LORVIQUA 25 mg film kapl� tablet (30 tablet) K�sa �r�n Bilgisi
{ Lorlatinib }
1. BE�ER� TIBB� �R�N�N ADI
LORV�QUA 25 mg film kapl� tablet
2. KAL�TAT�F VE KANT�TAT�F B�LE��M
Etkin madde
Lorlatinib 25 mg
Yard�mc� maddeler
Laktoz monohidrat (s���r kaynakl�) 1,58 mg
Yard�mc� maddeler i�in 6.1'e bak�n�z.
3. FARMAS�T�K FORMU
Film Kapl� Tablet.
Bir y�z�nde “Pfizer”, di�er y�z�nde “25” ve “LLN” bask�l� yuvarlak sar�ms� kahverengi h�zl� sal�ml� film kapl� tablet
4. KL�N�K �ZELL�KLER
4.1. Terap�tik endikasyonlar
LORV�QUA, monoterapi olarak daha �nce bir ALK (anaplastik lenfoma kinaz) inhibit�r� ile tedavi edilmemi� veya bir veya daha fazla ALK tirozin kinaz inhibit�r� (TKI) ile tedavi edilmi� ALK pozitif, metastatik k���k h�creli d��� akci�er kanseri (KHDAK) olan yeti�kinlerin tedavisinde endikedir.
4.2. Pozoloji ve uygulama �ekli
PozolojiLorlatinib tedavisi, antikanser t�bbi �r�nlerin kullan�m�nda deneyimli bir doktor taraf�ndan ba�lat�lmal� ve g�zetim alt�nda tutulmal�d�r.
Yarar sa�lad��� g�sterilen tek hasta grubu oldu�undan, ALK pozitif KHDAK'nin saptanmas� lorlatinib tedavisinde hasta se�imi i�in gereklidir. ALK pozitif KHDAK de�erlendirmesi, yeterlili�i kan�tlanm�� laboratuvarlar taraf�ndan belirli teknoloji kullan�larak yap�lmal�d�r. Uygun olmayan test performans�, g�venilir olmayan test sonu�lar�na yol a�abilir.
�nerilen doz oral yoldan g�nde bir defa 100 mg'dir.
Uygulama s�kl��� ve s�resi:
Hastal�k ilerleyene veya kabul edilemez toksisite meydana gelene kadar lorlatinib ile tedaviye
devam edilmelidir.
Geciken ya da unutulan dozlar
Bir dozun unutulmas� halinde bir sonraki doza 4 saatten az bir zaman kalmad��� s�rece unutulan doz hat�rlan�r hat�rlanmaz al�nmal�d�r. Bir sonraki doza 4 saatten daha az s�re kalm��sa unutulan tablet atlanmal�d�r. Unutulan dozu tamamlamak i�in 2 doz ayn� anda al�nmamal�d�r.
Dozaj Modifikasyonlar�
Bireysel g�venlilik ve toleransa ba�l� olarak dozun azalt�lmas� veya kesilmesi gerekebilir.
Lorlatinib doz azaltma seviyeleri a�a��da �zetlenmi�tir:
�lk doz azaltma: Oral yoldan g�nde bir defa LORV�QUA 75 mg
�kinci doz azaltma: Oral yoldan g�nde bir defa LORV�QUA 50 mg
Oral yoldan g�nde bir defa 50 mg'lik dozu tolere edemeyen hastalarda LORV�QUA'n�n kullan�m�na kal�c� olarak son verilmelidir.
Toksisiteler ve atriyoventrik�ler (AV) blok geli�en hastalar i�in �nerilen dozaj de�i�iklikleri Tablo 1'de verilmi�tir.
Tablo 1. Advers Reaksiyonlar i�in �nerilen LORV�QUA Dozaj Modifikasyonlar�
Advers Reaksiyon | Lorlatinib dozaj� |
Hiperkolesterolemi veya hipertrigliseridemi | |
Hafif hiperkolesterolemi (kolesterol ULN ile 300 mg/dL veya ULN ile 7.75 mmol/L aras�nda)
VEYA
Orta derecede hiperkolesterolemi (kolesterol 301 ile 400 mg/dL veya 7,76 ile 10,34 mmol/L aras�nda) VEYA Hafif hipertrigliseridemi (trigliserit 150 ve 300 mg/dL veya 1,71 ve 3,42 mmol/L aras�nda) VEYA Orta derecede hipertrigliseridemi (trigliserit 301 ile 500 mg/dL veya 3,43 ile 5,7 mmol/L aras�nda) | �lgili re�eteleme bilgilerine g�re lipid d���r�c� tedaviye ba�lay�n veya lipid d���r�c� tedaviyi modifiye edin / ayarlay�n; lorlatinibe ayn� dozda devam edin. |
�iddetli hiperkolesterolemi (kolesterol 401 ile 500 mg/dL veya 10,35 ile 12,92 mmol/L aras�nda) VEYA �iddetli hipertrigliseridemi | Lipid d���r�c� tedaviye ba�lay�n. Halihaz�rda lipid d���r�c� tedavi uygulan�yorsa, ilgili re�ete bilgilerine g�re bu tedavinin dozunu artt�r�n veya yeni bir lipid d���r�c� tedaviye ge�in. Ara vermeden lorlatinibe ayn� dozda devam edin. |
(trigliserit 501 and 1,000 mg/dL or 5,71 ve 11,4 mmol/L aras�nda) |
|
Hayat� tehdit eden hiperkolesterolemi (kolesterol 500 mg/dL'nin veya 12,92 mmol/L'nin �zerinde)
VEYA
Hayat� tehdit eden hipertrigliseridemi (trigliserit 1,000 mg/dL'nin veya 11,4 mmol/L'nin �zerinde) | �lgili re�eteleme bilgilerine g�re lipid d���r�c� tedaviye ba�lay�n veya bu tedavinin dozunu artt�r�n ya da yeni bir lipid d���r�c� tedaviye ge�i� yap�n. Hiperkolesterolemi ve/veya hipertrigliseridemi orta veya hafif dereceye iyile�ene kadar lorlatinibe ara verin.
�lgili re�eteleme bilgilerine g�re lipid d���r�c� tedaviyi maksimuma ��kar�rken ayn� lorlatinib dozu ile devam edin.
�iddetli hiperkolesterolemi ve/veya hipertrigliseridemi, ilgili re�ete bilgilerine g�re en y�ksek lipid d���r�c� tedavisine ra�men tekrarlarsa, lorlatinib dozunu bir seviye azalt�n. |
Merkezi Sinir Sistemi (MSS) etkileri (bili�sel, ruh hali, zihinsel durum veya konu�madaki psikotik etkileri ve de�i�iklikleri i�erir) | |
Derece 2: Orta VEYA Derece 3: Ciddi | Toksisite Derece 1'e e�it veya daha d���k olana kadar doza ara verin. Ard�ndan bir seviye azalt�lm�� dozda lorlatinibe devam edin. |
Derece 4: Ya�am� tehdit eden | Kal�c� olarak lorlatinib kullan�m�na son verin. |
Lipaz/Amilaz art��� | |
Derece 3: �iddetli VEYA Derece 4: Ya�am� tehdit eden/Acil m�dahale gerektiren |
Lipaz veya amilaz ba�lang�� seviyesine d�n�nceye kadar lorlatinibe ara verin. Ard�ndan bir seviye azalt�lm�� dozda lorlatinibe devam edin. |
�nterstisyel Akci�er Hastal��� (ILD)/Akci�er �ltihab� | |
Derece 1: Hafif VEYA Derece 2: Orta | Semptomlar ba�lang�� seviyesine d�n�nceye kadar lorlatinibe ara verin ve kortikosteroidlere ba�lamay� d���n�n. Ard�ndan bir seviye azalt�lm�� dozda lorlatinibe devam edin.
6 haftal�k lorlatinib kesilmesi ve steroid tedavisinden sonra ILD/ akci�er iltihab� tekrarlarsa ya da iyile�mezse lorlatinibi kal�c� olarak b�rak�n. |
Derece 3: �iddetli VEYA Derece 4: Ya�am� tehdit eden/Acil m�dahale gerektiren |
Kal�c� olarak lorlatinib kullan�m�na son verin. |
PR aral��� uzamas�/Atrioventrik�ler (AV) Blok | |
Birinci derecede AV blo�u: Asemptomatik | Lorlatinibe ara vermeden ayn� dozda devam edin. E� zamanl� t�bbi �r�nlerin etkilerini g�z �n�nde bulundurun. PR aral���n� uzatabilecek elektrolit dengesizli�ini de�erlendirin ve d�zenleyin. AV blo�u ile potansiyel olarak ili�kili olan EKG/semptomlar� yak�ndan izleyin. |
Birinci derecede AV blo�u: Semptomatik | Lorlatinibe ara verin. E� zamanl� t�bbi �r�nlerin etkilerini g�z �n�nde bulundurun. PR aral���n� uzatabilecek elektrolit dengesizli�ini de�erlendirin ve d�zenleyin. AV blo�u ile potansiyel olarak ili�kili olan EKG/semptomlar� yak�ndan izleyin. Semptomlar d�zelirse, bir seviye azalt�lm�� dozda lorlatinibe devam edin. |
�kinci derecede AV blo�u: Asemptomatik | Lorlatinibe ara verin. E� zamanl� t�bbi �r�nlerin etkilerini g�z �n�nde bulundurun. PR aral���n� uzatabilecek elektrolit dengesizli�ini de�erlendirin ve d�zenleyin. AV blo�u ile potansiyel olarak ili�kili EKG/semptomlar� yak�ndan izleyin. Sonraki EKG'de ikinci derecede AV blo�u g�r�lmezse, bir seviye azalt�lm�� dozda lorlatinibe devam edin. |
�kinci derecede AV blo�u: Semptomatik | Lorlatinibe ara verin. E� zamanl� t�bbi �r�nlerin etkilerini g�z �n�nde bulundurun. PR aral���n� uzatabilecek elektrolit dengesizli�ini de�erlendirin ve d�zenleyin. Kardiyak g�zlem ve takibe ba�vurun. Semptomatik AV blo�u devam ederse kalp pili tak�lmas�n� de�erlendirin. Semptomlar ve ikinci derecede AV blo�u d�zelirse veya hastalar asemptomatik birinci derece AV blo�una geri d�nerse, bir seviye azalt�lm�� dozda lorlatinibe devam edin. |
Tam AV blo�u | Lorlatinibe ara verin. E� zamanl� t�bbi �r�nlerin etkilerini g�z �n�nde bulundurun. PR aral���n� uzatabilecek elektrolit dengesizli�ini de�erlendirin ve d�zenleyin. Kardiyak g�zlem ve takibe ba�vurun. AV blo�u ile ili�kili �iddetli semptomlar i�in kalp pili tak�lmas� gerekebilir. AV blo�u ��z�lmezse, kal�c� bir kalp pili tak�lmas� d���n�lebilir.
