KYPROLIS 60 mg IV enjeksiyonluk çözelti için toz içeren flakon (1 flakon) Kısa Ürün Bilgisi
{ Karfilzomib }
1. BEŞERİ TIBBİ ÜRÜNÜN ADI
KYPROLİS® 60 mg IV enjeksiyonluk çözelti için toz içeren flakon Steril
2. KALİTATİF VE KANTİTATİF BİLEŞİM
Etkin madde
Her bir flakon 60 mg karfilzomib içerir.
Sulandırıldıktan sonra, 1 mL çözelti, 2 mg karfilzomib içerir.
Yardımcı maddeler
Sulandırıldıktan sonra KYPROLİS®'in her mL'si, 7 mg sodyuma karşılık gelen 0,3 mmol sodyum içerir.
Yardımcı maddeler
3. FARMASÖTİK FORMU
Enjeksiyonluk çözelti için toz.
Steril, beyaz veya beyazımsı renkte liyofilize toz, tek kullanımlık flakon olarak mevcuttur.
derece ve üstü olgu gelişmiştir), giderilmiş, nadiren tedavinin kesilmesi ile sonuçlanmış ve çalışmanın erken bir döneminde (< 3 döngü) ortaya çıkma özelliği göstermiştir. KYPROLİS® tedavisi sırasında dispnenin klinik tedavisi için bkz. bölüm 4.4.
Her bir KYPROLİS® flakonunu yalnızca Steril Enjeksiyonluk Su ile aşağıda açıklanan hacmi kullanarak aseptik şekilde sulandırın. 21 gauge veya daha büyük bir iğne (dış çapı 0,8 mm veya daha küçük olan iğne) kullanarak her flakonu yavaşça 29 mL steril enjeksiyonluk su enjekte ederek hazırlayın, köpüklenmeyi minimize etmek için Steril Enjeksiyonluk Suyu tıpa üzerinden FLAKONUN İÇ DUVARINA doğru yönlendirin. KYPROLİSile kapalı sistem transfer cihazlarının kullanımını destekleyen veri mevcut değildir.

4. KLİNİK ÖZELLİKLER
4.1. Terapötik endikasyonlar
KYPROLİS® daha önce 1-3 seri tedavi almış yetişkin relaps veya refrakter multipl miyelom hastalarının tedavisinde aşağıdakiler ile kombinasyon halinde kullanımda endikedir:
Lenalidomid ve deksametazon ya da
Deksametazon
KYPROLİS® daha önce bortezomib ve immunmodülatuar ilaç (IMID) içeren en az 1-3 sıra kombinasyon tedavisi uygulanmış relaps ve refrakter multipl miyelom hastalarında Daratumumab ve deksametazon ile kombinasyon halinde kullanımda endikedir.
KYPROLİS® daha önce 1 veya daha fazla seri tedavi almış yetişkin relaps veya refrakter multipl miyelom hastalarının tedavisinde tek başına kullanımda endikedir.
4.2. Pozoloji ve uygulama şekli
KYPROLİS® tedavisi anti-kanser tedavilerinde deneyimli bir doktor tarafından izlenmelidir.
Yardımcı madde siklodekstrinin süreli kullanımının taşıdığı güvenlik riskleri nedeniyle, ürünün 14 günlük tedavi sonrasında, hekim tarafından gerekli değerlendirmenin yapılıp, kullanıma devam edilip edilmeyeceğinin belirlenmesi gerekmektedir.
Pozoloji/uygulama sıklığı ve süresi:
Uygulamada dikkat edilmesi gereken konular
Hidrasyon - 1. döngüde dozlama öncesinde, özellikle tümör lizis sendromu (TLS) veya renal toksisite riski yüksek olan hastalarda yeterli hidrasyon gereklidir. Hem oral sıvılar (1. döngü, 1. günden en az 48 saat önce 30 ml/kg) hem de intravenöz sıvılar (1. döngüde her doz öncesinde 250 mL ila 500 mL uygun intravenöz sıvı) ile hidrasyon düşünülmelidir. KYPROLİS® uygulamasının ardından, eğer gerekirse 250 mL ila 500 mL ilave intravenöz sıvı verilebilir. Sonraki döngülerde oral ve/veya intravenöz sıvı ile hidrasyona gerektikçe devam edilir. Özellikle kalp yetmezliği olan veya kalp yetmezliği riski taşıyan hastalar aşırı miktarda sıvı yüklemesi açısından izlenmelidir ve hidrasyon her hastanın gereksinimlerine göre ayarlanmalıdır (bkz. bölüm 4.4).
Elektrolit izleme - KYPROLİS® ile tedavi süresince serum potasyum düzeyleri düzenli olarak izlenmelidir (bkz. bölüm 4.4).
Premedikasyonlar ve Eşlik Eden İlaçlar - Monoterapi için deksametazonun önerilen dozu ile veya kombinasyon tedavisinin parçası olarak uygulanan deksametazon ile premedikasyon uygulanmalıdır (bkz. bölüm 4.2). İnfüzyonla ilişkili reaksiyonlarının insidansını ve şiddetini azaltmak amacıyla 1. döngü sırasında KYPROLİS®'in tüm dozlarınının uygulanmasından en az 30 dakika, en fazla 4 saat öncesinde oral veya intrevanöz yoldan deksametazon uygulanmalıdır (bkz. bölüm 4.4). Sonraki döngüler sırasında bu semptomlar ortaya çıkarsa tekrar deksametazon premedikasyonu uygulanmalıdır.
KYPROLİS®'in diğer tedaviler ile birlikte kombinasyon halinde uygulanacağı hastalarda tromboprofilaksi sağlanmalıdır (bkz. bölüm 4.4).
Herpes zoster reaktivasyonu riskini azaltmak için antiviral profilaksi düşünülmelidir (bkz. bölüm 4.8).
Doz hesaplama - Vücut yüzey alanı (VYA) değeri 2,2 m2'den az veya bu değere eşit olan hastalar için KYPROLİS® dozu (bkz. bölüm 4.2), mutlak VYA kullanılarak hesaplanmalıdır. Vücut ağırlığında %20 oranında veya daha az değişiklikler için doz ayarlaması yapılması gerekli değildir. VYA değeri 2,2 m2'den fazla olan hastalar için VYA 2,2 m2 kullanılarak KYPROLİS® dozu hesaplanmalıdır.
Önerilen dozlama
Deksametazon ile kombinasyon halinde KYPROLİS
![]()
30 dakikalık infüzyonla haftada bir 20/70 mg/m2 rejimi
KYPROLİS® Tablo 1'de gösterildiği gibi, hastalık progresyonuna veya kabul edilemez toksisite meydana gelene kadar deksametazon ile kombinasyon halinde her 28 günlük döngünün 1, 8 ve 15. Günlerinde 30 dakikalık infüzyon şeklinde intravenöz yolla uygulanır.
Döngünün 1. Gününde, KYPROLİS® için önerilen başlangıç dozu 20 mg/m2'dir. Tolere
edilirse, 1. Döngünün 8. Gününde doz 70 mg/m2'ye çıkarılır. Deksametazon, KYPROLİS®'ten 30 dakika ila 4 saat önce uygulanır. Ek dozaj bilgileri için deksametazonun Ürün Bilgilerine bakın.
Tablo 1: Deksametazonla kombinasyon halinde haftada bir KYPROLİS20/70 mg/m(30 dakikalık infüzyon)
| ||||||||||||
|
|
|
|
|
|
|
|
|
|
|
| |
| ||||||||||||
| ||||||||||||
| ||||||||||||
![]()
30 dakikalık infüzyonla haftada iki defa 20/56 mg/m2 rejimi
KYPROLİS® Tablo 2'de gösterildiği gibi, hastalık progresyonuna veya kabul edilemez toksisite meydana gelene kadar deksametazon ile kombinasyon halinde her 28 günlük döngünün 1, 2, 8, 9, 15 ve 16. Günlerinde 30 dakikalık infüzyon şeklinde intravenöz yolla uygulanır. 1. Döngünün 1. ve 2. Günlerinde, KYPROLİS® için önerilen başlangıç dozu 20 mg/m2'dir. Tolere edilirse, 1. Döngünün 8. Gününde doz 56 mg/m2'ye çıkarılır. 28 günlük
döngülerin her birinin 1, 2, 8, 9, 15, 16, 22 ve 23. Günlerinde ağızdan veya intravenöz yolla 20 mg deksametazon alınır. Deksametazon, KYPROLİS®'ten 30 dakika ila 4 saat önce uygulanır. Ek dozaj bilgileri için deksametazonun Ürün Bilgilerine bakın.
Tablo 2: Deksametazonla kombinasyon halinde haftada iki defa KYPROLİS20/56 mg/m(30 dakikalık infüzyon)
| ||||||||||||
| ||||||||||||
|
|
|
|
|
|
|
|
|
| |||
Lenalidomid ve deksametazon ile kombinasyon halinde KYPROLİS
KYPROLİS® Tablo 3'te gösterildiği gibi, 12. Döngüye kadar lenalidomid ve deksametazon ile kombinasyon halinde her 28 günlük döngünün 1, 2, 8, 9, 15 ve 16. Günlerinde 10 dakikalık infüzyon şeklinde intravenöz yolla uygulanmalıdır. KYPROLİS® için önerilen başlangıç dozu
1. döngünün 1. ve 2. günlerinde 20 mg/m2'dir. Tolere edilirse, 1. döngünün 8. gününde doz
mg/m2'ye çıkartılmalıdır. KYPROLİS®'i 13. döngüden 18. döngüye kadar 1., 2., 15. ve 16. günlerde uygulayınız. 18. döngüden sonra KYPROLİS® uygulaması kesilmelidir. Lenalidomid ve deksametazona, hastalık progresyonuna veya kabul edilemez toksisite meydana gelene kadar devam edilmelidir. Ek dozaj bilgileri için lenalidomid ve deksametazonun Ürün Bilgilerine bakın.
Tablo 3: Lenalidomid ve deksametazon ile kombinasyon halinde haftada iki defa KYPROLİS20/27 mg/m(10 dakikalık infüzyon)
| |||||||||||
| |||||||||||
| |||||||||||
edilmelidir.
İntravenöz Daratumumab ve Deksametazon ile Kombinasyon Halinde KYPROLİS
![]()
Haftada iki defa 30 dakikalık infüzyonla 20/56 mg/m2 rejimi
KYPROLİS® Tablo 4'te gösterildiği gibi, hastalık progresyonuna veya kabul edilemez toksisite meydana gelene kadar intravenöz daratumumab ve deksametazon ile kombinasyon halinde her 28 günlük döngünün 1, 2, 8, 9, 15 ve 16. Günlerinde 30 dakikalık infüzyon şeklinde intravenöz yolla uygulanır. KYPROLİS® için önerilen başlangıç dozu 1. Döngünün 1 ve
4.3. Kontrendikasyonlar
Etkin madde
Emziren kadınlar (bkz. bölüm 4.6)
4.4. Özel kullanım uyarıları ve önlemleri
KYPROLİS® diğer tıbbi ürünler ile kombinasyon halinde uygulandığından KYPROLİS® ile tedaviye başlamadan önce söz konusu diğer tıbbi ürünlere ait kısa ürün bilgileri incelenmelidir. KYPROLİS® ile kombinasyon halinde lenalidomid kullanılabileceği için lenalidomid gebelik testi ve önleme gerekliliklerine özellikle dikkat edilmesi gerekmektedir (bkz. Bölüm 4.6).
Kardiyak toksisiteler
KYPROLİS® uygulamasının ardından, yeni başlayan veya önceden mevcut olup kötüleşen kalp yetmezliği (örneğin konjestif kalp yetmezliği, pulmoner ödem, düşük ejeksiyon fraksiyonu), kardiyomiyopati ve fataliteler dahil miyokard iskemisi ve miyokard enfarktüsü meydana gelmiştir. Bazı olaylar başlangıç ventrikül fonksiyonu normal olan hastalarda meydana gelmiştir. KYPROLİS® ile yapılan klinik çalışmalarda, bu olaylar KYPROLİS® tedavisinin uygulanması boyunca meydana gelmiştir. KYPROLİS® uygulamasından sonraki bir gün içerisinde kardiyak areste bağlı ölüm meydana gelmiştir. Kombinasyon tedavilerine ilişkin randomize, açık etiketli, çok merkezli çalışmalarda kalp yetmezliği olaylarının insidansı
%8 ve aritmilerin insidansı (büyük bir kısmı atriyal fibrilasyon ve sinüs taşikardisi olan) %8 olmuştur (bkz. bölüm 4.8).
Hastalar kalp yetmezliği veya kardiyak iskemi klinik belirtileri veya semptomları için izlenmelidir. Kardiyak toksisite şüphesi varsa hemen değerlendirme yapılmalıdır. 3. veya
4. derece kardiyak advers reaksiyonların görülmesi durumunda KYPROLİS® bu olaylar düzelene kadar durdurulmalıdır ve yarar/risk değerlendirmesinin sonucuna göre KYPROLİS®'i 1 doz düzeyinde azaltarak tekrar başlanıp başlanılmamasına karar verilmelidir (bkz. bölüm 4.2).
