam radyoaktivitenin %1'inden az�n� olu�turmu�tur.
Do�rusall�k/do�rusal olmayan durum:
Tek ve �oklu doz �al��malar�nda doz oransall��� g�sterilmi�tir.
Hastalardaki karakteristik �zellikler
V�cut y�zey alan�, ya�, cinsiyet veya �rk�n etkileri
Sa�l�kl� g�n�ll�lerde yap�lan �al��malara g�re, cinsiyet ve �rka g�re ruksolitinib farmakokineti�inde anlaml� herhangi bir farkl�l�k g�zlenmemi�tir. MF hastalar�nda yap�lan bir pop�lasyon farmakokinetik de�erlendirmesinde, oral klirens ile hastan�n ya�� veya �rk� aras�nda herhangi bir ili�ki g�r�lmemi�tir. �ng�r�len oral klirens, MF hastalar�nda hastalar aras� %39 de�i�kenlik ile kad�nlarda 17,7 L/saat ve erkeklerde 22,1 L/saat olmu�tur. PV hastalar�nda klirens 12,7 L/saat olup hastalar aras� de�i�kenlik %42 bulunmu� ve PV hastalar�nda yap�lan bir pop�lasyon farmakokinetik de�erlendirmesine g�re oral klirens ile cinsiyet, hastan�n ya�� veya �rk� aras�nda herhangi bir ili�ki g�r�lmemi�tir. Klirens, akut GvHD'li hastalarda 10.4 L/saat ve kronik GvHD'li hastalarda 7,8 L/saat olup, hastalar aras�nda %49 de�i�kenlik g�stermi�tir. GvHD hastalar�nda bir pop�lasyon farmakokinetik de�erlendirmesine dayal� olarak, oral klirens ile cinsiyet, hastan�n ya�� veya �rk� aras�nda herhangi bir ili�ki g�r�lmemi�tir. D���k v�cut y�zey alan�na sahip GvHD hastalar�nda maruziyet artm��t�r. V�cut y�zey alan� 1 m2, 1,25 m2 ve 1,5 m2 olan hastalarda �ng�r�len ortalama maruziyet (EAA) tipik bir yeti�kinden (1,79 m2) s�ras�yla %31, %22 ve %12 daha y�ksek olmu�tur.
Pediyatrik pop�lasyon
MF ve PV'li 18 ya��ndan k���k pediyatrik hastalarda JAKAVI'nin farmakokineti�i belirlenmemi�tir. Akut veya kronik GvHD'li ad�lesan hastalarda g�zlenen farmakokinetik profil, genel hasta pop�lasyonu ile benzer olmu�tur (bkz. B�l�m 5.1, “Pediyatrik pop�lasyon”). Ruksolitinib, 12 ya��n alt�ndaki akut veya kronik GvHD'li pediyatrik hastalarda hen�z de�erlendirilmemi�tir.
B�brek yetmezli�i:
B�brek fonksiyonu hem B�brek Hastal���nda Diyet Modifikasyonu (MDRD) hem de �riner
kreatinin kullan�larak tespitedilmi�tir.Ruksolitinibin25mg'l�k tek dozundan sonra
ruksolitinibe maruziyet �e�itli derecede b�brek yetmezli�i olan hastalar ile normal b�brek fonksiyonuna sahip olanlar aras�nda benzer olmu�tur. Ancak ruksolitinib metabolitlerinin plazma EAA de�erleri, b�brek yetmezli�inin �iddeti artt�k�a y�kselme e�ilimi g�stermi�, bu durum �iddetli b�brek yetmezli�i olan hastalarda belirginlik kazanm��t�r. Artm�� metabolit maruziyetinin bir g�venlilik endi�esi te�kil edip etmedi�i bilinmemektedir. �iddetli b�brek yetmezli�i ve son d�nem b�brek yetmezli�i hastalar� i�in doz plan�nda d�zenleme �nerilmektedir (bkz. B�l�m 4.2). Dozlar�n sadece diyaliz g�nlerinde uygulanmas� metabolit maruziyetini azalt�r fakat ba�ta diyaliz aras� g�nlerde olmak �zere farmakodinamik etkiyi de azaltmaktad�r.