Kalp pili tak�lm��sa, lorlatinibe tam dozda devam edin. Kalp pili tak�lmam��sa, yaln�zca semptomlar d�zeldi�inde ve PR aral��� 200 ms'den az oldu�unda bir seviye azalt�lm�� dozda lorlatinibe devam edin. |
Hipertansiyon | |
Derece 3 (160 mmHg'ye e�it veya daha y�ksek SKB ya da 100 mmHg'ye e�it veya daha y�ksek DKB; t�bbi m�dahale gerektiren; birden fazla antihipertansif ila� veya daha �nce kullan�landan daha yo�un tedavi gerektiren) | Derece 1 veya daha d���k hipertansiyon (SKB 140 mmHg'den d���k ve DKB 90 mmHg'den d���k) g�zlenene kadar lorlatinibe ara verin ve ard�ndan ayn� dozda devam edin.
Derece 3 hipertansiyon tekrarlarsa, derece 1 veya daha d���k seviyeye iyile�inceye kadar lorlatinibe ara verin ve azalt�lm�� bir dozda devam edin. Optimal t�bbi tedavi ile yeterli hipertansiyon kontrol� sa�lanamazsa, lorlatinib kal�c� olarak kesilmelidir. |
Derece 4 (Ya�am� tehdit eden sonu�lar� olan, acil m�dahale gerektiren) | Derece 1 veya daha d���k seviyeye gelinceye kadar lorlatinibe ara verin ve azalt�lm�� bir dozda devam edin ya da lorlatinibi kal�c� olarak kesin.
Derece 4 hipertansiyon tekrar ederse, lorlatinib kal�c� olarak kesilmelidir. |
Hiperglisemi | |
Derece 3 VEYA Derece 4 (Optimal anti hiperglisemik tedaviye ra�men 250 mg/dL'den y�ksek kal�c� hiperglisemi) | Hiperglisemi yeterince kontrol edilinceye kadar lorlatinibe ara verin, ard�ndan bir seviye azalt�lm�� dozda lorlatinibe devam edin.
Optimal t�bbi tedavi ile yeterli hiperglisemik kontrol sa�lanamazsa, lorlatinib kal�c� olarak kesilmelidir. |
Di�er advers reaksiyonlar |
|
Derece 1: Hafif VEYA Derece 2: Orta |
Klinik duruma g�re doz modifikasyonu yapmamay� ve dozu bir seviye azaltmay� de�erlendirin. |
Derece 3 veya daha y�ksek: �iddetli | Semptomlar Derece 2'ye veya ba�lang��takine e�it ya da daha d���k oluncaya kadar lorlatinibe ara verin. Ard�ndan bir seviye azalt�lm�� dozda lorlatinibe devam edin. |
K�saltmalar: MSS: merkezi sinir sistemi; CTCAE Advers Olaylar i�in Genel Terminoloji Kriterleri; DKB: diastolik kan bas�nc�; EKG: elektrokardiyogram; HMG CoA: 3-hidroksi-3- metilglutaril koenzim A; NCL: Ulusal Kanser Enstit�s�; SKB: sistolik kan bas�nc�; ULN: normalin �st s�n�r�.
G��l� sitokrom P-450 (CYP) 3A4/5 inhibit�rleri
Lorlatinibin g��l� CYP 3A4/5 inhibit�r� olan t�bbi �r�nler ve greyfurt suyu �r�nleri ile e� zamanl� kullan�m�, lorlatinib plazma konsantrasyonunu artt�rabilir. CYP 3A4/5'i inhibe etme potansiyeli daha d���k olan alternatif bir e� zamanl� t�bbi �r�n d���n�lmelidir (bkz. B�l�m 4.5). G��l� bir CYP 3A4/5 inhibit�r� birlikte uygulanmak zorundaysa, g�nde bir kez 100 mg olan ba�lang�� lorlatinib dozu, g�nde bir kez 75 mg'a d���r�lmelidir (bkz. B�l�m 4.5 ve 5.2). G��l� bir CYP 3A4/5 inhibit�r�n�n e� zamanl� kullan�m� kesilirse, g��l� CYP3A4/5 inhibit�r�n�n 3 ile 5 yar�lanma �mr� kadar bir ar�nma d�neminden sonra, g��l� CYP 3A4/5 inhibit�r�ne ba�lamadan �nce kullan�lan dozda lorlatinibe devam edilmelidir.
Uygulama �ekli:
LORV�QUA oral kullan�m i�indir.
Hastalara dozlar�n� yakla��k olarak g�n�n ayn� saatinde yiyeceklerle veya a� almalar� �nerilmelidir (Bkz. B�l�m 5.2). Tabletler b�t�n olarak yutulmal�d�r (tabletler yutulmadan �nce �i�nenmemeli, k�r�lmamal� veya b�l�nmemelidir). K�r�lm��, �atlam�� veya ba�ka �ekilde zarar g�rm�� tabletler al�nmamal�d�r.
�zel pop�lasyonlara ili�kin ek bilgiler:
Karaci�er Yetmezli�i:
Hafif karaci�er yetmezli�i olan hastalar i�in doz ayarlamas� �nerilmez. Orta veya �iddetli karaci�er yetmezli�i olan hastalar i�in bir bilgi mevcut de�ildir. Bu nedenle orta veya �iddetli karaci�er yetmezli�i olan hastalarda lorlatinib kullan�m� �nerilmez (bkz. B�l�m 5.2).
B�brek Yetmezli�i:
B�brek fonksiyonu normal olan ve hafif veya orta derecede b�brek yetmezli�i [mutlak tahmini glomer�ler filtrasyon h�z� (eGFR): ≥ 30 mL/dk] olan hastalarda doz ayarlamas� yap�lmas� gerekli de�ildir. �iddetli b�brek yetmezli�i olan hastalarda (mutlak eGFR < 30 mL/dk), azalt�lm�� lorlatinib dozu, �rne�in g�nde bir kez oral al�nan 75 mg'l�k ba�lang�� dozu, �nerilir (bkz. B�l�m 5.2). Renal diyaliz hastalar� i�in bilgi mevcut de�ildir.
Pediyatrik pop�lasyon:
Lorlatinibin 18 ya� alt� pediatrik hastalarda g�venlili�i ve etkilili�i belirlenmemi�tir. Veri mevcut de�ildir.
Geriyatrik pop�lasyon:
Bu pop�lasyona ili�kin verilerin s�n�rl� olmas� nedeniyle 65 ya� ve �zeri hastalar i�in doz �nerisi yap�lamamaktad�r (bkz. B�l�m 5.2).
4.3. Kontrendikasyonlar
Lorlatinibe veya 6.1'de listelenen yard�mc� maddelerden herhangi birine kar�� a��r� duyarl�l��� olanlarda kontrendikedir.
LORV�QUA ile g��l� CYP3A4/5 ind�kleyicilerinin e� zamanl� kullan�m� kontrendikedir (Bkz.
B�l�m 4.4 ve 4.5).
4.4. �zel kullan�m uyar�lar� ve �nlemleri
Hiperlipidemi
Lorlatinib kullan�m� serum kolesterol ve trigliserid art��lar� ile ili�kili bulunmu�tur (bkz. B�l�m 4.8). Serum kolesterol ve trigliserid seviyelerinde a��r art�� olu�mas�na kadar ge�en medyan s�re s�ras�yla 104 g�n (aral�k: 29- 518 g�n) ve 120 g�nd�r (aral�k: 15-780 g�n). Serum kolesterol ve trigliserid seviyeleri lorlatinib ba�lanmadan �nce ve ba�land�ktan 2, 4 ve 8. haftalarda ve ard�ndan d�zenli olarak izlenmelidir. E�er endike ise lipid d���r�c� bir ilaca ba�lanmal� ya da kullan�lmakta olan ilac�n dozu artt�r�lmal�d�r (Bkz. B�l�m 4.2).
Merkezi Sinir Sistemi Etkileri
Lorlatinib kullanan hastalarda psikotik etkiler ve bili�sel fonksiyon, duygudurum, zihinsel durum veya konu�ma de�i�iklikleri dahil merkezi sinir sistemi (MSS) etkileri g�zlenmi�tir (Bkz. B�l�m 4.8). MSS etkileri geli�en hastalar i�in doz de�i�ikli�i ya da lorlatinib tedavisinin b�rak�lmas� gerekebilir (Bkz. B�l�m 4.2).
Atriyoventrik�ler blok
Lorlatinib ikinci derece ya da ���nc� derece AV blo�u (pacemaker tak�l� olmad��� s�rece) olan ya da PR aral���n�n >220 milisaniye oldu�u herhangi bir AV blo�u olan hastalar�n d��land��� bir hasta pop�lasyonunda incelenmi�tir. Lorlatinib alan hastalarda PR aral���nda uzama ve AV blok bildirilmi�tir (Bkz. B�l�m 5.2). �zellikle klinik �nemi olan kalp olaylar�n�n olu�mas�na zemin sa�layan hastal�klar� olan hastalarda olmak �zere lorlatinib ba�lamadan �nce ve ard�ndan ayda bir elektrokardiyografi (EKG) takibi yap�lmal�d�r. AV blok geli�en hastalarda doz de�i�iklikleri gerekebilir (Bkz. B�l�m 4.2).
Sol ventrik�l ejeksiyon fraksiyonunda d��me
Lorlatinib alan ve ba�lang��ta ve en az bir takip sol ventrik�l ejeksiyon fraksiyonu (LVEF) de�erlendirmesi olan hastalarda sol ventrik�l ejeksiyon fraksiyonunda d��me bildirilmi�tir. Mevcut klinik �al��ma verilerine g�re kalbin kas�lma kapasitesindeki de�i�iklikler �zerindeki etki ve lorlatinib aras�nda bir neden ili�kisi belirlemek olanaks�zd�r. Kardiyak risk fakt�rleri olan ve LVEF seviyesini etkileyebilecek hastal�klar� olan hastalarda ba�lang��ta ve tedavi s�ras�nda LVEF de�erlendirmesi dahil olmak �zere kalp a��s�ndan takip yap�lmas� d���n�lmelidir. Tedavi s�ras�nda kardiyak belirti ve semptomlar� geli�en hastalarda LVEF de�erlendirmesi dahil olmak �zere kalp a��s�ndan takip yap�lmas� d���n�lmelidir.