1. döngüde her dozdan önce yeterli sıvı yüklemesi gereklidir, fakat tüm hastalar ve özellikle kalp yetmezliği olan hastalar fazla miktarda sıvı yüklenmesine karşı da izlenmelidir. Başlangıçta kalp yetmezliği olan veya kalp yetmezliği riski taşıyan hastalarda total sıvı alımı klinik olarak uygun şekilde düzenlemelidir (bkz. bölüm 4.2).
75 yaş ve üzerindeki hastalarda kalp yetmezliği riski daha genç hastalara göre daha yüksektir. New York Kalp Derneği (New York Heart Association (NYHA)) sınıflamasına göre Sınıf III ve IV kalp yetmezliği olan, yakın zamanda miyokard enfarktüsü geçirmiş olan ve tıbbi ilaçlarla kontrol edilemeyen ileti anomalileri, angina veya aritmileri olan hastalar klinik çalışmalara dahil edilmemiştir. Bu hastalarda kardiyak komplikasyonların gelişme riski daha yüksektir. Bu hastalar için KYPROLİS® ile tedaviye başlamadan önce kapsamlı bir tıbbi değerlendirme (kan basıncı kontrolü ve sıvı yönetimini içeren) tamamlanmalı ve bu hastalar yakından takip edilmelidir.
Elektrokardiyografik değişiklikler
Klinik çalışmalarda QT intervalinde uzama saptanan olgular bildirilmiştir. KYPROLİS®'in QT intervali üzerindeki etkisi gözardı edilmemelidir (bkz. bölüm 5.1).
Akut böbrek yetmezliği
KYPROLİS® kullanmakta olan hastalarda akut böbrek yetmezliği olguları meydana gelmiştir. Bu olgulardan bazıları ölümle sonuçlanmıştır. KYPROLİS® kullanan hastaların yaklaşık
%9'unda böbrek yetersizliği (böbrek yetmezliğini de içeren) meydana gelmiştir. KYPROLİS® monoterapisi uygulanan ileri evre relaps ve refrakter multipl miyelomlu hastalarda akut böbrek yetmezliğinin daha sık görüldüğü bildirilmiştir. Başlangıç tahmini kreatinin klerensi düşük (Cockcroft-Gault denklemi kullanarak hesaplanır) olan hastalarda ölümcül böbrek yetmezliği riski daha yüksektir. Renal fonksiyon, serum kreatininin ve/veya tahmini kreatinin klerensinin ölçümüyle düzenli olarak izlenmelidir. Doz uygun şekilde azaltılmalı veya kesilmelidir (bkz. bölüm 4.2).
Tümör lizis sendromu
KYPROLİS® ile tedavi gören hastalarda ölümcül sonuçlar da görülebilen TLS olguları bildirilmiştir. Yüksek tümör yükü olan multipl miyelomlu hastalarda TLS riskinin daha fazla olduğu dikkate alınmalıdır. 1. döngüde ve gerektiğinde takip eden döngülerde, KYPROLİS® verilmeden önce intravenöz sıvılar uygulanmalıdır. TLS riski altında olan hastalarda ürik asit azaltıcı ilaçlar düşünülmelidir. Tedavi sırasında hastalar TLS yönünden izlenmeli ve bunların bulunması durumunda TLS düzelene kadar KYPROLİS®'e ara vermek de dahil hızlıca tedavi edilmelidir (bkz. bölüm 4.2).
Pulmoner toksisite
KYPROLİS® alan hastaların yaklaşık %2'sinde Akut Solunum Sıkıntısı Sendromu (Acute Respiratory Distress Syndrome (ARDS)) ve akut solunum yetmezliği meydana gelmiştir. Ayrıca, KYPROLİS® kullanan hastaların yaklaşık %2'sinde pnömoni ve interstisyel akciğer rahatsızlığı gibi akut diffüz infiltratif pulmoner hastalıklar da görülmüştür. Bu olgulardan bazıları ölümle sonuçlanmıştır. İlaca bağlı pulmoner toksisite durumunda KYPROLİS® kesilmelidir.
Pulmoner hipertansiyon
KYPROLİS® kullanan hastaların yaklaşık %2'sinde pulmoner arteriyel hipertansiyon rapor edilmiştir; %1'inden azında 3. derece veya üzerindedir. Hastalar kardiyak görüntüleme ve/veya gereken diğer testlerle değerlendirilmelidir. Pulmoner hipertansiyon durumunda olay düzelene veya başlangıç durumuna dönene kadar KYPROLİS® durdurulmalı ve yarar/risk değerlendirmesi esas alınarak KYPROLİS®'in yeniden başlanıp başlanmamasına karar verilmelidir.
Dispne
KYPROLİS® ile tedavi edilen hastaların %25'inde dispne rapor edilmiştir; %4'ünde 3. derece veya üzerindedir. Kalp yetmezliği ve pulmoner sendromlar gibi kardiyopulmoner hastalıkları dışlamak için dispne değerlendirmesi yapılmalıdır. 3 veya 4. derece dispne olduğunda düzelene veya başlangıç durumuna dönene kadar KYPROLİS® tedavisi durdurulmalıdır. Yarar/risk değerlendirmesi esas alınarak KYPROLİS®'in yeniden başlanıp başlanmamasına karar verilmelidir (bkz. bölüm 4.4 ve 4.8).
Hipertansiyon
KYPROLİS® tedavisi ile hipertansif kriz ve acil hipertansif tablo dahil olmak üzere hipertansiyon gözlenmiştir. ASPIRE çalışmasında hipertansiyon olaylarının insidansı KRd kolunda %17, Rd kolunda ise %9 olmuştur. ENDEAVOR çalışmasında hipertansiyon olaylarının insidansı Kd kolunda %34, Vd kolunda ise %11 olmuştur. CANDOR çalışmasında
hipertansiyon olaylarının insidansı DKd kolunda %31, Kd kolunda ise %28 olmuştur. Bu olgulardan bazılarının ölümcül olduğu kaydedilmiştir. KYPROLİS®'e başlamadan önce kan basıncı optimize edilmelidir. KYPROLİS® almakta olan tüm hastalarda kan basıncı düzenli olarak izlenmelidir. Hipertansiyonun yeterince kontrol edilemediği durumlarda KYPROLİS® kesilmeli ve değerlendirme yapılmalıdır. Yarar/risk değerlendirmesi esas alınarak KYPROLİS®'e yeniden başlayıp başlamamak düşünülmelidir.
Venöz tromboz
KYPROLİS® ile venöz tromboembolik olaylar (derin ven trombozunu ve pulmoner emboliyi içerecek şekilde) gözlenmiştir. Her iki kolda tromboprofilaksi kullanılan ASPIRE çalışmasında ilk 12 döngüde venöz tromboembolik olayların insidansı KRd kolunda %13, Rd kolunda ise
%6 olmuştur. ENDEAVOR çalışmasında venöz tromboembolik olayların insidansı 1 ila 6 aylarda Kd kolunda %9, Vd kolunda ise %2 olmuştur. KYPROLİS® monoterapisi ile venöz tromboembolik olayların insidansı %2 olmuştur.
KYPROLİS®'in lenalidomid ve deksametazon; deksametazon veya intravenöz daratumumab ve deksametazon ile kombinasyon halinde uygulanacağı hastalar için tromboprofilaksi uygulanmalıdır. Tromboprofilaksi rejimi, hastanın altta yatan riskleri esas alınarak seçilmelidir.
Oral kontraseptifler veya tromboz riskiyle ilişkili hormonal gebelikten korunma yöntemi kullanan hastalar için kombinasyon halinde KYPROLİS® uygulandığında, tedavi sırasında gebelikten korunmak için hormonal olmayan bir yöntem düşünülmelidir (bkz. bölüm 4.6).
İnfüzyonla ilişkili reaksiyonlar
KYPROLİS® alan hastalarda yaşamı tehdit eden reaksiyonlar da dahil, infüzyonla ilişkili reaksiyonları meydana gelmiştir. Belirti ve semptomlar arasında ateş, ürperme, artralji, miyalji, yüzde ateş basması, yüzde ödem, laringeal ödem, kusma, güçsüzlük, nefes darlığı, hipotansiyon, senkop, göğüs sıkışması veya angina yer alır. Bu reaksiyonlar, KYPROLİS® uygulamasından hemen sonra veya 24 saat içinde meydana gelebilir. İnfüzyonla ilişkili reaksiyonlarının insidansını ve şiddetini azaltmak amacıyla KYPROLİS® öncesinde deksametazon uygulanmalıdır (bkz. bölüm 4.2 ve 4.8).
Hemoraji
KYPROLİS® ile tedavi edilen hastalarda ölümcül veya ciddi hemoraji vakaları rapor edilmiştir (bkz. bölüm 4.8). Hemorajik olaylar gastrointestinal, pulmoner ve intrakraniyal hemorajiyi ve epistaksisi içermiştir. Kanama spontan olabilir ve intrakraniyal hemoraji travma olmadan meydana gelmiş olabilir. Hemoraji trombosit sayımları düşük veya normal olan hastalarda bildirilmiştir. Hemoraji ayrıca antitrombosit tedavisi veya antikoagülasyon uygulanmayan hastalarda da bildirilmiştir. Kan kaybına ilişkin belirtiler ve semptomlar hemen değerlendirilmelidir. Doz uygun şekilde azaltılmalı veya kesilmelidir (bkz. bölüm 4.2).
Trombositopeni
KYPROLİS®, en düşük trombosit değerinin her bir 28 günlük döngünün 8. ve 15. günleri arasında gözlendiği ve trombosit sayısının genellikle bir sonraki döngünün başından önce başlangıç değerine döndüğü trombositopeniye neden olmaktadır (bkz. bölüm 4.8). KYPROLİS® ile yapılan klinik çalışmalarda hastaların yaklaşık %32'sinde trombositopeni rapor edilmiştir. Hemoraji meydana gelebilir (bkz. bölüm 4.4 ve 4.8). KYPROLİS® ile tedavi sırasında trombosit sayımları sık sık izlenmelidir. Doz uygun şekilde azaltılmalı veya kesilmelidir (bkz. bölüm 4.2).
Hepatik toksisite ve hepatik yetmezlik
KYPROLİS® ile tedavi sırasında ölümcül olgular dahil olmak üzere, hepatik yetmezlik olguları bildirilmiştir (%2). KYPROLİS® serum transaminazlarının yükselmesine neden olabilir (bkz. bölüm 4.8). Başlangıç değerleri dikkate alınmadan, karaciğer enzimleri düzenli olarak izlenmelidir. Doz uygun şekilde azaltılmalı veya kesilmelidir (bkz. bölüm 4.2).
Trombotik mikroanjiyopati
KYPROLİS® ile tedavi gören hastalarda trombotik trombositopenik purpura ve hemolitik üremik sendrom (TTP/HUS) da dahil olmak üzere trombotik mikroanjiyopati olguları bildirilmiştir. Bu olgulardan bazılarının ölümcül olduğu kaydedilmiştir. TTP/HUS belirtileri ve semptomları izlenmelidir. Tanıdan şüpheleniliyorsa KYPROLİS® kullanımı sonlandırılmalı ve değerlendirme yapılmalıdır. TTP/HUS tanısı dışlandığında KYPROLİS® kullanımına tekrar başlanabilir. Önceden TTP/HUS gelişmiş olan hastalarda KYPROLİS® tedavisinin yeniden başlatılmasının güvenliliği bilinmemektedir.
Posterior geri dönüşlü ensefalopati sendromu
KYPROLİS® ile tedavi gören hastalarda posterior geri dönüşlü ensefalopati sendromu (PRES) olguları rapor edilmiştir. Daha önce geri dönüşlü posterior lökoensefalopati sendromu (RPLS) olarak adlandırılmış olan PRES bir nörolojik bozukluk olup nöbet, başağrısı, letarji, konfüzyon, körlük, bilinç düzeyinde değişimler ve başka görme bozuklukları ve nörolojik rahatsızlıklarla beraber hipertansiyonla kendini belli edebilmektedir. Tanı nöro-radyolojik görüntülemeyle (MRI) doğrulanmaktadır. PRES'ten kuşkulanıldığında KYPROLİS® kesilmeli ve değerlendirilmelidir. Daha önce PRES gelişen hastalarda KYPROLİS® terapisine yeniden başlanmanın güvenliliği bilinmemektedir.
Progresif multifokal lökoensefalopati
KYPROLİS® ile ölümcül olabilen progresif multifokal lökoensefalopati (PML) bildirilmiştir. KYPROLİS® dışında, bu duruma katkıda bulunabilecek diğer olası faktörler arasında immünosüpresyona yol açabilecek immünosüpresif tedavinin önceden veya eşzamanlı kullanımı yer almaktadır. Yeni başlangıçlı nörolojik bulgu veya semptomları olan ya da önceden var olan nörolojik bulgu veya semptomlarında değişiklik görülen tüm hastalarda PML düşünülmelidir. PML şüphesi olduğu takdirde, KYPROLİS® kesilmeli ve nöroloji konsültasyonunu da içeren PML değerlendirmesi başlatılmalıdır.