Karaci�er yetmezli�i:
�e�itli derecelerde karaci�er yetmezli�i olan hastalarda 25 mg'l�k tek bir ruksolitinib dozunun uygulanmas�n�n ard�ndan; ruksolitinibin ortalama EAA de�eri,hafif, orta ve �iddetli karaci�er yetmezli�i olan hastalarda normal karaci�er fonksiyonu olan hastalara k�yasla s�ras�yla %87,
%28 ve %65 oranlarda artm��t�r.Child-Pugh skorlar�na dayal� karaci�er yetmezli�i derecesi ile a��k bir ili�ki bulunmamaktad�r. Sa�l�kl� kontrollere k�yasla karaci�er yetmezli�i olan hastalarda terminal eliminasyon yar� �mr� uzam��t�r (2,8 saate kar��l�k 4,1-5,0 saat). Karaci�er yetmezli�i olan MF ve PV hastalar� i�in dozun %50 oran�nda azalt�lmas� �nerilmektedir (bkz. B�l�m 4.2).
GvHD ile ili�kili olmayan karaci�er yetmezli�i olan GvHD hastalar�nda, ruksolitinibin ba�lang�� dozu %50 oran�nda azalt�lmal�d�r.
5.3. Klinik �ncesi g�venlilik verileri
Ruksolitinib; g�venlilik farmakolojisi, tekrarl� doz toksisitesi, genotoksisite, �reme toksisitesi �al��malar�nda ve bir karsinojenisite �al��mas�nda de�erlendirilmi�tir. Tekrarl� doz �al��malar�nda ruksolitinibin farmakolojik etkisi ile ili�kili hedef organlar kemik ili�ini, periferik kan� ve lenfoid dokular� i�ermi�tir. K�peklerde, genellikle imm�n sistemin bask�lanmas�na ba�l� enfeksiyonlar g�zlenmi�tir. K�pek telemetri �al��mas�nda kan bas�nc�nda, kalp h�z�nda art��lar�n e�lik etti�i tersine d����ler ve s��anlarda solunum �al��mas�nda dakika hacminde bir tersine d���� g�r�lm��t�r. K�pek ve s��an �al��malar�ndaki advers olmayan d�zeydeki marjlar (ba�lanmam�� Cbaz�nda), g�nde iki kez 25 mg �eklindeki maksimum �nerilen insan dozundan s�ras�yla 15,7 ve 10,4 kat fazlad�r. Ruksolitinibin n�rofarmakolojik etkilerine y�nelik bir de�erlendirmede herhangi bir etki g�r�lmemi�tir.
J�venil s��an �al��malar�nda ruksolitinib uygulamas� b�y�me ve kemik �l��mleri �zerinde etkili olmu�tur. ≥5 mg/kg/g�n dozlar�nda tedaviye postnatal 7. g�nde (insanda yenido�an ile kar��la�t�r�labilir) ba�land���nda ve ≥ 15 mg/kg/g�n dozlar�nda tedaviye postnatal 14 veya 21. g�nlerde (insanda 1-3 ya� bebek ile kar��la�t�r�labilir) ba�lad���nda azalm�� kemik b�y�mesi g�zlenmi�tir. ≥30 mg/kg/g�n dozlar�nda tedaviye postnatal 7. g�nde ba�land���nda s��anlarda k�r�klar ve erken terminasyon g�zlemlenmi�tir. Ba�lanmam�� EAA de�erine dayan�larak, postnatal 7. g�n gibi erken bir d�nemde tedavi edilen j�venil s��anlarda NOAEL (advers etkinin g�zlenmedi�i d�zey) d�zeyinde maruziyet, g�nde iki kez 25 mg dozunda eri�kin hastalarda g�r�lenin 0.3 kat� olurken g�nde iki kez 25 mg dozunda eri�kin hastalarda g�r�lenin s�ras�yla 1.5 ve 13 kat� maruziyetlerde azalm�� kemik b�y�mesi ve k�r�klar g�r�lm��t�r. Etkiler, tedavi postnatal d�nemde daha erken ba�land���nda genellikle daha �iddetli olmu�tur. Kemik geli�imi d���nda j�venil s��anlarda ruksolitinibin etkisi yeti�kin s��anlardakine benzer olmu�tur. J�venil s��anlar, yeti�kin s��anlara g�re ruksolitinib toksisitesine kar�� daha hassast�r.