Lipaz ve amilaz art���
Lorlatinib alan hastalarda lipaz ve/veya amilaz art��lar� olu�mu�tur (Bkz. B�l�m 4.8). Serum lipaz ve amilaz seviyelerinde art�� olu�mas�na kadar ge�en medyan s�re s�ras�yla 141 g�n (aral�k: 1- 1091 g�n) ve 138 g�nd�r (aral�k: 1-1112 g�n). Lorlatinib alan hastalarda e� zamanl� hipertrigliseridemi ve/veya olas� intrensek bir mekanizma nedeniyle pankreatit riski dikkate al�nmal�d�r. Hastalar lorlatinib ba�lanmadan �nce ve klinik olarak endike ise ba�land�ktan sonra d�zenli olarak lipaz ve amilaz a��s�ndan takip edilmelidir (Bkz. B�l�m 4.2).
�nterstisyel akci�er hastal���/Pn�monit
Lorlatinib ile ILD/pn�monit ile uyumlu a��r ya da ya�am� tehdit eden pulmoner advers reaksiyonlar olu�mu�tur (Bkz. B�l�m 4.8). Solunum semptomlar�nda ILD/pn�monit d���nd�ren (�rne�in dispne, �ks�r�k ve ate�) k�t�le�me olan t�m hastalar derhal ILD/pn�monit a��s�ndan de�erlendirilmelidir. Tablonun �iddetine g�re lorlatinibe ara verilmeli ya da kesin olarak b�rak�lmal�d�r (Bkz. B�l�m 4.2).
Hipertansiyon
Lorlatinib alan hastalarda hipertansiyon bildirilmi�tir (bkz. B�l�m 4.8). Lorlatinib tedavisine ba�lamadan �nce kan bas�nc� kontrol edilmelidir. Lorlatinib tedavisi ba�lang�c�ndan 2 hafta sonra ve devam�nda en az ayda bir kez kan bas�nc� izlenmelidir. Tablonun �iddetine g�re lorlatinibe ara verilmeli ve azalt�lm�� bir dozda yeniden ba�lanmal� ya da kal�c� olarak b�rak�lmal�d�r (Bkz. B�l�m 4.2).
Hiperglisemi
Lorlatinib alan hastalarda hiperglisemi meydana gelmi�tir (bkz. B�l�m 4.8). Lorlatinib tedavisine ba�lamadan �nce a�l�k serum glukozu �l��lmeli ve sonras�nda ulusal k�lavuzlara g�re periyodik olarak izlenmelidir. Tablonun �iddetine g�re lorlatinibe ara verilmeli ve azalt�lm�� bir dozda yeniden ba�lanmal� ya da kal�c� olarak b�rak�lmal�d�r (Bkz. B�l�m 4.2).
�la� etkile�imleri
Sa�l�kl� g�n�ll�lerde yap�lan bir �al��mada lorlatinib ile e� zamanl� olarak g��l� bir CYP3A4/5 ind�kleyicisi olan rifampin kullan�m� total bilir�bin ve alkalin fosfataz art��� olmaks�z�n alanin aminotransferaz (ALT) ve aspartat aminotransferaz (AST) art��lar� ile ili�kili bulunmu�tur (Bkz. B�l�m 4.5). E� zamanl� olarak g��l� bir CYP3A4/5 ind�kleyicisi kullan�m� kontrendikedir (Bkz. B�l�mler 4.3 ve 4.5).
Lorlatinib ile orta derece CYP3A4/5 ind�kleyicisi modafinil kombinasyonu alan sa�l�kl� g�n�ll�lerde karaci�er fonksiyon testlerinde klinik olarak anlaml� de�i�iklikler g�r�lmemi�tir (Bkz. B�l�m 4.5).
Lorlatinib e� zamanl� olarak verildi�inde alfentanil, siklosporin, dihidroergotamin, ergotamin, fentanil, hormonal kontraseptifler, pimozid, kinidin, sirolimus ve takrolimus dahil ancak bunlarla k�s�tl� olmamak �zere terap�tik indeksi dar CYP3A4/5 substratlar�n�n konsantrasyonlar�n� d���rebilece�inden bu ila�lar�n lorlatinib ile e� zamanl� verilmesinden ka��nmal�d�r (Bkz. B�l�m 4.5).
�reme ve gebelik
Lorlatinib ile tedavi s�ras�nda ve son dozdan sonra en az 14 hafta boyunca gebe kalma olas�l��� olan kad�n partneri bulunan erkek hastalar kondom kullan�m�n� i�eren etkili bir gebelikten korunma y�ntemi ve partneri gebe olan erkek hastalar kondom kullanmal�d�r (Bkz. B�l�m 4.6). Erkek �reme fonksiyonu lorlatinib tedavisi s�ras�nda olumsuz etkilenebilir (Bkz. B�l�m 5.3). Erkekler tedaviden �nce �reme fonksiyonunun etkili bir �ekilde korunmas� konusunda bir doktora dan��mal�d�r. Gebe kalma olas�l��� olan kad�nlara lorlatinib kullan�rken gebe kalmaktan ka��nmalar� s�ylenmelidir. Lorlatinib hormonal kontraseptifleri etkisizle�tirebilece�inden lorlatinib tedavisi s�ras�nda kad�n hastalar i�in hormonal olmayan y�ksek etkili bir kontraseptif gerekir (Bkz. B�l�mler 4.5 ve 4.6). E�er hormonal bir gebelikten korunma y�nteminin kullan�lmas� ka��n�lmazsa bu durumda hormonal y�ntemle birlikte kondom kullan�lmal�d�r. Etkili gebelikten korunma tedaviyi tamamlad�ktan sonra en az 35 g�n s�rd�r�lmelidir (Bkz. B�l�m 4.6). Lorlatinibin kad�n �reme fonksiyonunu etkileyip etkilemedi�i bilinmemektedir.
Laktoz intolerans�
Bu t�bbi �r�n laktoz i�ermektedir. Nadir kal�t�msal galaktoz intolerans�, Lapp laktaz yetmezli�i ya da glukoz-galaktoz malabsorpsiyon problemi olan hastalar�n bu ilac� kullanmamalar� gerekir.
Sodyum
Bu t�bbi �r�n her “doz”unda (25 mg veya 100 mg tablette) 1 mmol (23 mg)'dan daha az sodyum ihtiva eder. D���k sodyum diyeti uygulayan hastalar, bu �r�n�n asl�nda “sodyum i�ermedi�i” konusunda bilgilendirilmelidir.
4.5. Di�er t�bbi �r�nler ile etkile�imler ve di�er etkile�im �ekilleri
Farmakokinetik etkile�imleri
�n vitro veriler lorlatinibin ba�l�ca CYP3A4 ve �ridin difosfat-glukunosiltransferaz (UGT) 1A4 ile metabolize edildi�ini g�stermektedir. CYP2C8, CYP2C19, CYP3A5 ve UGT1A3 min�r katk�da bulunmaktad�r.
�la�lar�n lorlatinib �zerindeki etkisi
CYP3A4/5 ind�kleyicileri
G��l� bir CYP3A4/5 ind�kleyici olan rifampin sa�l�kl� g�n�ll�lere 12 g�n boyunca g�nde bir kez oral yolla 600 mg dozunda verildi�inde tek bir 100 mg oral lorlatinib dozunun ortalama e�ri alt�ndaki alan�n� (EAA) %85 ve Cde�erini %76 azaltm��t�r; AST ve ALT art��lar� da g�zlenmi�tir. Lorlatinibin g��l� CYP3A4/5 ind�kleyicileri (�rne�in rifampisin, karbamazepin, enzalutamid, mitotan, fenitoin ve sar� kantaron otu) ile e� zamanl� verilmesi lorlatinibin plazma konsantrasyonlar�n� azaltabilir. Lorlatinibin g��l� CYP3A4/5 ind�kleyicileri ile kullan�m� kontrendikedir (Bkz. B�l�mler 4.3 ve 4.4). Sa�l�kl� g�n�ll�lerde tek bir 100 mg oral lorlatinib dozu ile orta derecede CYP3A4/5 ind�kleyici olan modafinil (19 g�n boyunca g�nde bir kez 400 mg) kombinasyonunun uygulanmas�ndan sonra karaci�er fonksiyon testi sonu�lar�nda klinik olarak anlaml� bir de�i�iklik g�r�lmemi�tir. Modafinilin e� zamanl� kullan�m�, lorlatinibin farmakokineti�i �zerinde klinik olarak anlaml� bir etkiye sahip olmam��t�r.
CYP3A4/5 inhibit�rleri
G��l� bir CYP3A4/5 inhibit�r� olan itrakonazol sa�l�kl� g�n�ll�lere 5 g�n boyunca 200 mg
oral g�nde tek doz verildi�inde tek bir 100 mg oral lorlatinib dozunun ortalama EAAde�erini
%42 ve tek bir 100 mg oral lorlatinib dozunun Cde�erini %24 azaltm��t�r. G��l� CYP3A4/5 inhibit�rleri (�rne�in boseprevir, kobisistat, itrakonazol, ketakonazol, posakonazol, troleandomisin, vorikonazol, ritonavir, ritonavir ile kombine paritaprevir ve ombitasvir ve/veya dasabuvir ve elvitegravir, indinavir, lopinavir ya da tipranavir ile kombine ritonavir) lorlatinibin plazma konsantrasyonlar�n� artt�rabilir. Greyfurt �r�nleri de lorlatinibin plazma konsantrasyonlar�n� artt�rabilir ve ka��n�lmal�d�r. CYP3A4/5 inhibisyonu yapma olas�l��� daha d���k olan alternatif bir ilac�n e� zamanl� verilmesi d���n�lmelidir. E�er g��l� bir CYP3A4/5 inhibit�r�n�n e� zamanl� olarak verilmesi zorunlu ise lorlatinib dozunun azalt�lmas� �nerilir (Bkz. B�l�m 4.2).
Lorlatinibin di�er ila�lar �zerindeki etkisi
CYP3A4/5 substratlar�
�n vitro �al��malar lorlatinibin CYP3A4/5'nin zamana ba��ml� bir inhibit�r� ve bunun yan� s�ra ind�kleyicisi oldu�unu g�stermi�tir. Lorlatinib 15 g�n boyunca g�nde tek 150 mg dozunda oral yolla verildi�inde tek bir 2mg midazolam (duyarl� bir CYP3A substrat�) dozunun EAAde�erini ve Cde�erini s�ras�yla %61'e ve %50'ye kadar azaltm��t�r; dolay�s�yla, lorlatinib orta g��te CYP3A ind�kleyicisidir. Bu nedenle lorlatinib e� zamanl� olarak verildi�inde alfentanil, siklosporin, dihidroergotamin, ergotamin, fentanil, hormonal kontraseptifler, pimozid, kinidin, sirolimus ve takrolimus dahil ancak bunlarla k�s�tl� olmamak �zere terap�tik indeksi dar CYP3A4/5 substratlar�n�n konsantrasyonlar�n� d���rebilece�inden bu ila�lar�n lorlatinib ile e� zamanl� verilmesinden ka��nmal�d�r (bkz. B�l�m 4.4).