Transplant için uygun olmayan yeni tanı konmuş hastalarda melfalan ve prednizon ile kombinasyon halinde ölümcül ve ciddi toksisitelerde artış
Transplant için uygun olmayan yeni tanı konmuş multipl miyelomlu 955 hastanın KYPROLİS® (20/36 mg/m2, altı haftalık siklusların dört haftasında haftada iki kez 30 dakikalık infüzyon olarak) melfalan ve prednizon (KMP) veya bortezomib, melfalan ve prednizon (VMP) kullanacak şekilde randomize edildiği bir klinik çalışma olan CLARION çalışmasında KMP kolunda VMP kolundaki hastalarla karşılaştırıldığında sırasıyla ölümcül advers reaksiyonlar (%7'ye karşı %4) ve ciddi advers reaksiyonların (%50'ye karşı %42) insidansının daha yüksek olduğu gözlenmiştir. KMP kolundaki hastalarda kalp yetmezliği (%11'e karşı %4), hipertansiyon (%25'e karşı %8), akut böbrek yetmezliği (%14'e karşı %6) ve dispneyi (%18'e karşı %9) de içeren herhangi bir derecede advers reaksiyonların insidansının daha yüksek olduğu gözlenmiştir. Bu çalışmada KMP kolunda primer sonuç ölçütü olan progresyonsuz sağkalım (PFS) üstünlüğü karşılanmamıştır. Transplant için uygun olmayan yeni tanı konmuş multipl miyelomlu hastalarda KYPROLİS® melfalan ve prednizon ile kombine kullanımda endike değildir.
Embriyo-fetal toksisite
Etki mekanizması ve hayvanlarda elde edilmiş bulgular ışığında KYPROLİS®, hamile kadınlara uygulandığında fetüse zarar verebilir. Karfilzomib organogenez döneminde gebe tavşanlara VYA esas alınıp 27 mg/m2'lik klinik dozun yaklaşık %40'ı düzeyinde bir dozda intravenöz yolla uygulandığında implantasyon sonrası kayba ve fetüs ağırlığında azalmaya neden olmuştur.
Gebe kadınlar fetüse yönelik potansiyel risk konusunda bilgilendirilmelidir. Üreme potansiyeli olan kadınlara KYPROLİS® tedavisi sırasında ve son dozu takip eden 6 ay süresince doğum kontrol yöntemi uygulamaları tavsiye edilmelidir. Üreme potansiyeline sahip kadın partneri olan erkeklere KYPROLİS® ile tedavileri sırasında ve son dozu takip eden 3 ay süresince doğum kontrol yöntemi uygulamaları tavsiye edilmelidir (bkz. bölüm 4.6 ve 5.3).
Kontrasepsiyon
Çocuk doğurma potansiyeli bulunan kadın hastalar, gebeliği önlemek için tedavi sırasında ve tedavinin tamamlanmasının ardından 6 ay süreyle etkili doğum kontrol yöntemleri kullanmalı veya cinsel ilişkiden kaçınmalıdır. Üreme potansiyeli bulunan erkek hastalar, gebeliği önlemek için tedavi sırasında ve tedavinin tamamlanmasının ardından 3 ay süreyle etkili doğum kontrol yöntemleri kullanmalı veya cinsel ilişkiden kaçınmalıdır (bkz. bölüm 4.6). Karfilzomib oral kontraseptiflerin etkililiğini azaltabilir (bkz. bölüm 4.5).
Sodyum içeriği
Bu tıbbi ürün sulandırılmış çözeltinin her mL'sinde 0,3 mmol (7 mg) sodyum içermektedir. Bu durum, kontrollü sodyum diyetinde olan hastalar için göz önünde bulundurulmalıdır.
Yardımcı madde siklodekstrinin süreli kullanımının taşıdığı güvenlik riskleri nedeniyle, ürünün 14 günlük tedavi sonrasında, hekim tarafından gerekli değerlendirmenin yapılıp, kullanıma devam edilip edilmeyeceğinin belirlenmesi gerekmektedir.
4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri
Karfilzomib ağırlıklı olarak peptidaz ve epoksit hidrolaz aktiviteleri aracılığıyla metabolize edilir ve sonuç olarak, karfilzomibin farmakokinetik profilinin sitokrom P450 inhibitörleri ve indükleyicilerinin eş zamanlı uygulamasından etkilenmesi olası değildir.
İn vitro çalışmalar karfilzomibin kültürlenmiş insan hepatositlerinde insan CYP3A4'ü indüklemediğini göstermiştir. CYP3A probu olarak oral midazolamın kullanıldığı bir klinik çalışma, midazolamın farmakokinetiğinin eş zamanlı karfilzomib uygulamasından etkilenmediğini ortaya koyarak karfilzomibin CYP3A4/5 substratlarının metabolizmasını inhibe etmesinin beklenmediğini ve insan gönüllülerde CYP3A4 indükleyicisi olmadığını göstermiştir. Bununla birlikte, karfilzomibin terapötik konsantrasyonlarda CYP1A2, 2C8, 2C9, 2C19 ve 2B6'nın indükleyicisi olup olmadığı bilinmemektedir. Karfilzomib, bu enzimlerin substratları olan oral kontraseptifler gibi tıbbi ürünlerle birlikte uygulandığında dikkatli olunmalıdır. Gebeliği önlemeye yönelik etkili önlemler alınmalıdır (bkz. bölüm 4.6), hasta oral kontraseptifler kullanıyorsa alternatif bir etkili doğum kontrol yöntemi kullanılmalıdır.
Karfilzomib CYP1A2, 2B6, 2C8, 2C9, 2C19 ve 2D6'yı in vitro inhibe etmez ve bu nedenle, inhibisyon sonucu olarak bu enzimlerin substratları olan tıbbi ürünlerin maruziyetini etkilemesi beklenmez.
Karfilzomib bir P-glikoprotein (P-gp) substratıdır ancak BCRP substratı değildir. Bununla birlikte, KYPROLİS®'in intravenöz olarak uygulandığı ve büyük ölçüde metabolize olduğu dikkate alındığında, karfilzomibin farmakokinetik profilinin P-gp veya BCRP inhibitörleri veya indükleyicilerinden etkilenmesi olası değildir. İn vitro, terapötik dozlarda beklenenlerden daha düşük konsantrasyonlarda (3 µM), karfilzomib P-gp substratı olan digoksinin hücre dışına taşınmasını %25 oranında inhibe eder. Karfilzomib, P-gp substratlarıyla (örn. digoksin, kolşisin) birlikte uygulandığında dikkatli olunmalıdır.
İn vitro, karfilzomib OATP1B1'i IC50 = 2,01 µM oranında inhibe eder, halbuki karfilzomibin sistemik düzeyde OATP1B3, OAT1, OAT3, OCT2 ve BSEP'nin diğer taşıyıcılarını inhibe edebileceği veya edemeyeceği bilinmemektedir. Karfilzomib insan UGT2B7'yi inhibe etmez ancak insan UGT1A1'i 5,5 µM IC50 oranında inhibe eder. Bununla birlikte, karfilzomibin hızlı eliminasyonu, özellikle infüzyon bitiminden 5 dakika sonra sistemik konsantrasyondaki hızlı düşüş göz önünde bulundurulduğunda, OATP1B1 ve UGT1A1 substratları ile klinik olarak anlamlı etkileşim riski muhtemelen düşüktür.
Özel popülasyonlara ilişkin ek bilgiler:
Özel popülasyonlara ilişkin etkileşim çalışması yapılmamıştır.
Pediyatrik popülasyon:
Pediyatrik popülasyona ilişkin etkileşim çalışması yapılmamıştır.
4.6. Gebelik ve laktasyon
Gebelik kategorisi: D (bkz. bölüm 4.4)
Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon)
Üreme potansiyeli olan kadınlara KYPROLİS® tedavisi sırasında ve son dozu takip eden en az
6 ay boyunca etkili doğum kontrolü uygulamaları tavsiye edilmelidir.
Karfilzomib tedavisi sırasında oral kontraseptiflerin etkililiğinin azalma olasılığı dışlanamaz (bkz. bölüm 4.5). Ayrıca, karfilzomib ile ilişkili venöz tromboembolik olaylar riskindeki artış nedeniyle, kadınlar karfilzomib ile tedavi sırasında tromboz riski taşıyabilen hormonal kontraseptiflerin kullanımından kaçınmalıdır (bkz. bölüm 4.4 ve 4.8). Hasta mevcut durumda oral kontraseptifler veya tromboz riski taşıyabilen hormonal kontrasepsiyon kullanıyorsa, alternatif etkili doğum kontrol yöntemine geçmelidir.
Üreme potansiyeli bulunan ve kadın cinsel partneri olan erkeklere, KYPROLİS® tedavisi sırasında ve son dozu takip eden en az 3 ay boyunca etkili doğum kontrolü uygulamaları tavsiye edilmelidir.
Gebelik dönemi
Karfilzomibin gebelik ve/veya fetus/yeni doğan üzerinde zararlı farmakolojik etkileri bulunmaktadır.
Gebelik testi, üreme potansiyeli bulunan kadınlara, KYPROLİS® tedavisi başlanmadan uygulanmalıdır.
KYPROLİS® gerekli olmadıkça gebelik döneminde kullanılmamalıdır. Etki mekanizmasına ve hayvanlardaki bulgulara dayanarak KYPROLİS® fetüse zarar verebilir. İlaçla ilişkili riskleri değerlendirmek üzere gebe kadınlarda KYPROLİS® kullanımına ilişkin herhangi bir veri bulunmamaktadır. KYPROLİS® tavşanlarda klinik dozdan daha düşük dozlarda embriyo-fetal ölümlere neden olmuştur. Gebe kadınlara fetüs açısından söz konusu olan potansiyel risk konusunda bilgi verilmelidir.
Endikasyon bulunan popülasyonun majör doğum kusurları ve düşüğe ilişkin tahmini arka plan riski bilinmemektedir. Tüm hamileliklerde doğum kusuru, kayıp veya diğer advers sonuçlara ilişkin bir arka plan risk söz konusudur.
Lenalidomid, yapısal açıdan talidomid ile bağlantılıdır. Talidomid, ciddi ve hayatı tehdit edici doğum kusurlarına sebep olduğu bilinen bir insan teratojenik aktif maddedir. Lenalidomid gebelik sırasında alınırsa lenalidomidin insanlarda teratojenik etki göstermesi beklenir. Hastanın çocuk doğurma potansiyelinin bulunmadığına dair güvenilir kanıt olmadığı takdirde tüm hastalar lenalidomid için Gebeliği Önleme Programının şartlarına uymalıdır. Lütfen güncel lenalidomid kısa ürün bilgisine bakınız.
Laktasyon dönemi
KYPROLİS®‘in anne sütündeki varlığı, emzirilen çocuğa ve ilacın süt üretimine etkisi hakkında veri bulunmamaktadır. Emzirilen çocukta ciddi advers reaksiyon potansiyelinden dolayı bir önlem olarak, KYPROLİS® ile tedavi sırasında ve tedaviden sonra 2 hafta süreyle emzirme kontrendikedir.
Üreme yeteneği/Fertilite
Etki mekanizması ışığında KYPROLİS®, erkek veya kadın fertilitesi üzerinde etki gösterebilir (bkz. bölüm 5.1 ve 5.3). KYPROLİS®'in insan fertilitesi üzerindeki etkisine ilişkin veri mevcut değildir.
4.7. Araç ve makine kullanımı üzerindeki etkiler
KYPROLİS®'in araç ve makine kullanımı üzerine minör etkisi vardır.
Klinik çalışmalarda yorgunluk, baş dönmesini de içeren sersemlik hali, baygınlık, bulanık görme, uyku hali ve/veya tansiyon düşmesi gözlenmiştir. KYPROLİS® tedavisi gören hastalar bu semptomlardan biri ile karşılaşırlarsa araç veya makine kullanmamaları tavsiye edilmelidir.
4.8. İstenmeyen etkiler
Güvenlilik profilinin özeti
KYPROLİS® tedavisi sırasında meydana gelebilecek ciddi advers reaksiyonlar şunları içerir: kalp yetmezliği, miyokard enfarktüsü, kardiyak arest, miyokard iskemisi, interstisyel akciğer hastalığı, pnömonit, akut solunum sıkıntısı sendromu, akut solunum yetmezliği, pulmoner hipertansiyon, dispne, hipertansif krizler dahil hipertansiyon, akut böbrek hasarı, tümör lizis sendromu, infüzyonla ilişkili reaksiyon, gastrointestinal kanama, intrakraniyal kanama, pulmoner kanama, trombositopeni, karaciğer yetmezliği, PRES, trombotik mikroanjiyopati ve TTP/HUS. KYPROLİS® ile yapılan klinik çalışmalarda kardiyak toksisite ve dispne tipik olarak KYPROLİS® tedavisinin başlarında meydana gelmiştir (bkz. bölüm 4.4). En yaygın advers olaylar (gönüllülerin %20'sinden fazlasında meydana gelen) şunlar olmuştur: anemi,
yorgunluk, trombositopeni, bulantı, ishal, pireksi, dispne, solunum yolu enfeksiyonu, öksürük ve nötropeni.
20 mg/m2 düzeyindeki ilk karfilzomib dozlarının ardından, doz ASPIRE çalışmasında 27 mg/m2'ye ve ENDEAVOR çalışmasında 56 mg/m2'ye yükseltilmiştir (bkz. bölüm 5.1). ENDEAVOR çalışmasının KYPROLİS® ve deksametazon (Kd) kolunda meydana gelen advers reaksiyonlar ile ASPIRE çalışmasının KYPROLİS®, lenalidomid ve deksametazon (KRd) kolunda meydana gelen advers reaksiyonlar arası karşılaştırması, aşağıdaki advers reaksiyonlarda dozla potansiyel ilişki olabileceğini ortaya koymuştur: kalp yetmezliği (Kd
%8,2, KRd %6,4), dispne (Kd %30,9, KRd %22,7), hipertansiyon (Kd %25,9, KRd %15,8) ve
pulmoner hipertansiyon (Kd %1,3, KRd %0,8).