d����ler ile ili�kilendirilmi�tir. S��anlarda ve tav�anlarda teratojenik etki y�n�nde bir kan�t s�z konusu olmam��t�r. Di�er yandan, en y�ksek klinik doz ile kar��la�t�r�ld���nda maruziyet marjlar� d���k bulunmu�tur ve dolay�s�yla sonu�lar, insanlar a��s�ndan s�n�rl� �neme sahiptir. Fertilite �zerinde herhangi bir etki g�zlenmemi�tir. Prenatal ve postnatal geli�im �al��mas�nda gestasyon s�resinde hafif uzama, implantasyon b�lgelerinin say�s�nda azalma ve do�urulan yavru say�s�nda d���� g�zlenmi�tir. Yavru hayvanlarda ilk beden a��rl�klar� ortalamas�nda azalma ve k�sa s�reli azalm�� ortalama beden a��rl��� art��� g�zlenmi�tir. Emziren s��anlarda ruksolitinib ve/veya metabolitleri, maternal plazma konsantrasyonlar�n�n 13 kat� daha y�ksek bir konsantrasyonla s�te at�lm��t�r. Ruksolitinib, mutajenik ya da klastojenik etki g�stermemi�tir. Ruksolitinib, Tg.rasH2 transjenik fare modelinde karsinojenik etki g�stermemi�tir.
6. FARMAS�T�K �ZELL�KLER
6.1. Yard�mc� maddelerin listesi
Mikrokristalin sel�loz Magnezyum stearatKoloidal susuz silika
Sodyum ni�asta glikolat (Tip A) Hidroksipropilsel�loz (300-600 cps) Povidon K30
Laktoz monohidrat (s���r kaynakl�)
6.2. Ge�imsizlikler
Yeterli veri yoktur.
6.3. Raf �mr�
36 ay
6.4. Saklamaya y�nelik �zel tedbirler
tedbirler25°C'nin alt�ndaki oda s�cakl���nda saklay�n�z.
6.5. Ambalaj�n niteli�i ve i�eri�i
PVC/PCTFE blister ambalajlar
Ambalaj b�y�kl���: 14, 56 ve 168 tablet i�eren blister ambalaj
Blister ambalajlar �s�yla kapat�labilir lakla kaplanm�� al�minyum folyoyla kapat�lm��t�r
6.6. Be�eri t�bbi �r�nden arta kalan maddelerin imhas� ve di�er �zel �nlemler
Kullan�lmam�� olan �r�nler ya da at�k materyaller “T�bbi At�klar�n Kontrol� Y�netmeli�i” ve
“Ambalaj At�klar�n�n Kontrol� Y�netmeli�i”ne uygun olarak imha edilmelidir.
 Diyabet Hastal���
Diyabet, ins�lin hormonu ile ilgili problemlerden kaynaklanan bir hastal�kt�r.
Diyabet Hastal���
Diyabet, ins�lin hormonu ile ilgili problemlerden kaynaklanan bir hastal�kt�r. |
 Tiroid Kanseri
En s�k g�r�len tiroid kanseri t�r� olan papiller tiroid kanseri, t�m tiroid kanserlerinin yakla��k %70'ini olu�turur.
Tiroid Kanseri
En s�k g�r�len tiroid kanseri t�r� olan papiller tiroid kanseri, t�m tiroid kanserlerinin yakla��k %70'ini olu�turur. |
 |
A��r� Alkol Kullan�m�, Alkolizm Alkol ba��ml�l���, alkol kullan�m� ve alkol sorunlar� aras�ndaki fark� a��klamak g��t�r. �rne�in, ge�mi�te alkol kullanm�� olan bir kimsenin mutlaka alkol ba��ml�s� olmas� gerekmez. |
 |
Ast�m Ast�ml� ki�ilerin akci�erlerindeki hava borular� (bron�lar) hassast�r. Bu ki�iler belirli tetikleyici fakt�rlere maruz kald�klar�nda, hava borular� nefes almalar�n� g��le�tirecek �ekilde daral�r. |
 |
L�semi Kan Kanseri L�semi, kan kanseridir ve v�cudunun kan olu�turan dokular�n�n hastalanmas� anlam�na gelir. Bir�ok l�semi t�r� vard�r; baz� l�semi t�rleri �ocuklarda baz�lar� da yeti�kinlerde s�k g�r�l�r. |
�LA� GENEL B�LG�LER�
Novartis Sa�l�k,G�da ve Tar�m �r�nleri San. Tic. A.�.
| Geri �deme Kodu | A15608 |
| Sat�� Fiyat� | TL |
| �nceki Sat�� Fiyat� | |
| Original / Jenerik | Original �la� |
| Re�ete Durumu | Normal Re�eteli bir ila�d�r. |
| Barkodu | 8699504012149 |
| Etkin Madde | Ruksolitinib |
| �thal ( ref. �lke : Isvicre ) ve Be�eri bir ila�d�r. |