CYP2B6 substrat�
Lorlatinib 15 g�n boyunca g�nde tek 100 mg dozunda oral yolla verildi�inde tek bir oral 100 mg bupropion (CYP2B6 ve CYP3A4 substrat� kombinasyonu) dozunun EAAde�erini ve Cde�erini s�ras�yla %49,5'e ve %53'e kadar azaltm��t�r. Dolay�s�yla, lorlatinib zay�f bir CYP2B6 ind�kleyicisidir ve lorlatinib ba�l�ca CYP2B6 ile metabolize edilen ila�larla kombine kullan�ld���nda doz ayarlamas� gerekmez.
CYP2C9 substrat�
Lorlatinib 15 g�n boyunca g�nde tek 100 mg dozunda oral yolla verildi�inde tek bir oral 500 mg tolbutamid (duyarl� bir CYP2C9 substrat�) dozunun EAAde�erini ve Cde�erini s�ras�yla %43'e ve %15'e kadar azaltm��t�r. Dolay�s�yla, lorlatinib zay�f bir CYP2C9 ind�kleyicisidir ve lorlatinib ba�l�ca CYP2C9 ile metabolize edilen ila�larla kombine kullan�ld���nda doz ayarlamas� gerekmez.
Bununla birlikte CYP2C9 ile metabolize edilen dar terap�tik indekse sahip ila�larla (�rne�in kumarin antikoag�lanlar) e� zamanl� tedavi durumunda hastalar takip edilmelidir.
UGT substratlar�
Lorlatinib 15 g�n boyunca g�nde tek 100 mg dozunda oral yolla verildi�inde tek bir oral 500 mg asetaminofen (bir UGT, SULT ve CYP1A2, 2A6, 2D6 ve 3A4 substrat�) dozunun EAAde�erini ve Cde�erini s�ras�yla %45'e ve %28'e kadar azaltm��t�r. Dolay�s�yla, lorlatinib zay�f bir UGT ind�kleyicisidir ve lorlatinib ba�l�ca UGT ile metabolize edilen ila�larla kombine kullan�ld���nda doz ayarlamas� gerekmez. Bununla birlikte UGT ile metabolize edilen dar terap�tik indekse sahip ila�larla e� zamanl� tedavi durumunda hastalar takip edilmelidir.
P-glikoprotein substratlar�
Lorlatinib 15 g�n boyunca g�nde tek 100 mg dozunda oral yolla verildi�inde tek bir oral 60 mg feksofenadin [duyarl� bir P-glikoprotein (P-gp) substrat�] dozunun EAAde�erini ve Cde�erini s�ras�yla %67'ye ve %63'e kadar azaltm��t�r. Dolay�s�yla, lorlatinib orta g��te bir P- gp ind�kleyicisidir. P-gp substrat� olan ve dar terap�tik indekse sahip ila�lar (�rne�in digoksin, dabigatran eteksilat) bu substratlar�n�n plazma konsantrasyonlar�n� d���rme olas�l��� nedeniyle lorlatinib ile kombine kullan�l�rken dikkatli olunmal�d�r.
Di�er CYP enzimlerinin in vitro inhibisyon ve ind�ksiyon �al��malar�
�n vitro olarak lorlatinibin CYP1A2 ind�ksiyonu yoluyla ila�-ila� etkile�imlerine neden olma olas�l��� d���kt�r.
P-gp d���ndaki ila� ta��y�c�lar� ile in vitro �al��malar
�n vitro �al��malar lorlatinibin klinik konsantrasyonlarda BCRP (gastrointestinal sistem), OATP1B1, OATP1B3, OCT1, MATE1 ve OAT3 substratlar�n�n plazma seviyelerinde klinik olarak anlaml� de�i�ikliklere yol a�ma olas�l��� oldu�unu g�stermi�tir. BCRP, OATP1B1, OATP1B3, OCT1, MATE1 ve OAT3 substratlar�n�n plazma seviyelerinde klinik olarak anlaml� de�i�ikliklere yol a�ma olas�l��� d��lanamayaca��ndan lorlatinib bu substratlarla kullan�l�rken dikkatli olunmal�d�r.
�zel pop�lasyonlara ili�kin ek bilgiler
�zel pop�lasyonlara ili�kin hi�bir klinik etkile�im �al��mas� y�r�t�lmemi�tir.
Pediyatrik pop�lasyon:
Pediyatrik pop�lasyona ili�kin hi�bir klinik etkile�im �al��mas� y�r�t�lmemi�tir.
4.6. Gebelik ve laktasyon
Genel tavsiye
Gebelik kategorisi: C
�ocuk do�urma potansiyeli bulunan kad�nlar/Do�um kontrol� (Kontrasepsiyon)
Gebe kalma potansiyeli olan kad�nlara lorlatinib kullan�rken gebe kalmaktan ka��nmalar� s�ylenmelidir. Lorlatinib hormonal kontraseptifleri etkisizle�tirebilece�inden lorlatinib tedavisi s�ras�nda kad�n hastalar�n y�ksek etkili hormonal olmayan bir gebelikten korunma y�ntemi kullanmalar� gerekir (Bkz. B�l�mler 4.4 ve 4.5). E�er hormonal kontraseptif kullan�lmas� zorunlu ise hormonal y�ntemle birlikte kondom kullan�lmal�d�r. Tedavi tamamland�ktan en az 35 g�n sonras�na kadar etkili bir gebelikten korunma y�ntemi kullan�lmaya devam edilmelidir.
Gebe kalma potansiyeli olan kad�n partnerleri olan erkek hastalar lorlatinib tedavisi s�ras�nda ve son dozdan en az 14 hafta sonras�na kadar kondomu da i�eren etkili gebelikten korunma y�ntemleri kullanmal� ve partnerleri gebe olan erkek hastalar kondom kullanmal�d�r.
Gebelik d�nemi
Lorlatinibin gebe kad�nlarda kullan�m�na ili�kin yeterli veri mevcut de�ildir. Hayvanlar �zerinde yap�lan ara�t�rmalarda embriyo-fetal toksisite g�sterilmi�tir (Bkz. B�l�m 5.3). �nsanlara y�nelik potansiyel risk bilinmemektedir.
Lorlatinib gebe bir kad�na verildi�inde fet�se zarar verebilir. Lorlatinibin gebelik s�ras�nda ve gebe kalma potansiyeli olup gebelikten korunma y�ntemi kullanmayan kad�nlarda kullan�lmas� �nerilmez.
Laktasyon d�nemi
Lorlatinibin ve metabolitlerinin anne s�t�ne ge�ip ge�medi�i bilinmemektedir. Yeni do�anlarda ve s�t �ocuklar�nda risk d��lanamaz.
Lorlatinib emzirme s�ras�nda kullan�lmamal�d�r. Lorlatinib tedavisi s�ras�nda ve son dozdan 7 g�n sonras�na kadar emzirmeye ara verilmelidir.
�reme yetene�i/Fertilite
Preklinik g�venlilik bulgular�na g�re lorlatinib tedavisi s�ras�nda erkek �reme fonksiyonu olumsuz etkilenebilir (Bkz. B�l�m 5.3). Kad�n �reme fonksiyonunun etkilenip etkilenmedi�i bilinmemektedir. Tedaviye ba�lamadan �nce erkekler �reme fonksiyonunun korunmas� a��s�ndan doktora dan��mal�d�r.
4.7. Ara� ve makine kullan�m� �zerindeki etkiler
Lorlatinibin ara� ve makine kullan�m� �zerine orta d�zeyde etkisi bulunmaktad�r. Hastalar santral sinir sistemi etkileri ya�ayabilece�inden, ara� veya makine kullan�l�rken dikkatli olunmal�d�r (Bkz. B�l�m 4.8).
4.8. �stenmeyen etkiler
G�venlilik profili �zeti
En s�k bildirilen advers reaksiyonlar hiperkolesterolemi (%81,1), hipertrigliseridemi (%67,2), �dem (%55,7), periferik n�ropati (%43,7), kilo art��� (%30,9), bili�sel etkiler (%27,7), fatigue
(%27,3), artralji (%23,5), ishal (%22,9) ve duygudurum etkileri (%21) olmu�tur.
Lorlatinib alan hastalar�n %7,4'�nde ciddi yan etkiler bildirilmi�tir. En s�k g�r�len ciddi advers reaksiyonlar, bili�sel etkiler ve pn�monidir.
Lorlatinib alan hastalar�n %20'sinde advers reaksiyonlara ba�l� olarak doz azalt�lm��t�r. Dozun azalt�lmas�na en s�k neden olan advers reaksiyonlar �dem ve periferik n�ropatidir. Lorlatinib alan hastalar�n %3,2'si geli�en advers reaksiyonlara ba�l� olarak tedaviyi b�rakm��t�r. Tedavinin kesin olarak b�rak�lmas�na en s�k neden olan advers reaksiyonlar bili�sel etkiler, periferik n�ropati, pn�moni ve psikotik etkilerdir.
Advers reaksiyonlar�n tablo listesi
Tablo 2 �al��ma A'da (N=327) ve CROWN �al��mas�nda (N=149) ileri evre KHDAK nedeniyle g�nde bir kez 100 mg lorlatinib ile tedavi edilen 476 eri�kin hastada olu�an advers reaksiyonlar� sunmaktad�r.
Advers reaksiyonlar sistem organ s�n�f�na uygun olarak listelenmi�tir. Advers reaksiyonlar her sistem organ s�n�f� alt�nda a�a��daki d�zen kullan�larak, en s�k g�r�len reaksiyonlar en ba�ta olmak �zere g�r�lme s�kl���na g�re s�ralanm��t�r: �ok yayg�n (≥1/10); yayg�n (≥1/100 ila 1/10); yayg�n olmayan (≥1/1.000 ila <1/100); seyrek (≥1/10.000 ila < 1/1.000); �ok seyrek (< 1/10.000). Her bir s�kl�k grupland�rmas� i�inde istenmeyen etkiler azalan t�bbi ciddiyet s�ras�na g�re verilmi�tir.