KYPROLİS®'in daratumumab ve deksametazon (KdD) ile kombinasyon halinde uygulanmasının, deksametazon ile kombinasyon halindeki KYPROLİS® (Kd) ile karşılaştırıldığı CANDOR çalışmasında (bkz. bölüm 5.1), herhangi bir çalışma tedavisinin son dozundan sonraki 30 gün içinde gerçekleşen advers olaylara bağlı ölümler, KdD kolundaki hastaların %10'unda ve Kd kolundaki hastaların ise %5'inde meydana gelmiştir. İki koldaki (KdD'ye karşı Kd) hastalarda gerçekleşen ölüm olaylarının en yaygın nedeni, enfeksiyonlar (%5'e karşı %3) olmuştur. Tedavi ile ortaya çıkan ölümcül advers olay riski, 65 yaş ve üzeri hastalarda daha yüksek olmuştur. KdD kolundaki hastaların %56'sında ve Kd kolundaki hastaların %46'sında ciddi advers olaylar bildirilmiştir. Kd koluna kıyasla KdD kolunda bildirilen en yaygın ciddi advers olaylar; anemi (%2'ye karşı %1), ishal (%2'ye karşı %0), pireksi (%4'e karşı %2), pnömoni (%12'ye karşı %9), influenza (%4'e karşı %1), sepsis (%4'e karşı %1) ve bronşit (%2'ye karşı %0) olmuştur.
Advers reaksiyonların tablo halinde listesi
Advers reaksiyonlar aşağıdaki sistem organ sınıfı ve sıklık kategorisine göre sunulmaktadır (bkz. Tablo 10). Sıklık kategorileri havuzlanmış klinik çalışmalardan oluşan veri kümesinde her advers reaksiyon için rapor edilen kaba insidans oranından belirlenmiştir (n = 3.878). Her sistem organ sınıfı ve sıklık kategorisinde advers olaylar azalan ciddiyet sırasına göre sunulmaktadır.
Advers reaksiyonların insidansı, sistem organ sınıfları ve sıklık esas alınarak aşağıda listelenmiştir. Sıklık şu şekilde tanımlanır: Çok yaygın (≥ 1/10); yaygın (≥ 1/100 ila < 1/10); yaygın olmayan (≥ 1/1.000 ila < 1/100); seyrek (≥ 1/10.000 ila < 1/1.000); çok seyrek (< 1/10.000); bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor).
Tablo 10: Advers reaksiyonların tablo halinde listesi
MedDRA sistem organ sınıfı | Çok yaygın | Yaygın | Yaygın olmayan | Seyrek |
Enfeksiyonlar ve | Pnömoni | Sepsis | Clostridium difficile |
|
enfestasyonlar | Solunum yolu | Akciğer enfeksiyonu | koliti | |
| enfeksiyonu | Grip | Sitomegalovirüs | |
|
| Herpes zoster* | enfeksiyonu | |
|
| İdrar yolu enfeksiyonu | Hepatit B | |
|
| Bronşit | reaktivasyonu | |
|
| Gastroenterit |
| |
|
| Viral enfeksiyon |
| |
|
| Nazofarenjit |
| |
|
| Rinit |
|
MedDRA sistem organ sınıfı | Çok yaygın | Yaygın | Yaygın olmayan | Seyrek |
Bağışıklık sistemi hastalıkları |
|
| İlaca aşırı duyarlılık |
|
Kan ve lenfatik sistem hastalıkları | Trombositopeni Nötropeni Anemi Lenfopeni Lökopeni | Febril nötropeni | HUS TTP | Trombotik mikroanjiyopati |
Metabolizma ve beslenme hastalıkları | Hipokalemi İştahta azalma | Dehidrasyon Hiperkalemi Hipomagnezemi Hiponatremi Hiperkalsemi Hipokalsemi Hipofosfatemi Hiperürisemi Hipoalbüminemi Hiperglisemi | Tümör lizis sendromu |
|
Psikiyatrik hastalıklar | Uykusuzluk | Anksiyete Zihin karışıklığı |
|
|
Sinir sistemi hastalıkları | Sersemlik hissi Periferik nöropati Baş ağrısı | Parestezi Hipoestezi | İntrakraniyal hemoraji Serebrovasküler olay PRES |
|
Göz hastalıkları |
| Katarakt Bulanık görme |
|
|
Kulak ve iç kulak hastalıkları |
| Tinnitus |
|
|
Kardiyak hastalıklar |
| Kalp yetmezliği Miyokard enfarktüsü Atriyal fibrilasyon Taşikardi Çarpıntı Düşük ejeksiyon fraksiyonu | Kardiyak arest Kardiyomiyopati Miyokard iskemisi Perikardit Perikard efüzyonu Ventriküler taşikardi |
|
Vasküler hastalıklar | Hipertansiyon | Derin ven trombozu Hipotansiyon Kızarma | Hipertansif kriz Hemoraji | Hipertansif acil durum |
Solunum, göğüs bozuklukları ve mediastinal hastalıklar | Dispne Öksürük | Pulmoner emboli Pulmoner ödem Epistaksi Orofarengeal ağrı Disfoni Hırıltı Pulmoner hipertansiyon | ARDS Akut solunum yetmezliği Pulmoner hemoraji İnterstisyel akciğer hastalığı Pnömoni |
|
Gastrointestinal hastalıklar | Kusma İshal Kabızlık Karın ağrısı Bulantı | Gastrointestinal kanama Dispepsi Diş ağrısı | Gastrointestinal perforasyon Akut pankreatit |
|
Hepatobiliyer hastalıklar |
| Yüksek alanin aminotransferaz Yüksek aspartat aminotransferaz Yüksek gama glutamiltransferaz Hiperbilirubinemi | Karaciğer yetmezliği Kolestaz |
|
MedDRA sistem organ sınıfı | Çok yaygın | Yaygın | Yaygın olmayan | Seyrek |
Deri ve deri altı doku hastalıkları |
| Döküntü Pruritus Eritem Hiperhidroz |
| Anjioödem |
Kas-iskelet ve bağ dokusu hastalıkları | Sırt ağrısı Artralji Ekstremitede ağrı Kas spazmları | Kas-iskelet ağrısı Kas-iskelet göğüs ağrısı Kemik ağrısı Miyalji Kas güçsüzlüğü |
|
|
Böbrek ve idrar yolu hastalıkları | Kanda yüksek kreatinin | Akut böbrek hasarı Böbrek yetmezliği Böbrek bozukluğu Düşük kreatinin renal klerensi |
|
|
Genel bozukluklar ve uygulama bölgesine ilişkin hastalıklar | Pireksi Periferik ödem Asteni Yorgunluk Ürperme | Göğüs ağrısı Ağrı İnfüzyon yeri reaksiyonları Grip benzeri hastalık Kırıklık | Çoklu organ disfonksiyon sendromu |
|
Araştırmalar |
| Yüksek c-reaktif protein Kanda yüksek ürik asit |
|
|
Yaralanma, zehirlenme ve prosedür ile ilgili komplikasyonlar |
| İnfüzyonla ilişkili reaksiyon |
|
|
*Sıklık, hastaların çoğunun profilaksi tedavisinde olduğu klinik çalışmalardan elde edilen verilere dayanılarak hesaplanmaktadır.
Seçilmiş advers reaksiyonların tanımlanması
Kalp yetmezliği, miyokard enfarktüsü ve miyokard iskemisi
KYPROLİS® ile ilgili klinik çalışmalarda hastaların yaklaşık %5'inde kalp yetmezliği (hastaların yaklaşık %3'ünde 3. derece ve üstü olgu gelişmiştir), yaklaşık %1'inde miyokard enfarktüsü (hastaların yaklaşık %1'inde 3. derece ve üstü olgu gelişmiştir) ve %1'inde ve daha azında miyokard iskemisi (hastaların %1'inden daha azında 3. derece ve üstü olgu gelişmiştir) bildirilmiştir. Bu olgular tipik olarak KYPROLİS® tedavisinin erken döneminde (< 5 döngü) meydana gelmiştir. KYPROLİS® tedavisi sırasında kardiyak bozuklukların klinik tedavisi için bkz. bölüm 4.4.
CANDOR çalışmasında, başlangıçta vasküler bozuklukları veya başlangıçta hipertansiyonu olan hastalardan oluşan alt gruptaki kardiyak bozuklukların (tüm ve herhangi bir derece olgu) genel insidansı, sırasıyla %29,9'a karşı %19,8 (KdD'ye karşı Kd) ve %30,6'ya karşı %18,1 olmuştur. Ölümcül kardiyak olgular için insidans, sırasıyla %1,9'a karşı %0,0 (KdD'ye karşı Kd) ve %1,5'e karşı %0,0 olmuştur. Başlangıçta vasküler bozuklukları veya başlangıçta hipertansiyonu olan hastalardan oluşan alt grupta KdD ve Kd kolları arasında bildirilen farkta tek bir kardiyak olgu tipi dikkate alınmamıştır.
Dispne
KYPROLİS® ile ilgili klinik çalışmalarda hastaların yaklaşık %30'unda dispne bildirilmiştir. Dispne advers reaksiyonlarının çoğunluğu ciddi olmamış (hastaların %5'inden daha azında
derece ve üstü olgu gelişmiştir), giderilmiş, nadiren tedavinin kesilmesi ile sonuçlanmış ve çalışmanın erken bir döneminde (< 3 döngü) ortaya çıkma özelliği göstermiştir. KYPROLİS® tedavisi sırasında dispnenin klinik tedavisi için bkz. bölüm 4.4.
4.9. Doz aşımı ve tedavisi
Flakonu yaklaşık 1 dakika kadar veya tam olarak çözünene kadar nazikçe döndürün ve/veya yavaşça ters çevirin. Köpük oluşmasından kaçınmak için flakonu ÇALKALAMAYIN. Köpük oluşursa, flakondaki köpüklenme çökelene (yaklaşık 5 dakika) ve çözelti berrak hale gelene kadar çözeltinin oturmasını bekleyin.
5. FARMAKOLOJİK ÖZELLİKLER
5.1. Farmakodinamik özellikler
Farmakoterapötik grup: Antineoplastik ajanlar/Proteazom inhibitörü ATC kodu: L01XG02
Etki mekanizması
Karfilzomib 26S proteazomunun içerisindeki proteolitik çekirdek partikül olan 20S proteazomunun N-terminal treonin-içeren aktif yerlerine geri dönüşsüz olarak bağlanan tetrapeptit yapısındaki bir epoksiketon proteazom inhibitörüdür. Karfilzomib solid ve hematolojik tümör hücreleri üzerinde in vitro antiproliferatif ve proapoptotik aktivitelere sahiptir. Hayvanlarda, karfilzomib kan ve dokulardaki proteazom aktivitesini inhibe etmiş ve multipl miyelom, hematolojik ve solid tümör modellerinde tümörün büyümesini geciktirmiştir.
Farmakodinamik etkiler
İntravenöz karfilzomib uygulanması, kanda ilk dozdan 1 saat sonra ölçüldüğünde, proteazom kimotripsin-benzeri (chymotrypsin like (CT-L)) aktivitesinin süpresyonuyla sonuçlanmıştır. Lenalidomid ve deksametazon ile veya tek başına ≥ 15 mg/m2 düzeyinde karfilzomib dozları proteazomun CT-L aktivitesinin ≥ %80 inhibisyonunu indüklemiştir. Buna ek olarak, tek ajan olarak intravenöz yoldan 20 mg/m2 karfilzomib, proteazomun düşük moleküler kütleli polipeptid 2 (low molecular mass polypeptide 2 (LMP2)) ve multikatalitik endopeptidaz kompleksi benzeri 1 (multicatalytic endopeptidase complex-like 1 (MECL1)) alt birimlerinin sırasıyla %26 ila %32 ve %41 ila %49 ortalama inhibisyonuyla sonuçlanmıştır. Proteazom inhibisyonu, dozlamanın her haftasında, karfilzomibin ilk dozunu takiben ≥ 48 saat süreyle devam etmiştir.