Tablo 2.Advers reaksiyonlar
Sistem organ s�n�f� | Advers reaksiyon | S�kl�k | T�m dereceler % | Derece 3-4 % |
Kan ve lenf sistemi hastal�klar� | Anemi | �ok yayg�n | 18,5 | 4,2 |
Metabolizma ve beslenme hastal�klar� | Hiperkolesterolemiᵃ | �ok yayg�n | 81,1 | 18,3 |
Hipertrigliseridemiᵇ | �ok yayg�n | 67,2 | 19,3 | |
Hiperglisemi | Yayg�n | 9,2 | 3,2 | |
Psikiyatrik hastal�klar | Duygudurum �zerindeki etkilerᶜ | �ok yayg�n | 21 | 1,5 |
Psikotik etkilerᵈ | Yayg�n | 6,5 | 0,4 | |
Zihinsel durum de�i�iklikleri | Yayg�n | 2 | 1,7 | |
Sinir sistemi hastal�klar� | Bili�sel etkilerᵉ | �ok yayg�n | 27,7 | 2,9 |
Periferik n�ropatiᶠ | �ok yayg�n | 43,7 | 2,7 | |
Ba� a�r�s� | �ok yayg�n | 17,9 | 0,6 | |
Konu�ma �zerinde etkilerᶢ | Yayg�n | 8,2 | 0,6 | |
G�z hastal�klar� | G�rme bozukluklar�ʰ | �ok yayg�n | 17,2 | 0,2 |
Vask�ler hastal�klar� | Hipertansiyon | �ok yayg�n | 13 | 6,1 |
Solunum, g���s bozukluklar� ve mediastinal hastal�klar | Pn�monitᶦ | Yayg�n | 1,9 | 0,6 |
Gastrointestinal hastal�klar� | �shal | �ok yayg�n | 22,9 | 1,5 |
Bulant� | �ok yayg�n | 17,6 | 0,6 | |
Kab�zl�k | �ok yayg�n | 17,4 | 0,2 | |
Deri ve deri alt� doku hastal�klar� | D�k�nt�ʲ | �ok yayg�n | 13,7 | 0,2 |
Kas-iskelet bozukluklar�, ba� doku ve kemik hastal�klar� | Artalji | �ok yayg�n | 23,5 | 0,8 |
Miyaljiᵏ | �ok yayg�n | 19,3 | 0,2 | |
Genel bozukluklar ve uygulama b�lgesine ili�kin hastal�klar | �demˡ | �ok yayg�n | 55,7 | 2,7 |
Fatigueᵐ | �ok yayg�n | 27,3 | 1,3 | |
Ara�t�rmalar | Kilo art��� | �ok yayg�n | 30,9 | 10,1 |
Lipaz art��� | �ok yayg�n | 12,4 | 6,9 | |
Amilaz art��� | �ok yayg�n | 11,3 | 2,7 | |
Elektrokardiyogram PR uzamas� | Yayg�n olmayan | 0,8 | 0 |
Ayn� t�bbi kavram ya da durumu temsil eden advers reaksiyonlar birlikte grupland�r�lm��t�r ve yukar�daki tabloda tek bir advers reaksiyon olarak bildirilmi�tir. �al��malarda halen bildirilen ve ilgili advers reaksiyona katk�da bulunan terimler a�a��daki listede belirtildi�i �ekilde parantez i�inde belirtilmi�tir.
ᵃ Hiperkolesterolemi (kan kolesterol seviyesinde art��, hiperkolesterolemi dahil). ᵇ Hipertrigliseridemi (kan trigliseridlerinde art��, hipertrigliseridemi dahil).
ᶜ Duygudurum etkileri (duygulan�m bozuklu�u, duygusal de�i�kenlik, sald�rganl�k, ajitasyon, �fke, anksiyete, Tip I bipolar bozukluk, depresif duygudurum, depresyon, depresif semptom, �forik duygudurum, sinirlilik, mani, duygudurum de�i�ikli�i, duygudurum dalgalanmalar�, panik atak, ki�ilik de�i�ikli�i, stres dahil).
ᵈ Psikotik etkiler (i�itsel hal�sinasyonlar, hal�sinasyon, g�rsel hal�sinasyonlar dahil).
ᵉ Bili�sel etkiler (SOC sinir sistemi bozukluklar� olaylar�n� kapsar: amnezi, bili�sel bozukluk, demans, dikkat bozuklu�u, haf�za bozuklu�u, mental bozukluk; ve ayn� zamanda SOC psikiyatrik bozukluk olaylar�n� da kapsar: dikkat bozuklu�u, hiperaktivite bozuklu�u, konf�zyon durumu, deliryum, y�nelim bozuklu�u, okuma bozuklu�u). Bu etkiler i�inde SOC sinir sistemi bozukluklar� terimleri SOC psikiyatrik bozukluk terimlerinde daha s�k olarak bildirilmi�tir.
ᶠ Periferik n�ropati (yanma duygusu, disestezi, kar�ncalanma, y�r�y�� bozuklu�u, hipoestezi, motor i�lev bozuklu�u, kas g��s�zl���, n�ralji, n�ropati, periferik n�rotoksisite, parestezi, periferik motor n�ropati, periferik duysal n�ropati, peroneal sinir felci, duysal bozukluk dahil). ᶢ Konu�ma etkileri (dizartri, yava� konu�ma, konu�ma bozuklu�u).
ʰ G�rme bozuklu�u (�ift g�rme, ���ktan rahats�z olma, ���k �akmalar�, bulan�k g�rme, g�rme keskinli�inin azalmas�, g�rme kayb�, u�u�an cisimler g�rme).
ͥ Pn�monit (interstisyel akci�er hastal���, akci�er opasitesi, pn�monit dahil).
ʲ D�k�nt� (akneiform dermatit, mak�lopap�ler d�k�nt�, ka��nt�l� d�k�nt�, d�k�nt� dahil). ᵏ Miyalji (kas iskelet a�r�s�, kas a�r�s� dahil).
ˡ �dem (genel �dem, periferik �dem, periferik �i�lik, �i�lik dahil). ᵐ Fatigue (asteni, bitkinlik dahil).
Se�ili advers reaksiyonlar�n tan�m�
Hiperkolesterolemi/ hipertrigliseridemi
Serum kolesterol ya da trigliserid seviyelerinde art�� advers reaksiyonlar� hastalar�n s�ras�yla
%81,1'inde ve %67,2'sinde bildirilmi�tir. Bunlardan hafif ya da orta �iddette hiperkolesterolemi ya da hipertrigliseridemi advers reaksiyonlar� hastalar�n s�ras�yla
%62,8'inde ve %47,9'unda geli�mi�tir (Bkz B�l�m 4.4). Hem hiperkolesterolemi hem de hipertrigliseridemi i�in ba�lang�ca kadar ge�en medyan s�re 15 g�n olmu�tur (hiperkolesterolemi aral���: 1- 784 g�n; hipertrigliseridemi aral���: 1-796 g�n). Hiperkolesteroleminin ve hipertrigliserideminin medyan s�resi s�ras�yla 451 ve 427 g�n olmu�tur.
Merkezi sinir sistemi etkileri
Ba�l�ca MSS advers reaksiyonlar� bili�sel etkiler (%27,7), duygudurum etkileri (%21) , konu�ma etkileri (%8,2) ve psikotik etkiler (%6,5) olup genellikle hafif, ge�ici olmu� ve dozun ge� verilmesi ya da doz azalt�lmas� ile spontan olarak d�zelmi�tir (Bkz. B�l�mler 4.2 ve 4.4). Herhangi bir derecedeki en s�k bili�sel etki bellek bozuklu�udur (%11,3) ve en s�k Derece 3 ya da 4 reaksiyonlar konf�zyon durumu ve bili�sel bozukluktur (s�ras�yla %1,7 ve %0,8). Herhangi bir derecedeki en s�k duygudurum etkisi anksiyete (%6,5) ve en s�k g�r�len Derece 3 ve 4 reaksiyonlar� sinirlilik ve depresyondur (s�ras�yla %0,8 ve %0,4). Konu�ma �zerinde t�m derecelerde en s�k etki dizartri (%4) olurken Derece 3 ve 4 reaksiyonlar dizartri, yava� konu�ma ve konu�ma bozuklu�udur (her biri %0,2). T�m derecelerde en s�k psikotik etki hal�sinasyon (%3,7) olurken Derece 3 ve 4 reaksiyonlar hal�sinasyon, i�itsel hal�sinasyon ve g�rsel hal�sinasyondur (her biri %0,3). Bili�sel, duygudurum, konu�ma ve psikotik etkilerinin ortaya ��kmas�na kadar ge�en medyan s�re s�ras�yla 109, 43, 49 ve 23 g�nd�r. Bili�sel, duygudurum ve konu�ma etkilerinin medyan s�resi s�ras�yla 223, 143, 147 ve 74 g�nd�r.
Hipertansiyon
�al��ma A ve CROWN �al��mas�nda (B7461006) hastalar�n %13'�nde hipertansiyon yan etkisi bildirilmi�tir. Bu hastalar�n %6,9'unda hafif veya orta dereceli hipertansiyon advers reaksiyonu meydana gelmi�tir (bkz. B�l�m 4.4). Hipertansiyon ba�lang�c�na kadar ge�en medyan s�re 208 g�nd�r (aral�k: 1 ile 1028 g�n). Hipertansiyon medyan s�resi 219 g�nd�r.
Hiperglisemi
�al��ma A ve CROWN �al��mas�nda (B7461006) hastalar�n %9,2'sinde hiperglisemi yan etkisi bildirilmi�tir. Bu hastalar�n %6,1'inde hafif veya orta derecede hiperglisemi advers reaksiyonu meydana gelmi�tir (bkz. B�l�m 4.4). Hiperglisemi ba�lang�c�na kadar ge�en medyan s�re 145 g�nd�r (aral�k: 1 ile 1058 g�n). Hiperglisemi medyan s�resi 113 g�nd�r.
��pheli advers reaksiyonlar�n raporlanmas�
Ruhsatland�rma sonras� ��pheli ila� advers reaksiyonlar�n�n raporlanmas� b�y�k �nem ta��maktad�r. Raporlama yap�lmas�, ilac�n yarar / risk dengesinin s�rekli olarak izlenmesine olanak sa�lar. Sa�l�k mesle�i mensuplar�n�n herhangi bir ��pheli advers reaksiyonu T�rkiye Farmakovijilans Merkezi (T�FAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e- posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks: 0 312 218 35 99)
4.9. Doz a��m� ve tedavisi
T�bbi �r�nle doz a��m�n�n tedavisi genel destekleyici �nlemleri i�ermektedir. PR intervalinin doz-ba��ml� etkisi oldu�undan, EKG izlenmesi �nerilmektedir. Lorlatinibin antidotu bulunmamaktad�r.
5. FARMAKOLOJ�K �ZELL�KLER
5.1. Farmakodinamik �zellikler
Farmakoterap�tik grup: Antineoplastik �la�lar, Protein kinaz inhibit�rleri, anaplastik lenfoma
kinaz (ALK) inhibit�r�. ATC kodu: L01ED05
Etki mekanizmas�
Lorlatinib ALK ve c-ros onkogen (ROS1) tirozin kinazlar�n adenozin trifosfat (ATP) ile yar��an se�ici inhibit�r�d�r.
Preklinik �al��malarda rekombinant enzim ve h�cre temelli deneylerde lorlatinib mutasyona u�ramam�� ALK ve klinik olarak ilgili mutant ALK kinazlar�n katalitik etkinli�ini inhibe etmi�tir. Lorlatinib L1196M, G1269A, G1202R ve I1171T ALK mutasyonlar� dahil ALK varyant� 1 (v1) ile f�zyon yapan ekinoderm mikrot�b�l ile ili�kili protein benzeri 4 (EML4) eksprese eden t�m�r ksenograflar� ta��yan farede t�m�re kar�� belirgin etkinlik g�stermi�tir. Bu ALK mutantlar�ndan ikisinin (G1202R ve I1171T) alektinibe, brigatinibe, seritinibe ve krizotinibe kar�� diren� sa�lad��� bilinmektedir. Lorlatinib kan-beyin bariyerini ge�mi�tir. Lorlatinib ortotopik EML4-ALK ya da EML4-ALK beyin t�m�r implantlar�nda ta��yan farede etkinlik g�stermi�tir.
Klinik etkililik
Daha �nce Tedavi Almam�� ALK-Pozitif �lerlemi� KHDAK (CROWN �al��mas�)
Daha �nce metastatik hastal��a y�nelik olarak sistemik tedavi almam�� ALK-pozitif KHDAK hastalar�n�n tedavisinde lorlatinibin etkilili�i a��k-etiketli, randomize, aktif-kontroll�, �ok- merkezli B7461006 �al��mas�nda (CROWN �al��mas�) g�sterilmi�tir. Bu �al��mada hastalar�n Do�u Kooperatif Onkoloji Grubu (ECOG) performans stat�s�n�n 0-2 olmas�, VENTANA ALK (D5F3) CDx testi ile belirlendi�i �zere ALK-pozitif KHDAK olmas� gerekmekteydi. Leptomeningeal metastazlar dahil tedavi edilmi� ya da edilmemi� MSS metastazlar� bulunan n�rolojik a��dan stabil hastalar�n �al��maya kat�lmas� uygundu. Hastalar�n randomizasyondan �nceki 2 hafta i�inde stereotaktik veya k�smi beyin ���nlamas� dahil ���n tedavisini; randomizasyondan �nceki 4 hafta i�inde ise tam beyin ���nlamas� tedavisini bitirmi� olmalar� gerekmekteydi.
Hastalar g�nde bir kez oral yoldan 100 mg lorlatinib veya g�nde iki kez oral yoldan 250 mg krizotinib alacak �ekilde 1:1 oran�nda randomize edilmi�tir. Randomizasyon etnik k�kene (Asyal� veya Asyal�-olmayan) ve ba�lang��ta MSS metastazlar� varl��� ya da yoklu�una g�re katmanland�r�lm��t�r. Her iki tedavi grubunda tedaviye hastal�kta progresyon veya kabul edilmeyen toksisite g�r�l�nceye devam edilmi�tir. En �nemli etkililik sonu� �l��t� Solid T�m�rlerde Yan�t De�erlendirme Kriterleri (RECIST) versiyon 1.1'e (v1.1) g�re K�rlenmi� Ba��ms�z Merkezi �nceleme (BICR) taraf�ndan belirlendi�i �zere progresyonsuz sa�kal�md� (PFS). Ek etkililik sonu� �l��tleri, genel sa�kal�m (OS), ara�t�rmac� de�erlendirmesine g�re PFS, PFS2 ve genel yan�t oran� (ORR), yan�t s�resi (DOR) ve intrakraniyal progresyona kadar ge�en s�re (IC TTP) dahil olmak �zere BICR taraf�ndan belirlenen t�m�r de�erlendirmesiyle ilgili verilerdir. Ba�lang��ta MSS metastazlar� olan hastalarda ek sonu� �l��tleri, BICR taraf�ndan belirlenen intrakraniyal genel yan�t oran� (IC-ORR) ve intrakraniyal yan�t s�resi (IC- DOR) idi.
Toplam 296 hasta lorlatinib (n=149) veya krizotinib (n=147) alacak �ekilde randomize edilmi�tir. Genel �al��ma pop�lasyonunun demografik �zellikleri �u �ekildeydi: medyan ya� 59 (aral�k: 26 ila 90 ya� aras�), ≥65 ya� (%35), %59 kad�n, %49 beyaz, %44 Asyal� ve %0,3 siyah. Hastalar�n b�y�k bir �o�unlu�unda adenokarsinom vard� (%95) ve hi� sigara i�memi�lerdi (%59). BICR n�roradyologlar�n�n belirledi�i MSS metastazlar� hastalar�n %26's�nda (n=78) g�r�ld�: bunlardan 30 hastada �l��lebilir MSS lezyonlar� vard�.
CROWN �al��mas�ndan elde edilen sonu�lar Tablo 3'te �zetlenmektedir. Veri kesim
noktas�nda OS ve PFS2 verileri olgunla�m�� durumda de�ildi.
Tablo 3 CROWN �al��mas�nda Etkililik Sonu�lar�
Etkililik Parametresi | Lorlatinib N=149 | Krizotinib N=147 |
Medyan takip s�resi, ay (%95 GA) | 18 (16 - 20) | 15 (13 - 18) |
BICR'ye g�re progresyonsuz sa�kal�m | ||
Olay g�r�len hasta say�s�, n (%) | 41 (%28) | 86 (%59) |
Progresif hastal�k, n (%) | 32 (%22) | 82 (%56) |
�l�m, n (%) | 9 (%6) | 4 (%3) |
Medyan, ay (%95 GA) | NE (NE, NE) | 9 (8 - 11) |
Tehlike oran� (%95 GA) | 0,28 (0,19 - 0,41) | |
p-de�eri | <0,0001 | |
Genel sa�kal�m |
| |
Olay g�r�len hasta say�s�, n (%) | 23 (%15) | 28 (%19) |
Medyan, ay (%95 GA) | NE (NE, NE) | NE (NE, NE) |
Tehlike oran� (%95 GA) | 0,72 (0,41 - 1,25) | |
INV'ye g�re progresyonsuz sa�kal�m | ||
Olay g�r�len hasta say�s�, n (%) | 40 (%27) | 104 (%71) |
Progresif hastal�k, n (%) | 34 (%23) | 99 (%67) |
�l�m, n (%) | 6 (%4) | 5 (%3) |
Medyan, ay (%95 GA) | NE (NE, NE) | 9 (7 - 11) |
Tehlike oran� (%95 GA) | 0,21 (0,14 - 0,31) | |
p-de�eri | < 0,0001 | |
BICR'ye g�re genel yan�t | ||
Genel yan�t oran�, n (%) | 113 (%76) | 85 (%58) |
(%95 GA) | 68 – 83 | 49 - 66 |
�ntrakraniyal progresyona kadar ge�en zaman | ||
Medyan, ay (%95 GA) | NE (NE, NE) | 16,6 (11 - NE) |
Tehlike oran� (%95 GA) | 0,07 (0,03 - 0,17) | |
Yan�t s�resi | ||
Yan�t verenlerin say�s� | 113 | 85 |
Medyan, ay (%95 GA) | NE (NE, NE) | 11 (9 - 13) |
Ba�lang��ta �l��lebilir MSS lezyonlar� olan hastalarda intrakraniyal genel yan�t | N=17 | N=13 |
�ntrakraniyal yan�t oran�, n (%) | 14 (%82) | 3 (%23) |
(%95 GA)c | (57 - 96) | (5 - 54) |
Tam yan�t oran� | %71 | %8 |
Yan�t s�resi |
|
|
Yan�t verenlerin say�s� | 14 | 3 |
Medyan, ay (%95 GA)a | NE (NE - NE) | 10 (9 - 11) |
Ba�lang��ta �l��lebilir veya �l��lemeyen MSS lezyonlar� olan hastalarda intrakraniyal genel yan�t | N=38 | N=40 |
�ntrakraniyal yan�t oran�, n (%) | 25 (%66) | 8 (%20) |
(%95 GA)c | (49 - 80) | (9 - 36) |
Tam yan�t oran� | %61 | %15 |
Yan�t s�resi |
|
|
Yan�t verenlerin say�s� | 25 | 8 |
Medyan, ay (%95 GA)a | NE (NE - NE) | 9 (6 - 11) |
K�saltmalar: BICR=K�r Ba��ms�z Merkez �ncelemesi; GA=g�ven aral���; MSS=merkezi sinir sistemi; INV= ara�t�rmac� de�erlendirmesi; N=hasta say�s�; NE=tahmin edilemeyen.
![]()
�ekil 1: CROWN �al��mas�'nda BICR'ye G�re Progresyonsuz Sa�kal�ma Ait Kaplan- Meier Grafi�i
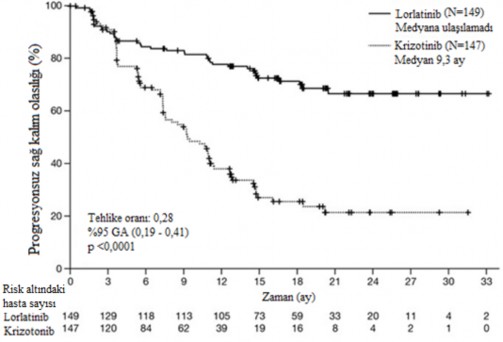
K�saltmalar: GA=g�ven aral���; N=hasta say�s�.
Lorlatinib tedavisinden sa�lanan fayda, ba�lang��ta MSS metastaz� olan (n=38, HR=0,2, %95 GA: 0,1-0,43) ve MSS metastaz� olmayan hastalar (n=111, HR=0,32, %95 GA: 0,2-0,49) dahil olmak �zere, ba�lang��taki hasta ve hastal�k �zelliklerinin alt gruplar� aras�nda kar��la�t�r�labilir olmu�tur.
Daha �nce bir ALK kinaz inhibit�r� ile tedavi edilmi� ALK-Pozitif �lerlemi� KHDAK
ALK- pozitif ileri evre KHDAK'nin tedavisinde en az bir ikinci nesil ALK TK� ile tedaviden sonra lorlatinib kullan�m� tek kollu, �ok merkezli faz 1/2 �al��ma olan �al��ma A'da ara�t�r�lm��t�r. Toplam 139 ALK pozitif ileri evre KHDAK hastas� en az bir ikinci nesil ALK TK� ile tedaviden sonra �al��man�n Faz 2 b�l�m�ne kaydedilmi�tir. Hastalar kesintisiz olarak oral yolla g�nde bir kez 100 mg dozunda lorlatinib kullanm��t�r.