Daha yüksek 56 mg/m2 dozunda CT-L alt birimlerinde 15 ila 20 mg/m2 dozuna kıyasla daha yüksek inhibisyon (≥%90) görülmesinin yanı sıra diğer proteazom alt birimlerinde (LMP7, MECL1 ve LMP2) de daha yüksek inhibisyon saptanmıştır. 56 mg/m2 dozunda LMP7, MECL1
ve LMP2 alt birimlerinde 15 ila 20 mg/m2 dozuna kıyasla inhibisyonda sırasıyla %8, %23 ve
%34 artış gözlemlenmiştir. Karfilzomib ile benzer proteazom inhibisyonu, test edildiği 2 doz
seviyesinde (20 ve 36 mg/m2) 2 ila 10 dakikalık ve 30 dakikalık infüzyonlar ile elde edilmiştir. Klinik etkililik ve güvenlilik
Relaps veya refrakter multipl miyelom için lenalidomid ve deksametazon ile kombinasyon halinde
ASPIRE
ASPIRE, 1 ila 3 seri tedavi (burada tedavi serisi, relaps veya progresif hastalık gibi etkililik eksikliği nedeniyle kesintiye uğramamış olan planlanmış bir tedavi kürüdür [sıralı indüksiyon, transplantasyon, konsolidasyon ve/veya idame dahildir]) almış olan relaps veya refrakter multipl miyelom hastalarında KYPROLİS®'in lenalidomid ve deksametazon (KRd) ile kombinasyonunu tek başına lenalidomid ve deksametazon (Rd) ile karşılaştırarak değerlendiren randomize, açık etiketli, çok merkezli çalışmadır. Aşağıdaki hastalar çalışmaya alınmamıştır: en son rejimde bortezomibe refrakter, en son rejimde lenalidomid ve deksametazona refrakter, önceki hiçbir rejime yanıt vermemiş olan, kreatinin klerensi
< 50 ml/dak, ALT/AST > 3,5 × ULN ve bilirubin > 2 × ULN; New York Kalp Derneği sınıf III ila IV konjestif kalp yetmezliği veya son 4 ay içinde miyokard enfarktüsü geçiren hastalar. KRd kolunda KYPROLİS® başlangıç dozu olarak 20 mg/m2 düzeyinde değerlendirilmiş, bu düzey 1. döngünün 8. gününde ve sonrasında 27 mg/m2'ye yükseltilmiştir. KYPROLİS®
1. döngüden 12. döngüye kadar her 28 günlük döngünün 1, 2, 8, 9, 15 ve 16. günlerinde 10 dakikalık infüzyon olarak uygulanmıştır. KYPROLİS®, 13. döngüden 18. döngüye kadar her 28 günlük döngünün 1, 2, 15 ve 16. günlerinde dozlanmıştır. Deksametazon 40 mg her
döngünün 1, 8, 15 ve 22. gününde oral veya intravenöz yoldan uygulanmıştır. Lenalidomid, her 28 günlük döngünün 1 ila 21. günlerinde 25 mg oral yoldan verilmiştir. Rd tedavi kolunda lenalidomid ve deksametazon rejimi KRd tedavi kolu ile aynıdır. KYPROLİS®, hastalık progresyonu veya kabul edilemez toksisite nedeniyle erken kesilene kadar en fazla 18 döngü uygulanmıştır. Lenalidomid ve deksametazon uygulamasına hastalık progresyonuna veya kabul edilemez toksisiteye kadar devam edilmiştir. Her iki kol için tromboprofilaksi ve bir proton pompa inhibitörünün eşzamanlı kullanımı ve KRd kolu için antiviral profilaksi gerekli görülmüştür.
ASPIRE çalışmasında 792 hasta KRd veya Rd koluna 1:1 oranında randomize edilmiştir. Demografik bilgiler ve başlangıç özellikleri iki grup arasında iyi bir dengededir (bkz. Tablo 11). Hastaların sadece %53'ünde genetik mutasyon için test yapılmıştır; KRd kolundaki hastaların %12'sinde ve Rd kolundaki hastaların %13'ünde yüksek riskli bir genetik mutasyon saptanmıştır.
Tablo 11: ASPIRE çalışmasında demografik bilgiler ve başlangıç özellikleri
Özellikler | KRd (N = 396) | Rd (N = 396) |
Yaş, medyan yıl (min, maks) | 64 (38; 87) | 65 (31; 91) |
Yaş ≥ 75, n (%) | 43 (11) | 53 (13) |
Erkekler, n (%) | 215 (54) | 232 (59) |
Irk, n (%) | ||
Beyaz | 377 (95) | 377 (95) |
Siyah | 12 (3) | 11 (3) |
Diğer veya bildirilmemiş | 7 (2) | 8 (2) |
Özellikler | KRd (N = 396) | Rd (N = 396) |
Önceki rejim sayısı, n (%) | ||
1 | 184 (46) | 157 (40) |
2 | 120 (30) | 139 (35) |
3 | 92 (23) | 100 (25) |
Önceki nakil, n (%) | 217 (55) | 229 (58) |
ECOG performans durumu, n (%) | ||
0 | 165 (42) | 175 (44) |
1 | 191 (48) | 186 (47) |
2 | 40 (10) | 35 (9) |
Çalışma başlangıcında ISS evresi, n (%) | ||
I | 167 (42) | 154 (39) |
II | 148 (37) | 153 (39) |
III | 73 (18) | 82 (21) |
Bilinmiyor | 8 (2) | 7 (2) |
Kreatinin Klerensi, ml/dak, medyan (min, maks) | 79 (39; 212) | 79 (30; 208) |
30 ila < 50, n (%) | 19 (5) | 32 (8) |
50 ila < 80, n (%) | 185 (47) | 170 (43) |
Son tedaviye refrakter, n (%) | 110 (28) | 119 (30) |
Herhangi bir zamanda aşağıdakilere refrakter, n (%): | ||
Bortezomib | 60 (15) | 58 (15) |
Lenalidomid | 29 (7) | 28 (7) |
Bortezomib + immünomodülatör ajan | 24 (6) | 27 (7) |
ECOG = Doğu Kooperatif Onkoloji Grubu (Eastern Cooperative Oncology Group); ISS = Uluslararası Evreleme Sistemi; KRd = KYPROLİS, lenalidomid ve deksametazon; Rd = lenalidomid ve deksametazon
Bağımsız İnceleme Komitesi (Independent Review Committee (IRC)) tarafından Standart Uluslararası Miyelom Çalışma Grubu (International Myeloma Working Group (IMWG))/Avrupa Kan ve Kemik İliği Nakli (European Blood and Marrow Transplantation (EBMT)) yanıt kriterleri kullanılarak belirlendiğine göre KRd kolundaki hastalar Rd kolundakilere göre daha iyi PFS sergilemiştir (HR = 0,69, 2 yanlı P-değeri = 0,0001 ile).
Medyan PFS süresi KRd kolunda 26,3 ay, Rd kolunda ise 17,6 ay olmuştur (bkz. Tablo 12 ve Şekil 1).
KRd kolunda 246 ölümün ve Rd kolunda 267 ölümün ardından önceden planlanmış bir genel sağkalım (OS) analizi gerçekleştirilmiştir. Medyan takip süresi yaklaşık 67 ay olmuştur. KRd kolundaki hastalarda, Rd kolundaki hastalara kıyasla OS'de istatistiksel açıdan anlamlı bir avantaj gözlenmiştir (bkz. Tablo 12 ve Şekil 2).
Tablo 12: ASPIRE çalışmasında etkililik sonuçları
| Kombinasyon tedavisi | |
KRd (N = 396) | Rd (N = 396) | |
PFS | ||
Medyan, ay (%95 CI) | 26,3 (23,3; 30,5) | 17,6 (15,0; 20,6) |
HR (%95 CI) | 0,69 (0,57; 0,83) | |
P-değeri (2 yanlı) | 0,0001 | |
Genel Sağkalım | ||
Medyan, Ay (%95 CI) | 48,3 (42,4; 52,8) | 40,4 (33,6; 44,4) |
HR (%95 CI) | 0,79 (0,67; 0, 95) | |
P değeri (2 yanlı) | 0,0091 | |
| Kombinasyon tedavisi | |
KRd (N = 396) | Rd (N = 396) | |
Genel yanıt | ||
Yanıt alınan hasta sayısı (N) | 345 | 264 |
ORR (%) (%95 CI) | 87 (83; 90) | 67 (62; 71) |
P-değeri (2 yanlı) | < 0,0001 | |
Yanıt kategorisi, n (%) | ||
sCR | 56 (14) | 17 (4) |
CR | 70 (18) | 20 (5) |
VGPR | 151 (38) | 123 (31) |
PR | 68 (17) | 104 (26) |
CI = güven aralığı; CR = tam yanıt; HR = Risk oranı; KRd = KYPROLİS, lenalidomid ve deksametazon; ORR = genel yanıt oranı; PFS = progresyonsuz sağkalım; PR = kısmi yanıt; Rd = lenalidomid ve deksametazon; sCR = kesin tam yanıt; VGPR = çok iyi kısmi yanıt
Medyan yanıt süresi (DOR), KRd kolunda yanıt elde eden 345 hasta için 28,6 ay (%95 CI: 24,9; 31,3) ve Rd kolunda yanıt elde eden 264 hasta için 21,2 ay (%95 CI: 16,7; 25,8) olmuştur. Yanıta kadar geçen medyan süre, KRd kolunda 1 ay (aralık: 1 ila 14 ay) ve Rd kolunda da 1 ay (aralık 1 ila 16 ay) olmuştur.

Şekil 1: ASPIRE Çalışmasında Progresyonsuz Sağkalımın Kaplan-Meier Eğrisi
| ||
| ||

|
|
CI = güven aralığı; EBMT = Avrupa Kan ve Kemik İliği Nakli (European Blood and Marrow Transplantation); IMWG = Uluslararası Miyelom Çalışma Grubu (International Myeloma Working Group);
HR = risk oranı; KRd = KYPROLİS, lenalidomid ve deksametazon kolu; PFS = Progresyonsuz Sağkalım;
Rd = lenalidomid ve deksametazon kolu
Not: Yanıt ve PH (progresif hastalık) sonuçları standart objektif IMWG/EBMT yanıt kriterleri kullanılarak tayin edilmiştir.
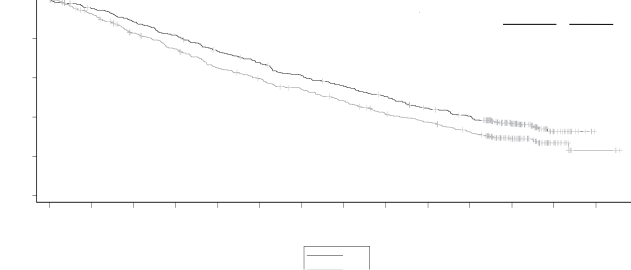
![]()
Şekil 2: ASPIRE Çalışmasında Genel Sağkalımın Kaplan-Meier Eğrisi
| ||
CI = güven aralığı; HR = risk oranı; KRd = KYPROLİS, lenalidomid ve deksametazon; OS = genel
sağkalım; Rd = lenalidomid ve deksametazon kolu
Relaps veya refrakter multipl miyelom için deksametazon ile kombinasyon
Deksametazon ile kombinasyon halindeki KYPROLİS®'in etkililiği, iki açık etiketli, randomize çalışmada (ENDEAVOR ve ARROW) değerlendirilmiştir
ENDEAVOR
ENDEAVOR, 1 ila 3 basamak tedavi almış olan relaps veya refrakter multipl miyelom hastalarında KYPROLİS® ve deksametazona (Kd) karşı bortezomib ve deksametazonu (Vd) karşılaştırarak değerlendiren randomize, açık etiketli, çok merkezli bir çalışmadır. Toplam 929 hasta kaydedilmiş ve randomize edilmiştir (464 hasta Kd koluna; 465 hasta Vd koluna). Randomizasyon önceki proteazom inhibitörü tedavisi (var veya yok), önceki tedavi basamakları (1'e karşılık 2 veya 3), güncel Uluslararası Evreleme Sistemi evresi (1'e karşılık 2 veya 3) ve planlanan bortezomib uygulama yoluna göre tabakalandırılmıştır. Önceki rejimlerde yanıtları PR'den daha az; kreatinin klerensi < 15 ml/dak; hepatik transaminazları
≥ 3 × ULN; sol ventrikül ejeksiyon fraksiyonu < %40 veya başka önemli bir kalp sorunu olan hastalar çalışmaya alınmamıştır. Bu çalışmada KYPROLİS® başlangıç dozu olarak 20 mg/m2 ile başlayıp, 1. döngünün 8. gününde ve sonrasında 56 mg/m2'ye yükseltilmesi değerlendirilmiştir. KYPROLİS® her 28 günlük döngünün 1, 2, 8, 9, 15 ve 16. gününde 30 dakikalık infüzyon olarak haftada iki kez uygulanmıştır. Deksametazon 20 mg her döngünün 1, 2, 8, 9, 15, 16, 22 ve 23. günlerinde oral veya intravenöz yoldan uygulanmıştır.
Vd kolunda bortezomib 21 günlük döngünün 1, 4, 8 ve 11. günlerinde 1,3 mg/m2 dozda
intravenöz veya subkutan yoldan dozlanmış, deksametazon 20 mg her döngünün 1, 2, 4, 5, 8, 9, 11 ve 12. günlerinde oral veya intravenöz yoldan uygulanmıştır. Eşzamanlı olarak tromboprofilaksi uygulanması isteğe bağlı olmuş, ve bir antiviral ajan ve proton pompa inhibitörü ile profilaksi ise gerekli görülmüştür. Vd kolundaki 465 hastanın 381'i subkutan yoldan bortezomib almıştır. Hastalık progresyonuna veya kabul edilemez toksisiteye kadar tedaviye devam edilmiştir.
Demografik bilgiler ve başlangıç özellikleri Tablo 13'te özetlenmektedir.