�al��man�n Faz 2 k�sm�nda birincil etkililik sonlan�m noktas� Ba��ms�z Merkezi �nceleme (ICR) ile solid t�m�rlerde modifiye RECIST v1.1'e g�re de�erlendirilen intrakranyal (�K)- ORR dahil objektif yan�t oran�d�r. �kincil sonlan�m noktalar� kapsam�nda DOR, �K-DOR, t�m�r yan�t�na kadar ge�en s�re (TTR) ve PFS vard�r.
En az bir ikinci nesil ALK TK� ile tedavi alm�� olan 139 ALK-pozitif ileri evre KHDAK hastas�n�n demografik �zellikleri a�a��da yer almaktad�r: %56 kad�n, %48 beyaz, %38 Asyal�, medyan ya� 53 ya� (aral�k 29-83 ya�) olup hastalar�n %16's� 65 ya� ve �st�d�r. ECOG performans durumu ba�lang��ta hastalar�n %96's�nda 0 ya da 1 bulunmu�tur. Hastalar�n
%67'sinde ba�lang��ta beyin metastazlar� vard�r. 139 hastan�n %20'si daha �nce krizotinib d���nda bir ALK TK� kullanm��ken %47'si daha �nce 2 ALK TK�, %33'� ise 3 ya da daha fazla ALK TK� alm��t�r.
A �al��mas�n�n ba�l�ca etkililik sonu�lar� Tablo 4 ve 5'te yer almaktad�r.
Tablo 4. A �al��mas�nda �nceki tedaviye g�re genel etkililik sonu�lar�
Etkililik parametresi | Daha �nce kemoterapi ile ya da tek ba��na bir ALK TK�ᵃ (N=28) | Daha �nce kemoterapi ile ya da tek ba��na iki ya da daha fazla ALK TK� (N=111) |
Objektif yan�t oran�ᵇ (%95 GA) Tam yan�t, n K�smi yan�t, n | %42,9 (24,5 - 62,8) 1 11 | %39,6 (30,5 - 49,4) 2 42 |
Yan�t�n s�resi Medyan, ay (%95 GA) |
5,6 (4,2 - NR) |
9,9 (5,7 - 24,4) |
Progresyonsuz sa�kal�m Medyan, ay (%95 GA) |
5,5 (2,9 - 8,2) |
6,9 5,4 - 9,5 |
K�saltmalar: ALK= anaplastik lenfoma kinaz; GA=g�ven aral���; ICR=Ba��ms�z Merkezi �nceleme; N/n=hasta say�s�; NR= eri�ilmedi; TKI= tirozin kinaz inhibit�r.
ᵃ Alektinib, brigatinib ya da seritinib. ᵇ ICR'ye g�re
Tablo 5. A �al��mas�nda �nceki tedaviye g�re intrakranyal* etkililik sonu�lar�
Etkililik parametresi | Daha �nce kemoterapi ile ya da tek ba��na bir ALK TK�ᵃ (N=9) | Daha �nce kemoterapi ile ya da tek ba��na iki ya da daha fazla ALK TK� (N=48) |
Objektif yan�t oran�ᵇ (%95 GA) Tam yan�t, n K�smi yan�t, n | %66,7 (29,9 - 92,5) 2 4 | %52,1 (37,2 – 66,7) 10 15 |
�ntrakranyal yan�t�n s�resi Medyan, ay (%95 GA) |
NR (4,1- NR) |
12,4 (6 - NR) |
K�saltmalar: ALK= anaplastik lenfoma kinaz; GA=g�ven aral���; ICR=Ba��ms�z Merkezi �nceleme; N/n=hasta say�s�; NR= eri�ilmedi; TKI= tirozin kinaz inhibit�r.
*Ba�lang��ta en az bir �l��lebilir beyin metastaz� olan hastalarda. ᵃ Alektinib, brigatinib ya da seritinib.
ᵇ ICR'ye g�re
139 hastadan olu�an genel etkililik pop�lasyonunda ICR'ye g�re 56 hastada do�rulanm�� objektif yan�t al�nm��, medyan TTR 1,4 ay (aral�k: 1,2 - 16,6 ay) olmu�tur. ORR Asyal� pop�lasyonda %49,1 (%95 GA: 35,1 - 63,2) ve Asyal� olmayan pop�lasyonda %31,5 (%95 GA: 21,1 - 43,4) olmu�tur. Do�rulanm�� objektif �K t�m�r yan�t� olan ve ICR'ye g�re ba�lang��ta en az bir �l��lebilir beyin metastaz� olan 31 hastada medyan �K-TTR 1,4 ay (aral�k= 1,2 - 16,2 ay) olmu�tur. �K-ORR Asyal� pop�lasyonda %54,5 (%95 GA: 32,2- 75,6) ve Asyal� olmayan
pop�lasyonda %46,4 (%95 GA: 27,5 - 66,1) olmu�tur.
5.2. Farmakokinetik �zellikler
Emilim
Lorlatinibin pik plazma konsantrasyonlar�na h�zla eri�ilmekte olup 100 miligraml�k tek dozu takiben medyan T1,2 saat ve g�nde bir kez 100 g dozunda birden fazla kez verildi�inde ise 2 saat bulunmu�tur.
�ntraven�z uygulama ile kar��la�t�r�ld���nda lorlatinib tabletlerin oral yolla verilmesinden sonra ortalama mutlak biyo-yararl�l��� %80,8 (%90 GA: 75,7 - 86,2) olmu�tur.
Lorlatinibin y�ksek ya� ve y�ksek kalori i�eren bir yemekle birlikte verilmesi a� karn�na verilmesine g�re kan seviyelerini %5 artt�rm��t�r.
Lorlatinib tok ya da a� karn�na verilebilir.
Kanser hastalar�nda g�nde bir kez 100 mg dozda uyguland���nda; geometrik ortalama (% varyasyon katsay�s� [CV]) pik plazma konsantrasyonu 577 (42) ng/ml ve EAA5,650 (39) ng·saat/ml bulunmu�tur. Geometrik ortalama (% CV) oral klirens 17,7 (39) l/saat bulunmu�tur.
Da��l�m
Lorlatinib insan plazma proteinlerine in vitro %66 oran�nda ba�lanmakta olup alb�mine ya da α-asit glikoproteine orta derecede ba�lanmaktad�r.
Biyotransformasyon
�nsanlarda lorlatinib birincil metabolik yolaklar olarak oksidasyona ve glukuronidasyona u�ramaktad�r. �n vitro veriler lorlatinibin birincil olarak CYP3A4 ve UGT1A4 taraf�ndan metabolize edildi�ini ve CYP2C8, CYP2C19, CYP3A5 ve UGT1A3'�n min�r katk�da bulundu�unu g�stermektedir.
Plazmada lorlatinibin amid ve aromatik ba�lar�n�n oksidatif ayr��mas�ndan kaynaklanan bir
benzoik asit metaboliti maj�r metabolit olarak belirlenmi� olup dola��mdaki radyoaktivitenin
%21'inden sorumludur. Oksidatif ayr��ma ile olu�an metabolit farmakolojik olarak inaktiftir.
Eliminasyon
Tek bir 100 miligraml�k dozdan sonra lorlatinibin plazma yar� �mr� 23,6 saattir. Otoind�ksiyonun tamamlanmas�n� takiben kararl� durumdaki tahmini lorlatinib efektif plazma yar� �mr� 14,83 saattir. 100 miligraml�k radyoaktif etiketli lorlatinib dozun oral yolla verilmesini takiben radyoaktivitenin ortalama %47,7'si idrardan, %40,9'u fe�esten ve genelde ortalama %88,6's� geri kazan�lm��t�r.
De�i�memi� lorlatinib insan plazmas�nda ve fe�este belirlenen maj�r bile�endir ve toplam radyoaktivitenin s�ras�yla %44'� ve %9,1'ini olu�turmaktad�r. Lorlatinibin %1'inden az� idrarda de�i�memi� olarak saptanm��t�r.
Ayr�ca lorlatinib insan pregnan-X-resept�r� (PXR) ve insan yap�sal androstan resept�r� (CAR) arac�l���yla bir ind�kleyicidir.
Do�rusall�k/Do�rusal Olmayan durum
Tek dozda maruz kal�nan lorlatinib seviyesi (EAAve C) 10 mg ile 200 mg doz aral���nda doza ba�l� olarak artm��t�r. 10 mg ile 200 mg doz aral��� d���nda az miktarda veri mevcuttur; bununla birlikte tek dozdan sonra EAAve Ci�in do�rusall�ktan sapma olmam��t�r.
G�nde bir kez doz birden fazla kez verildi�inde lorlatinib Cdozla orant�l� olarak artm��t�r ve EAAg�nde10 mg ile 200 mg doz aral��� d���nda dozlarda hafif�e daha az orant�l� olarak artm��t�r.
Ayn� �ekilde kararl� durumda maruz kal�nan plazma lorlatinib seviyeleri tek doz farmakokineti�i i�in beklenenden daha d���kt�r ve bu da net zamana ba�l� otoind�ksiyon etkisini g�stermektedir.
Hastalardaki Karakteristik �zellikler
Karaci�er yetmezli�i
Lorlatinibin karaci�erde metabolize edilmesi nedeniyle karaci�er yetmezli�i lorlatinibin plazma konsantrasyonlar�n� y�kseltme olas�l���n� artt�rmaktad�r. Yap�lan klinik �al��malar AST ya da ALT seviyelerinin normalin �st seviyesinin 2,5 kat�ndan fazla oldu�u ya da art�� altta yatan kansere ba�l�ysa normalin �st seviyesinin 5 kat�ndan fazla oldu�u ya da total bilir�bin seviyesinin normalin �st seviyesinin 1,5 kat�ndan fazla oldu�u hastalar� d��lam��t�r. Pop�lasyon farmakokinetik analizleri maruz kal�nan lorlatinib maruziyetinin hafif karaci�er yetmezli�i olan hastalarda klinik olarak anlaml� �l��de de�i�medi�ini g�stermi�tir (n = 50). Hafif karaci�er yetmezli�i olan hastalar i�in doz ayarlamalar� �nerilmemektedir. Orta ya da a��r karaci�er yetmezli�i olan hastalar hakk�nda bilgi yoktur.