Tablo 13: ENDEAVOR çalışmasında demografik bilgiler ve başlangıç özellikleri
Özellik | Kd (N = 464) | Vd (N = 465) |
Yaş, yıl | ||
Medyan (min, maks) | 65 (35; 89) | 65 (30; 88) |
< 65, n (%) | 223 (48) | 210 (45) |
65–74, n (%) | 164 (35) | 189 (41) |
≥ 75, n (%) | 77 (17) | 66 (14) |
Cinsiyet, n (%) | ||
Kadın | 224 (48) | 236 (51) |
Erkek | 240 (52) | 229 (49) |
Irk, n (%) | ||
Beyaz | 353 (76) | 361 (78) |
Siyah | 7 (2) | 9 (2) |
Asyalı | 56 (12) | 57 (12) |
Diğer veya bildirilmemiş | 48 (10) | 38 (8) |
ECOG performans durumu, n (%) | ||
0 | 221 (48) | 232 (50) |
1 | 210 (45) | 203 (44) |
2 | 33 (7) | 30 (6) |
Kreatinin klerensi (ml/dak) | ||
Medyan (min, maks) | 73 (14; 185) | 72 (12; 208) |
< 30, n (%) | 28 (6) | 28 (6) |
30 – < 50, n (%) | 57 (12) | 71 (15) |
50 – < 80, n (%) | 186 (40) | 177 (38) |
≥ 80, n (%) | 193 (42) | 189 (41) |
FISH, n (%) | ||
Yüksek risk | 97 (21) | 113 (24) |
Standart risk | 284 (61) | 291 (63) |
Bilinmeyen risk | 83 (18) | 61 (13) |
Çalışma başlangıcında ISS evresi, n (%) | ||
ISS I | 219 (47) | 212 (46) |
ISS II | 138 (30) | 153 (33) |
ISS III | 107 (23) | 100 (22) |
Önceki rejim sayısı, n (%) | ||
1 | 232 (50) | 231 (50) |
2 | 158 (34) | 144 (31) |
3 | 74 (16) | 88 (19) |
4 | 0 (0) | 2 (0,4) |
Önceki tedaviler, n (%) | 464 (100) | 465 (100) |
Bortezomib | 250 (54) | 252 (54) |
Multipl miyelom için nakil | 266 (57) | 272 (59) |
Talidomid | 212 (46) | 249 (54) |
Lenalidomid | 177 (38) | 178 (38) |
Bortezomib + immünomodülatör ajan | 159 (34) | 168 (36) |
Önceki son tedaviye refrakter, n (%) | 184 (40) | 189 (41) |
KYPROLİS® etkililiği, IMWG yanıt kriterleri kullanılarak IRC tarafından belirlenen PFS'ye göre değerlendirilmiştir. Çalışmada Kd kolunda 18,7 ay ve Vd kolunda 9,4 ay medyan PFS görülmüştür (bkz. Şekil 3 ve Tablo 14).
Şekil 3: ENDEAVOR Çalışmasında Progresyonsuz Sağkalımın Kaplan-Meier Eğrisi
| ||
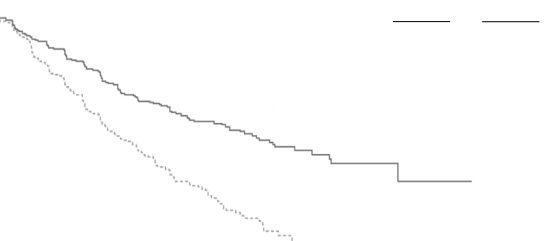
![]()
|
|
![]()
CI = güven aralığı; HR = risk oranı; Kd = KYPROLİS ve deksametazon; PFS = progresyonsuz sağkalım;
Vd = bortezomib ve deksametazon
Diğer sonlanım noktaları OS ve genel yanıt oranını (ORR) içermektedir.
Kd kolunda 189 ölümün ve Vd kolunda 209 ölümün ardından önceden planlanmış bir OS analizi gerçekleştirilmiştir. Medyan takip süresi yaklaşık 37 ay olmuştur. Kd kolundaki hastalarda, Vd kolundaki hastalarla karşılaştırıldığında anlamlı düzeyde daha uzun OS gözlenmiştir (HR = 0,79; %95 CI: 0,65; 0,96; P değeri = 0,01). Sonuçlar Tablo 14 ve Şekil 4'te sunulmaktadır.
Tablo 14: ENDEAVOR'da önemli sonuçların özeti (tedavisi amaçlanan popülasyon)
| Kd (N = 464) | Vd (N = 465) |
PFS | ||
Olay sayısı (%) | 171 (37) | 243 (52) |
Medyan, ay (%95 CI) | 18,7 (15,6; NE) | 9,4 (8,4; 10,4) |
HR (Kd/Vd) (%95 CI) | 0,53 (0,44; 0,65) | |
P-değeri (1 yanlı) | < 0,0001 | |
Genel Sağkalım | ||
Ölüm sayısı (%) | 189 (41) | 209 (45) |
Medyan , Ay (%95 CI) | 47,6 (42,5; NE) | 40,0 (32,6; 42.3) |
HR (Kd/Vd) (%95 CI) | 0,79 (0,65; 0,96) | |
P-değeri (1 yanlı) | 0,01 | |
| Kd (N = 464) | Vd (N = 465) |
Genel yanıt | ||
Yanıt verenler N | 357 | 291 |
ORR (%) (%95 CI) | 77 (73; 81) | 63 (58; 67) |
P-değeri (1 yanlı) | < 0,0001 | |
Yanıt kategorisi, n (%) | ||
sCR | 8 (2) | 9 (2) |
CR | 50 (11) | 20 (4) |
VGPR | 194 (42) | 104 (22) |
PR | 105 (23) | 158 (34) |
CI = güven aralığı; CR = tam yanıt; HR = risk oranı; Kd = KYPROLİS ve deksametazon; ORR = genel yanıt oranı; PFS = progresyonsuz sağkalım; PR = kısmi yanıt; NE = hesaplanabilir değildir; sCR = kesin tam yanıt; Vd = bortezomib ve deksametazon; VGPR = çok iyi kısmi yanıt;
Şekil 4: ENDEAVOR Çalışmasında Kaplan-Meier Genel Sağkalım Grafiği
| ||
| ||
![]()
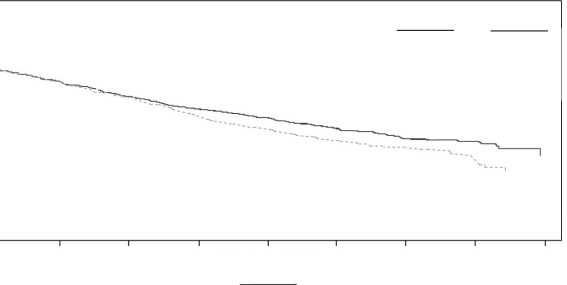
CI = güven aralığı; HR = risk oranı; Kd = KYPROLİS ve deksametazon; OS = genel sağkalım;
Vd = bortezomib ve deksametazon
PR veya daha iyi bir yanıt elde edilen gönüllülerde medyan DOR, Kd kolunda 21,3 ay (%95 CI: 21,3, tahmin edilemez) ve Vd kolunda 10,4 ay (%95 CI: 9,3; 13,8) olmuştur. Yanıta kadar geçen medyan süre her iki kolda 1 ay (aralık: < 1 ila 8 ay) olmuştur.
ARROW
ARROW, daha önce 2 ila 3 basamak tedavi görmüş olan relaps, refrakter multipl miyelom hastalarında haftada bir defa uygulanan KYPROLİS® ve deksametazon (Kd) (20/70 mg/m2) ile
haftada iki defa uygulanan Kd'nin (20/27 mg/m2) karşılaştırıldığı randomize, açık etiketli, çok merkezli ve üstünlük gösterilmesi hedeflenen bir çalışmadır. Daha önceki basamaklardan en az birine PR'nin altında yanıt vermiş olup; kreatinin klirensi < 30 mL/dak olan; hepatik transaminazları ≥ 3 × ULN olan; sol ventriküler ejeksiyon fraksiyonu < %40 olan veya başka anlamlı kardiyak durumları olan hastalar tedaviye alınmamıştır. Toplam 478 hasta kaydedilmiş ve randomize edilmiştir (20/70 mg/m2 kolunda 240; 20/27 mg/m2 kolunda 238). Randomizasyon, güncel Evrelendirme Sistemi evresine (evre 1 veya evre 2 ya da 3), bortezomib tedavisine dirençli durumuna (evet veya hayır) ve yaşa (< 65 veya ≥ 65 yaş) göre basamaklandırılmıştır. Çalışmanın 1. Kolunda 1. Döngünün 8. Gününden itibaren 70 mg/m2'ye yükseltilen 20 mg/m2'lik başlangıç dozunda KYPROLİS® değerlendirilmiştir.
1. Kolda KYPROLİS® haftada bir defa, 28 günlük döngülerin her birinin 1, 8 ve 15. günlerinde
30 dakikalık infüzyon şeklinde uygulanmıştır. Çalışmanın 2. Kolunda 1. Döngünün
8. Gününden itibaren 27 mg/m2'ye yükseltilen 20 mg/m2'lik başlangıç dozunda KYPROLİS® değerlendirilmiştir. 2. Kolda KYPROLİS® haftada iki defa, 28 günlük döngülerin her birinin 1, 2, 8, 9, 15 ve 16. günlerinde 10 dakikalık infüzyon şeklinde uygulanmıştır. Her iki rejimde
de tüm döngülerin 1, 8, 15. Günlerinde ve yalnızca 1 ila 9. Döngünün 22. Gününde oral yolla 40 mg deksametazon uygulanmıştır. Eşzamanlı tromboprofilaksi uygulaması isteğe bağlı bırakılmıştır, bir antiviral ajanla profilaksi önerilmiştir ve bir proton pompası inhibitorü ile profilaksi zorunlu kılınmıştır. Hastalık progresyonu veya kabul edilemez toksisite ortaya çıkana kadar tedaviye devam edilmiştir.
Demografik özellikler ve başlangıç özellikleri Tablo 15'te özetlenmektedir.
Tablo 15: ARROW Çalışmasındaki Demografik Özellikler ve Başlangıç Özellikleri
Özellikler | Haftada bir defa 20/70 mg/m Kd (N = 240) | Haftada iki defa 20/27 mg/m Kd (N = 238) |
Yaş, Yıl | ||
Medyan (min, maks) | 66 (39; 85) | 66 (35; 83) |
< 65, n (%) | 104 (43) | 104 (44) |
65–74, n (%) | 90 (38) | 102 (43) |
≥ 75, n (%) | 46 (19) | 32 (13) |
Cinsiyet, n (%) | ||
Kadın | 108 (45) | 110 (46) |
Erkek | 132 (55) | 128 (54) |
Irk, n (%) | ||
Beyaz | 200 (83) | 202 (85) |
Siyah | 3 (1) | 2 (1) |
Asyalı | 30 (13) | 15 (6) |
Diğer veya Bildirilmemiş | 7 (3) | 19 (8) |
ECOG Performans Durumu, n (%) | ||
0 | 118 (49) | 118 (50) |
1 | 121 (50) | 120 (50) |
2 | 1 (0,4) | 0 (0) |
Kreatinin Klirensi (mL/dak) | ||
Medyan (min, maks) | 70,80 (28;212) | 73,20 (29;181) |
< 30, n (%) | 2 (1) | 1 (0,4) |
30 – < 50, n (%) | 48 (20) | 34 (14) |
50 – < 80, n (%) | 91 (38) | 111 (47) |
≥ 80, n (%) | 99 (41) | 91 (38) |
Özellikler | Haftada bir defa 20/70 mg/m Kd (N = 240) | Haftada iki defa 20/27 mg/m Kd (N = 238) |
FISH, n (%) | ||
Yüksek risk | 34 (14) | 47 (20) |
Standart risk | 47 (20) | 53 (22) |
Bilinmeyen risk | 159 (66) | 138 (58) |
Çalışmanın Başlangıcındaki ISS Evresi, n (%) | ||
ISS I | 94 (39) | 99 (42) |
ISS II | 80 (33) | 81 (34) |
ISS III | 63 (26) | 54 (23) |
Önceki Rejimlerin Sayısı | ||
2 | 116 (48) | 125 (53) |
3 | 124 (52) | 112 (47) |
>3 | 0 (0) | 1 (0,4) |
Önceki Tedaviler, n (%) | ||
Bortezomib | 236 (98) | 237 (100) |
Transplantasyon | 146 (61) | 157 (66) |
Talidomid | 119 (50) | 119 (50) |
Lenalidomid | 207 (86) | 194 (82) |
ECOG = Doğu Kooperatif Onkoloji Grubu; FISH = Fluoresans in situ hibridizasyon; ISS = Uluslararası Evreleme Sistemi; Kd = KYPROLİS ve deksametazon
KYPROLİS®'in etkililiği IMWG yanıt kriterleri kullanılarak PFS'ye göre değerlendirilmiştir. Etkililik sonuçları Tablo 16 ve Şekil 5'te sunulmaktadır.
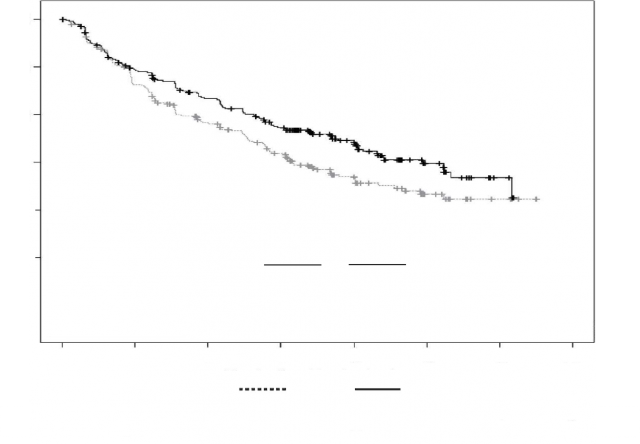
Şekil 5: ARROW Çalışmasında Progresyonsuz Sağkalıma İlişkin Kaplan-Meier Grafiği
| ||
| ||
|
|
CI = güven aralığı; HR = risk oranı; Kd = KYPROLİS ve deksametazon; PFS = progresyonsuz sağkalım
Tablo 16: ARROW Çalışmasındaki Kilit Bulguların Özeti (Tedavi Niyetli Popülasyon)
| Haftada bir defa 20/70 mg/m Kd (N = 240) | Haftada iki defa 20/27 mg/m Kd (N = 238) |
PFS | ||
Olay sayısı, n (%) | 126 (52,5) | 148 (62,2) |
Medyan, Ay (%95 CI) | 11,2 (8,6; 13,0) | 7,6 (5,8; 9,2) |
HR (%95 CI) | 0,69 (0,54; 0,88) | |
P değeri (1 yanlı) | 0,0014 | |
Genel Yanıt | ||
Yanıt verenlerin sayısı | 151 | 97 |
ORR (%) (%95 CI) | 62,9 (56,5; 69,0) | 40,8 (34,5; 47,3) |
P değeri (1 yanlı) | < 0,0001 | |
Yanıt Kategorisi, n (%) | ||
sCR | 4 (1,7) | 0 (0,0) |
CR | 13 (5,4) | 4 (1,7) |
VGPR | 65 (27,1) | 28 (11,8) |
PR | 69 (28,8) | 65 (27,3) |
CI = güven aralığı; CR = tam yanıt; HR = risk oranı; Kd = KYPROLİS ve deksametazon; ORR = genel yanıt oranı; PFS = progresyonsuz sağkalım; VGPR = çok iyi kısmi yanıt; PR = kısmi yanıt; sCR = kesin tam yanıt
PR veya daha iyi bir yanıt elde edilen gönüllülerde medyan DOR, 20/70 mg/m2 Kd kolunda 15 ay (%95 CI: 12,2, hesaplanabilir değildir), 20/27 mg/m2 Kd kolunda ise 13,8 ay (%95 CI:
9,5, hesaplanabilir değildir) olmuştur. Yanıt ortaya çıkana kadar geçen medyan süre 20/70 mg/m2 Kd kolunda 1,1 ay, 20/27 mg/m2 Kd kolunda ise 1,9 ay olmuştur.
KYPROLİS® tek başına deksametazon ile kombinasyon halinde haftada iki defa 20/27 mg/m2
şeklindeki uygulama için onaylı değildir.
Relaps veya Refrakter Multipl Miyelom için İntravenöz Daratumumab ve Deksametazon ile Kombinasyon Halinde
KYPROLİS®'in daratumumab ve deksametazon (DKd) ile kombinasyon halindeki etkililiği, iki açık etiketli klinik çalışmada (CANDOR ve EQUULEUS) değerlendirilmiştir.
CANDOR
CANDOR, daha önce 1 ila 3 basamak tedavi almış olan relaps veya refrakter multipl miyelom hastalarında haftada iki defa uygulanan KYPROLİS® 20/56 mg/m2'nin intravenöz daratumumab ve deksametazon (DKd) ile kombinasyonuna kıyasla haftada iki defa uygulanan KYPROLİS® 20/56 mg/m2 ve deksametazonu (Kd) değerlendiren randomize, açık etiketli, çok merkezli bir çalışmadır. Aşağıdaki hastalar çalışmaya alınmamıştır: son 2 yıl içinde bilinen orta ila şiddetli derecede inatçı astımı olan, FEV1'i öngörülen normalin %50'sinden az olan ve bilinen kronik obstrüktif akciğer hastalığı (COPD) bulunan veya aktif konjestif kalp yetmezliği olan hastalar. Randomizasyon taramasında ISS (evre 1 veya 2'ye karşılık evre 3), önceki proteazom inhibitörü maruziyeti (var veya yok), önceki tedavi basamağı sayısı (1'e karşılık 2 veya daha fazla) ya da önceki küme farklılaşma antijeni 38 (CD38) antikor tedavisine (var veya yok) göre sınıflandırma yapılmıştır.
KYPROLİS® 1. Döngünün 1 ve 2. Günlerinde 20 mg/m2'lik; 1. Döngünün 8, 9, 15 ve
16. Günlerinde ve ardından her 28 günlük döngünün 1, 2, 8, 9, 15 ve 16. Günlerinde
56 mg/m2'lik dozlar halinde intravenöz yoldan 30 dakika uygulanmıştır. 1, 2, 8, 9, 15 ve
16. Günlerde oral veya intravenöz yoldan deksametazon 20 mg ve ardından her 28 günlük
döngünün 22. Gününde oral veya intravenöz yoldan 40 mg uygulanmıştır. DKd kolunda,
1. Döngünün 1 ve 2. Günlerinde intravenöz yoldan 8 mg/kg daratumumab uygulanmıştır.
Ardından 1. Döngünün 8, 15 ve 22. Günlerinde; 2. Döngünün 1, 8, 15 ve 22. Günlerinde; 3 ila
6. Döngülerin 1 ve 15. Günlerinde ve kalan döngüler için veya hastalık progresyonuna kadar
1. Günde intravenöz yoldan 16 mg/kg daratumumab uygulanmıştır. 20 mg'a düşürülmüş deksametazon dozunu alan 75 yaş üzeri hastalar için 20 mg'lık dozun tamamı, daratumumabın uygulandığı günlerde daratumumab pre-infüzyon ilacı olarak verilmiştir. Her iki çalışma kolunda KYPROLİS® uygulandığında ise deksametazon dozajı günlere bölünmüştür. Hastalık progresyonuna veya kabul edilemez toksisiteye kadar her iki kolda tedaviye devam edilmiştir.
Toplam 466 hasta randomize edilmiştir (312 hasta DKd koluna ve 154 hasta Kd koluna). Demografik bilgiler ve başlangıç özellikleri Tablo 17'de özetlenmektedir.
Tablo 17: CANDOR Çalışmasında Demografik Bilgiler ve Başlangıç Özellikleri
Özellikler | DKd (N = 312) | Kd (N = 154) |
Randomizasyonda yaş (yıl) | ||
Medyan (min, maks) | 64 (29; 84) | 65 (29; 84) |
Özellikler | DKd (N = 312) | Kd (N = 154) |
Yaş grubu – n (%) | ||
18 – 64 yaş | 163 (52) | 77 (50) |
65 – 74 yaş | 121 (39) | 55 (36) |
75 yaş ve üzeri | 28 (9) | 22 (14) |
Cinsiyet – n (%) | ||
Erkek | 177 (57) | 91 (59) |
Kadın | 135 (43) | 63 (41) |
Irk – n (%) | ||
Asyalı | 46 (15) | 20 (13) |
Siyah veya Afro-Amerikan | 7 (2,2) | 2 (1,3) |
Beyaz | 243 (78) | 123 (80) |
Diğer | 16 (5) | 9 (6) |
Coğrafi bölge – n (%) | ||
Kuzey Amerika | 21 (7) | 12 (8) |
Avrupa | 207 (66) | 103 (67) |
Asya Pasifik | 84 (27) | 39 (25) |
ECOG performans durumu – n (%) | ||
0 veya 1 | 295 (95) | 147 (95) |
2 | 15 (4,8) | 7 (4,5) |
Eksik | 2 (0,6) | 0 (0,0) |
FISH ile belirlendiği şekilde risk grubu – n (%) | ||
Yüksek risk | 48 (15) | 26 (17) |
Standart risk | 104 (33) | 52 (34) |
Bilinmiyor | 160 (51) | 76 (49) |
Taramada I x RS uyarınca ISS evresi – n (%) | ||
I veya II | 252 (81) | 127 (82) |
III | 60 (19) | 27 (17) |
Önceki rejim sayısı – n (%) | ||
1 | 144 (46) | 70 (45) |
2 | 99 (32) | 46 (30) |
3 | 69 (22) | 37 (24) |
Önceki Tedaviler | ||
Lenalidomid | 123 (39) | 74 (48) |
Lenalidomide refrakter | 99 (32) | 55 (36) |
Bortezomib | 287 (92) | 134 (87) |
Önceki CD38 antikor tedavisi – n (%) | 1 (0,3) | 0 (0,0) |
Önceki kök hücre nakli (ASCT) – n (%) | 195 (62) | 75 (49) |
Etkililik, IMWG yanıt kriterleri kullanılarak IRC'nin PFS değerlendirmesine göre değerlendirmiştir. Etkililik sonuçları Tablo 18 ve Şekil 6'da sunulmaktadır. DKd kolunda medyan yanıt süresine ulaşılmamıştır ve medyan yanıt süresi Kd kolunda 16,6 ay (13,9; NE) olmuştur. Yanıta kadar geçen medyan (min, maks) süre, DKd kolu için 1,0 (1; 14) ay ve Kd kolu için 1,0 (1; 10) ay olmuştur.
Şekil 6: CANDOR Çalışmasında Progresyonsuz Sağkalıma İlişkin Kaplan-Meier Grafiği
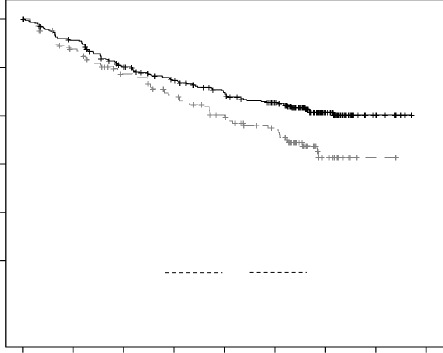
| ||
| ||
|
|
Tablo 18: CANDOR Çalışmasında Önemli Sonuçların Özeti (Tedavi Amaçlı Popülasyon)
| DKd (N = 312) | Kd (N = 154) |
PFS | ||
Olay sayısı (%) | 110 (%35) | 68 (%44) |
Medyan, Ay (%95 CI) | NE (NE, NE) | 15,8 (12,1; NE) |
HR (%95 CI) | 0,63 (0,46; 0,85) | |
P-değeri (1 yanlı) | 0,0014 | |
Genel Yanıt | ||
Yanıt Verenlerin Sayısı | 263 | 115 |
ORR (%) (%95 CI) | %84 (%80; %88) | %75 (%67; %81) |
P-değeri (1 yanlı) | 0,0040 | |
CR | 89 (%28) | 16 (%10) |
VGPR | 127 (%41) | 59 (%38) |
PR | 47 (%15) | 40 (%26) |
12. ayda MRD [-] CR oranı, n (%) (%95 CI) | 39 (%12) (%9; %17) | 2 (%1,3) (%0,2; %4,6) |
P-değeri (1 yanlı) | < 0,0001 | |
MRD [-] CR | 43 (%14) | 5 (%3,2) |
EQUULEUS
EQUULEUS, daha önce 1 ila 3 basamak tedavi almış olan relaps veya refrakter multipl miyelom hastalarında KYPROLİS®'in intravenöz daratumumab ve deksametazon ile kombinasyonunu değerlendiren açık etiketli, çok kohortlu bir çalışmadır. Aşağıdaki hastalar çalışmaya alınmamıştır: son 2 yıl içinde bilinen orta ila şiddetli inatçı astımı olan, FEV1'i öngörülen normalin %50'sinden az olan ve bilinen kronik obstrüktif akciğer hastalığı (COPD) bulunan veya aktif konjestif kalp yetmezliği (New York Kalp Derneği Sınıf III-IV olarak tanımlanır) olan hastalar.
KYPROLİS® haftada bir defa 1. Döngünün 1. Gününde 20 mg/m2'lik doz halinde intravenöz yoldan 30 dakika uygulanmış ve doz 1. Döngünün 8 ve 15. Günlerinde ve her 28 günlük
döngünün 1, 8 ve 15. Günlerinde 70 mg/m2'ye çıkarılmıştır. On hastaya 1. Döngünün
Gününde intravenöz yoldan 16 mg/kg daratumumab ve kalan hastalara 1. Döngünün 1 ve
5.2. Farmakokinetik özellikler
Genel özelliklerEmilim:
30 dakikalık infüzyon şeklinde uygulanan 20 mg/m2 ile 70 mg/m2 arasındaki dozlarda karfilzomib multipl miyelom hastalarında maksimum plazma konsantrasyonlarında (C) ve sonsuza giden zaman içinde eğri altında kalan alanda (AUC) doza bağımlı artış göstermiştir. Aynı zamanda relaps, refrakter multipl miyelom hastalarında 2 ila 10 dakikalık infüzyon şeklinde 20 mg/m ve 56 mg/m arası karfilzomible de Cve AUCdeğerlerinde doza bağımlı bir artış gözlenmiştir. 30 dakikalık bir infüzyon, aynı dozda 2 ila 10 dakikalık bir infüzyonla gözlenene benzer bir AUCfakat 2 ila 3 kat daha düşük bir Cortaya çıkarmıştır. Haftada bir 30 dakikalık infüzyon halinde 70 mg/m2 veya haftada iki defa 2 ila 10 dakikalık infüzyon halinde 15 ve 20 mg/m2 karfilzomibin tekrarlanan şekilde uygulanmasını takiben karfilzomib birikimine işaret eden kanıt ortaya çıkmamıştır.