B�brek yetmezli�i
Verilen dozun %1'inden az� idrarda de�i�memi� lorlatinib olarak saptanmaktad�r. Pop�lasyon farmakokinetik analizleri lorlatinib kararl� durum plazma maruziyet seviyesinin ve Cde�erinin k�t�le�en bazal b�brek fonksiyonu ile hafif artt���n� g�stermi�tir. Bir b�brek yetmezli�i �al��mas�na dayanarak, hafif veya orta �iddetteki b�brek yetmezli�i olan hastalar i�in ba�lang�� dozunda ayarlama yap�lmas� �nerilmemektedir [B�brek Hastal��� �al��mas�nda Diyet Modifikasyonu'na (MDRD) g�re t�retilen eGFR; eGFR (mL/dak/1,73 m) × �l��len v�cut y�zey alan�/1,73 ≥ 30 mL/dk]. Bu �al��mada, lorlatinib EAA, a��r b�brek yetmezli�i olan hastalarda (mutlak eGFR < 30 mL/dk), b�brek fonksiyonu normal olan g�n�ll�lere (mutlak eGFR ≥ 90 mL/dak) k�yasla %41 artm��t�r. A��r b�brek yetmezli�i olan hastalarda lorlatinib dozunun azalt�lmas� �nerilir, �rne�in g�nde bir kez 75 mg oral ba�lang�� dozu (bkz. B�l�m 4.2). Renal diyaliz hastalar� i�in bilgi mevcut de�ildir.
Ya�, cinsiyet, �rk, v�cut a��rl��� ve fenotip
�leri evre KHDAK olan ve sa�l�kl� g�n�ll�lerde yap�lan pop�lasyon farmakokinetik analizleri CYP3A5 ve CYP2C19 i�in ya��n, cinsiyetin, �rk�n, v�cut a��rl���n�n ve fenotipin klinik olarak anlaml� etkileri olmad���n� g�stermi�tir.
Kardiyak elektrofizyoloji
A �al��mas�nda 2 hastan�n (%0,7) mutlak Fridericia d�zeltme QTc (QTcF) de�erleri> 500 milisaniye olmu� ve 5 hastada (%1,8) QTcF de�i�ikli�i ba�lang�ca g�re> 60 milisaniye olmu�tur.
Ayr�ca, tek bir oral doz lorlatinibin (50 mg, 75 mg ve 100 mg) g�nde bir kez 200 mg itrakonazol ile ve tek ba��na etkisi 16 sa�l�kl� g�n�ll�de 2 y�nl� �apraz bir �al��mada de�erlendirilmi�tir. Bu �al��mada g�zlenen ortalama lorlatinib konsantrasyonlar�nda ortalama QTc de�erlerinde art�� g�zlenmemi�tir.
A �al��mas�nda �nerilen 100 miligraml�k dozda lorlatinib alan ve bir EKG incelemesi bulunan 295 hastadan QTc aral���n�n> 470 milisaniye olan hastalar�n d��land��� bir pop�lasyonda lorlatinib de�erlendirilmi�tir. �al��ma pop�lasyonunda PR aral���nda ba�lang�ca g�re maksimum ortalama de�i�iklik 16,4 milisaniye (2 y�nl� %90 �st GA 19,4 milisaniye) (Bkz.
B�l�mler 4.2, 4.4 ve 4.8) olmu�tur. Bu hastalardan yedisinde ba�lang�� PR> 200 milisaniyedir. PR aral���n�n <200 milisaniye oldu�u 284 hastan�n %14'�nde lorlatinib ba�land�ktan sonra PR aral���nda uzama ≥ 200 milisaniye olmu�tur. PR aral���ndaki uzama konsantrasyona ba��ml� bir �ekilde olu�mu�tur. Hastalar�n %1'inde atriyoventrik�ler blok olu�mu�tur.
PR uzamas� geli�en hastalarda doz de�i�ikli�i gerekebilir (Bkz B�l�m 4.2).
5.3. Klinik �ncesi g�venlilik verileri
Tekrarlayan doz toksisitesi
�nerilen pozolojide insanda maruz kal�nan klinik seviyelere e�de�er dozda g�zlenen ba�l�ca toksisiteler �e�itli dokularda inflamasyon (s��anlarda deri ve servikste, k�peklerde akci�erde, trakea, deride, lenf bezlerinde ve/veya mandibuler kemik dahil a��z bo�lu�unda; beraberinde beyaz kan h�creleri, fibrinojen ve/veya glob�lin seviyesinde art�� ve alb�min seviyesinde azalmalar) ve pankreas (beraberinde amilaz ve lipaz seviyelerinde art��), hepatobiliyer sistem (beraberinde karaci�er enzimlerinde art��lar), erkek �reme sistemi, kardiyovask�ler sistem, b�brekler ve gastrointestinal sistem, periferik sinirler ve MSS (bili�sel fonksiyonda bozulma) de�i�iklikleri olmu�tur. Hayvanlarda akut dozlama sonras� (Cde�erine g�re tek doz 100
mg ile insanda klinik olarak maruz kal�nan seviyenin yakla��k 2,6 kat�) kan bas�nc�nda ve kalp at�m h�z�nda ve QRS kompleksinde ve PR aral���nda de�i�iklikler de g�zlenmi�tir. Karaci�er safra kanal� hiperplazisi d���nda t�m hedef organ bulgular� tamam�yla geri d�n��l�d�r.
Genotoksisite
Lorlatinib in vitro ve in vivo olarak mutajenik de�ildir ancak an�jenik olup EAA'ya g�re 100 mg ile insan klinik olarak maruz kal�nan seviyenin yakla��k 16,5 kat� ile an�jenisite �zerinde herhangi bir seviyede etki g�zlenmemi�tir.
Karsinojenisite
Lorlatinib ile Karsinojenisite �al��malar� yap�lmam��t�r. �reme toksisitesi
S��anda ve k�pekte seminifer t�b�ler dejenerasyon ve/veya testis atrofisi ve epididim de�i�iklikleri (inflamasyon ve/veya vak�olle�me) g�zlenmi�tir. �nerilen pozolojide insanda maruz kal�nan seviyelere e�de�er dozda k�peklerde prostatta minimal-hafif gland�ler atrofi g�zlenmi�tir. Erkek �reme organlar�nda etkiler k�smen ya da tamamen geri d�n��l�d�r.
S��anlarda ve tav�anlarda yap�lan embriyofetal toksisite �al��malar�nda embriyo �l�m� ve d���k fet�s a��rl��� ve malformasyonlar g�zlenmi�tir. Fet�steki morfolojik anormallikler aras�nda kol ve bacaklarda rotasyon, parmak say�s�nda art��, gastro�izis, b�brek malformasyonu, kubbe kafa, y�ksek damak ve beyin ventrik�llerinde geni�leme vard�r. EAA'ya g�re hayvanlarda embriyofetal etkilerin geli�ti�i en d���k dozlarla maruz kal�nan seviye 100 mg dozu ile insanda klinik olarak maruz kal�nan seviyeye e�de�erdir.
6. FARMAS�T�K �ZELL�KLER
6.1. Yard�mc� maddelerin listesi
�ekirdek tablet:
Mikrokristalin Sel�loz
Dibazik Kalsiyum Fosfat, Susuz Sodyum Ni�asta Glikolat Magnezyum stearat
Film kaplama
HPMC 2910/Hipromelloz
Laktoz Monohidrat (s���r kaynakl�) Macrogol 4000/PEG 3350 Triasetin
Titanyum Dioksit
Ferrozoferrik Oksit/Siyah Demir Oksit Demir Oksit K�rm�z�
6.2. Ge�imsizlikler
Ge�erli de�il
6.3. Raf �mr�
36 ay
6.4. Saklamaya y�nelik �zel tedbirler
25°C'nin alt�ndaki oda s�cakl���nda saklay�n�z.
6.5. Ambalaj�n niteli�i ve i�eri�i
10 film kapl� tablet i�eren al�minyum folyo arka kaplama materyali olan OPA/Al/PVC folyo
blister
Ambalaj b�y�kl�kleri:
25 mg: 30, 60, 100, 120 film kapl� tablet: Her biri 10 tablet i�eren s�ras�yla 3, 6, 10, 12 blister
i�eren kutular
6.6. Be�eri t�bbi �r�nden arta kalan maddelerin imhas� ve di�er �zel �nlemler
�zel gereklilikler bulunmamaktad�r.
Kullan�lmam�� olan �r�nler ya da at�k materyaller ‘T�bbi At�klar�n Kontrol� Y�netmeli�i' ve ‘Ambalaj At�klar�n�n Kontrol� Y�netmelikleri'ne uygun olarak imha edilmelidir.
 Y�ksek Tansiyon
Hipertansiyon s�rekli anormal derecede y�ksek olan kan bas�nc�d�r. Tansiyon
atardamarlar�n�zdaki kan�n bas�nc�d�r.
Y�ksek Tansiyon
Hipertansiyon s�rekli anormal derecede y�ksek olan kan bas�nc�d�r. Tansiyon
atardamarlar�n�zdaki kan�n bas�nc�d�r. |
 Do�um Sonras� Depresyonu
Do�um sonras� depresyonu, do�umdan sonra her on kad�ndan biri taraf�ndan
tecr�be edilen stresli bir durumdur.
Do�um Sonras� Depresyonu
Do�um sonras� depresyonu, do�umdan sonra her on kad�ndan biri taraf�ndan
tecr�be edilen stresli bir durumdur. |
 |
�nme �nme, beynin hasar g�rmesinin sonucudur. Bu hasar, beynin bir k�sm�ndaki ya bir kanama ya da akut kan eksikli�i nedeniyle o k�sm�n ge�ici ya da kal�c� olarak i�levini yapamamas�na yol a�ar. |
 |
Pankreas Kanseri Pankreas karn�n alt k�sm�nda yatay �ekilde bulunan bir organd�r. Sindirime yard�mc� olan enzimleri ve kan �ekerini y�netmeye yard�mc� olan hormonlar� v�cuda da��tmakla g�revlidir. |
 |
S�rt A�r�s� S�rt a�r�s� birden bire ortaya ��k�p �iddetli (akut) olabilir veya zamanla geli�ip daha uzun s�reli sorunlara (kronik) neden olabilir. |
�LA� GENEL B�LG�LER�
Pfizer �la�lar� Ltd.�ti.
| Sat�� Fiyat� | 30398.57 TL [ 17 Dec 2024 ] |
| �nceki Sat�� Fiyat� | 30398.57 TL [ 2 Dec 2024 ] |
| Original / Jenerik | Original �la� |
| Re�ete Durumu | Normal Re�eteli bir ila�d�r. |
| Barkodu | 8681308092007 |
| Etkin Madde | Lorlatinib |
| ATC Kodu | L01ED05 |
| Birim Miktar | 25 |
| Birim Cinsi | MG |
| Ambalaj Miktar� | 30 |
| Antineoplastik ve �mm�nomod�lat�r Ajanlar |
| �thal ( ref. �lke : Almanya ) ve Be�eri bir ila�d�r. |