Tablo 26'de farklı doz rejimleri için birinci döngüde hesaplanan ortalama günlük eğri altındaki alan (AUC), kararlı durumdaki ortalama günlük eğri altındaki alan (AUC) ve birinci döngüdeki en yüksek dozda ortaya çıkan C(C) listelenmektedir.
Tablo 26: Farklı Doz Rejimleri için Karfilzomib Maruziyeti Parametreleri
Hesaplanan Parametreler (%CV) | 2 ila 10 dakikalık infüzyonla haftada iki defa 20/27 mg/m | 30 dakikalık infüzyonla haftada iki defa 20/56 mg/m | 30 dakikalık infüzyonla haftada bir defa 20/70 mg/m |
AUC(ng•sa/mL) | 95 (40) | 170 (35) | 114 (36) |
AUC(ng•sa/mL) | 111 (34) | 228 (28) | 150 (35) |
C(ng/mL) | 1282 (17) | 1166 (29) | 1595 (36) |
CV: Coefficient of variation
Dağılım:
Ortalama kararlı-durum dağılım hacmi 20 mg/m2 karfilzomib dozunda 28 L olmuştur. Karfilzomib %97 oranında insan plazma proteinlerine bağlanır ve 0,4 - 4 mikromolar aralığı içindeki konsantrasyonlarda in vitro ortalama %97‘dir.
Biyotransformasyon:
Karfilzomib peptidaz klivajı ile hızlı, şekilde metabolize olur ve epoksit hidrolizi metabolizmanın temel yolağıdır. Sitokrom P450-aracılıklı mekanizmalar genel karfilzomib metabolizmasına minör bir katkıda bulunur.
Eliminasyon:
Karfilzomibin yarı ömrü 1. Döngünün 1. Gününde ≥ 15 mg/m2'lik intravenöz dozları takiben
≤ 1 saattir. 30 dakikalık infüzyon veya 2 ila 10 dakikalık infüzyon şeklinde uygulandığında yarı ömrün benzer olduğu görülmüştür. Sistemik klirens, 151 ile 263 L/saat arasında değişmiştir.
Uygulanan karfilzomib dozunun yaklaşık %25'i 24 saat içinde idrarda metabolit olarak atılmıştır. Ana bileşiğin idrar ve dışkıyla atılımı önemsiz miktardadır (toplam dozun %0,3'ü).
Doğrusallık/Doğrusal olmayan durum:
Veri bulunmamaktadır.
Hastalardaki karakteristik özellikler
Spesifik popülasyonlar:
Yaş (35-89 yaş), cinsiyet, ırk veya etnisite (%80 Beyaz, %11 Siyah, %6 Asyalı, %3 Latin) ve hafif ila şiddetli böbrek yetmezliği (kreatinin klirensi 15-89 mL/dak) karfilzomibin farmakokinetiği üzerinde klinik açıdan anlamlı etki ortaya çıkarmamıştır.
Karaciğer yetmezliği
Hafif (total bilirubin 1 ila 1,5 × ULN ve herhangi bir AST veya total bilirubin ≤ ULN ve AST
> ULN) ve orta derece (total bilirubin > 1,5 ila 3 × ULN ve herhangi bir AST) karaciğer yetmezliği olan hastalarda, karfilzomibin AUC değeri karaciğer fonksiyonu normal olan hastalara göre yaklaşık %50 oranında artmıştır. Şiddetli karaciğer yetmezliği (total bilirubin > 3 × ULN ve herhangi bir AST) olan hastalarda karfilzomibin farmakokinetiği değerlendirilmemiştir.
Böbrek yetmezliği
Hemodiyaliz uygulanan SEBH hastalarında karfilzomibin AUC değeri böbrek fonksiyonu normal olan hastalara göre %33 oranında daha yüksek gözlenmiştir. KYPROLİS® konsantrasyonlarının hemodiyaliz klirensi araştırılmamış olduğundan, ilaç hemodiyaliz prosedüründen sonra uygulanmalıdır.
İlaç etkileşim çalışmaları
Klinik çalışmalar
Karfilzomibin hassas CYP3A substratı üzerine etkisi
Midazolamın (hassas bir CYP3A substratı) farmakokinetiği karfilzomib uygulamasından etkilenmemiştir.
In vitro çalışmalar
Karfilzomibin CYP450 enzimi üzerine etkisi
Karfilzomib direkt ve zamana bağımlı CYP3A inhibisyonu sergilemiştir fakat in vitro
CYP1A2 ve CYP3A4'ü indüklememiştir.
Taşıyıcıların karfilzomib üzerine etkisi
Karfilzomib in vitro bir P-glikoprotein (P-gp) substratıdır.
Karfilzomibin taşıyıcılar üzerine etkisi
Karfilzomib in vitro P-gp'yi inhibe eder. Bununla birlikte, KYPROLİS®'in intravenöz yoldan uygulandığı ve yaygın biçimde metabolize edildiği göz önüne alındığında, KYPROLİS®'in farmakokinetiğinin P-gp inhibitörlerinden veya indüktörlerinden etkilenmesi olası gözükmemektedir.
5.3. Klinik öncesi güvenlilik verileri
Gebelik
Gebe sıçanlar ve tavşanlara organogenez döneminde intravenöz yoldan uygulanan karfilzomib sıçanlarda 2 mg/kg/gün ve tavşanlarda 0,8 mg/kg/gün dozlarına kadar teratojenik olmamıştır. Karfilzomib test edilen herhangi bir dozda teratojenik olmamıştır. Tavşanlarda,
≥ 0,4 mg/kg/gün dozunda pre-implantasyon kaybında bir artış ve erken rezorpsiyonlarda ve post-implantasyon kayıplarında artış ve maternal toksik doz olan 0,8 mg/kg/gün dozunda fetal ağırlıkta bir azalma olmuştur. Tavşanlarda 0,4 ve 0,8 mg/kg/gün dozları VYA‘ya dayalı olarak insanlarda önerilen doz olan 27 mg/m2 dozunun sırasıyla, ortalama %20 ve %40'ına karşılık gelmektedir.
Karsinogenez, mutagenez, fertilitede bozulma Karfilzomib ile karsinojenisite çalışmaları yapılmamıştır.
Karfilzomib periferik kan lenfositlerinde in vitro kromozomal sapma testinde klastojenik olmuştur. Karfilzomib in vitro bakteriyel ters mutasyon (Ames) testinde mutajenik değildir ve in vivo fare kemik iliği mikronükleus tayininde klastojenik olmamıştır.
Karfilzomib ile fertilite çalışmaları yapılmamıştır. Üreme dokuları üzerinde 28 günlük yinelenen doz sıçan ve maymun toksisite çalışmalarında veya 6 aylık rat ve 9 aylık maymun kronik toksisite çalışmalarında etkiler saptanmamıştır.
Hayvan toksikolojisi ve/veya farmakolojisi
Kardiyovasküler toksisite: Tek bolus intravenöz doz olarak 3 mg/kg karfilzomib uygulanan maymunlarda (VYA'ya göre insanlarda önerilen doz olan 27 mg/m2 dozunun ortalama 1,3 katına karşılık gelmektedir) hipotansiyon, kalp hızında artış ve troponin-T serum düzeylerinde artış görülmüştür.
Kronik uygulama: Klinik olarak kullanılanlara benzer doz programı ile sıçanlarda karfilzomibin ≥ 2 mg/kg/dozunda ve maymunlarda 2 mg/kg/dozunda yinelenen bolus intravenöz uygulaması, kardiyovasküler (kalp yetmezliği, kardiyak fibroz, perikardiyal sıvı toplanması, kardiyak hemoraji/dejenerasyon), gastrointestinal (nekroz/hemoraji), renal (glomerülonefropati, tübüler nekroz, disfonksiyon) ve pulmoner (hemoraji/inflamasyon) sistemlerdeki toksisitelere bağlı mortalitelerle sonuçlanmıştır. Sıçanlardaki 2 mg/kg/dozu, VYA‘ya göre insanlarda önerilen doz olan 27 mg/m2 dozunun ortalama yarısı kadardır. Maymunlardaki 2 mg/kg/dozu, VYA‘ya göre insanlarda önerilen doza yaklaşık olarak eşdeğerdir.
Uygulamadan önce partikül madde veya renk değişimine karşı çıplak gözle inceleyin. Hazırlanmış ürün berrak, renksiz bir çözelti olmalı ve herhangi bir renk değişikliği veya partikül madde gözlenirse uygulanmamalıdır.
6. FARMASÖTİK ÖZELLİKLER
6.1. Yardımcı maddelerin listesi
Sülfobütileter beta-siklodekstrin Susuz sitrik asit
Sodyum hidroksit (pH ayarlaması için)
6.2. Geçimsizlikler
6.2. Geçimsizlikler
karıştırılmamalıdır.
KYPROLİS® enjektabl %0,9 sodyum klorürle karıştırılmamalıdır.
6.3. Raf ömrü
36 ay.
Rekonstitüye çözelti
Steril enjeksiyonluk su ile rekonstitüye edildikten sonra oda sıcaklığında (25°C) saklanmak koşuluyla 4 saat içerisinde veya 2°C-8°C arasında saklanmak koşuluyla 24 saat içerisinde kullanılmalıdır.
İlacın hazırlanmasından uygulanmasına kadar geçen toplam zaman 24 saati aşmamalıdır.
6.4. Saklamaya yönelik özel tedbirler
2°C-8°C arasında buzdolabında saklayınız. Dondurmayınız.Flakonu orijinal ambalajında ışıktan koruyarak saklayınız.
6.5. Ambalajın niteliği ve içeriği
50 mL Tip I şeffaf cam flakon, floropolimer lamine elastomerik tıpa ve mor renkli çıkarılabilir plastik kapaklı alüminyum conta ile kapatılmış.
Her bir ambalaj 1 adet tek kullanımlık flakon içerir.
6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler
KYPROLİS® flakonları antimikrobiyal koruyucu maddeler içermez ve sadece tek doz olarak kullanılması amaçlanmıştır.
Kullanılmamış olan ürünler ya da atık materyaller “Tıbbi Atıkların Kontrolü Yönetmeliği†ve “Ambalaj ve Ambalaj Atıklarının Kontrolü Yönetmelikleri†ne uygun olarak imha edilmelidir.
Hazırlama ve uygulama
Sulandırılmış çözelti 2 mg/mL konsantrasyonunda karfilzomib içermektedir. Hazırlamadan önce hazırlama talimatlarını eksiksiz okuyunuz. Parenteral ürünler kullanılmadan önce renkte değişiklik olup olmadığı ve partikül içerip içermediği, solüsyon ve kabı izin verdiği ölçüde kontrol edilmelidir.
Flakonu kullanımdan hemen önce buzdolabından çıkarın.
Flakonda kalan kullanılmayan kısım varsa atın. Flakonlardaki kullanılmayan kısımları BİRLEŞTİRMEYİN. Bir flakondan birden fazla doz UYGULAMAYIN.
 HIV ve Aids
HIV, Human Immunodeficiency Virus’dür (İnsanlarda Bağışıklık Sistemini Bozan
Virüsdür). Bu virüs AIDS hastalığına sebep olur.
HIV ve Aids
HIV, Human Immunodeficiency Virus’dür (İnsanlarda Bağışıklık Sistemini Bozan
Virüsdür). Bu virüs AIDS hastalığına sebep olur. |
 Depresyonu Anlamak
Depresyon farklı kişileri farklı biçimlerde etkiler. Duygusal veya fiziksel
olmak üzere geniş alanda belirtilere sebep olabilir.Depresyona neler sebep olur?
Depresyonu Anlamak
Depresyon farklı kişileri farklı biçimlerde etkiler. Duygusal veya fiziksel
olmak üzere geniş alanda belirtilere sebep olabilir.Depresyona neler sebep olur? |
 |
Doğum Sonrası Depresyonu Doğum sonrası depresyonu, doğumdan sonra her on kadından biri tarafından tecrübe edilen stresli bir durumdur. |
 Kanseri") |
Rahim Boyu ( Serviks ) Kanseri Rahim boynu (serviks) kanseri 35 yaş altı kadınlarda görülen vakalarda meme kanserinden sonra ikinci sırayı alır.Serviks kanserinin gelişmesi yıllarca sürebilir. |
 |
Travma Sonrası Bunalımı Travmatik bir olay, günlük olağan olayların dışında olan ve kişiyi derinden rahatsız eden bir olaydır.Birçok olay böyle bir etki gösterebilir. |
İLAÇ GENEL BİLGİLERİ
Amgen İlaç Tic. Ltd. Şti
| Geri Ödeme Kodu | A16943 |
| Satış Fiyatı | 24563.04 TL [ 8 Aug 2025 ] |
| Önceki Satış Fiyatı | 24563.04 TL [ 1 Aug 2025 ] |
| Original / Jenerik | Original İlaç |
| Reçete Durumu | Normal Reçeteli bir ilaçdır. |
| Barkodu | 8699862270014 |
| Etkin Madde | Karfilzomib |
| İthal ( ref. ülke : Hollanda ) ve Beşeri bir ilaçdır. |