LUCENTIS 10 mg/ml enjeksiyonluk çözelti Kısa Ürün Bilgisi
{ Ranibizumab }
1. BEŞERİ TIBBİ ÜRÜNÜN ADI
LUCENTIS 10 mg/ml enjeksiyonluk çözelti içeren flakon2. KALİTATİF VE KANTİTATİF BİLEŞİM
1 flakon (0.23 ml) içinde:
Ranibizumab.............................. 2.3 mg
Ranibizumab, rekombinan DNA teknolojisiyle Escherichia coli hücrelerinde üretilmiş bir insanlara uyarlanmış monoklonal antikor parçasıdır.
Yard
ı
mc
ı
maddeler:
Yardımcı maddeler için 6.1.’e bakınız.
3. FARMASÖTİK FORMU
Enjeksiyonluk çözelti.
Steril, koruyucu içermeyen, berrak, renksiz ile soluk sarı arasında sulu çözelti.
4.1. Terapötik endikasyonlar
4.2. Pozoloji ve uygulama şekli
Pozoloji:
Sadece intravitreal uygulama için tek kullanımlık flakon. LUCENTIS, bir "göz hastalıkları uzmanı" tarafından uygulanmalıdır. LUCENTIS için önerilen doz 0.5 mg’dır (0.05 ml’de).
Uygulama s
ı
kl
ı
ğ
ı
ve süresi:
LUCENTIS tedavisi birbirini takip eden 3 ay süreyle ayda bir enjeksiyonu içeren bir yükleme fazı ile başlatılmakta ve bunu hastaların aylık olarak görme keskinliğinin izlenmesi gereken bir idame fazı takip etmektedir. Eğer hastanın görme keskinliğinde > 5 harflik bir kayıp varsa (ETDRS ya da bir Snellen hattı eşdeğeri) LUCENTIS uygulanması gerekmektedir. İki doz arasındaki ara 1 aydan kısa olmamalıdır.
Uygulama şekli:
Parenteral kullanımlı bütün tıbbi ürünlerde olduğu gibi, uygulamadan önce LUCENTIS’in partiküllü madde ve renk değişimi açısından görsel olarak incelenmesi gerekmektedir.
4.3. Kontrendikasyonlar
’a bakınız). Perioküler cilt, göz kapağı ve oküler yüzey steril edilmelidir. Enjeksiyondan önce yeterli anestezi uygulanmalı ve steril ortam sağlanmalıdır.
Klinik çalışmalarda hastalara her bir enjeksiyondan önce ve sonra 3 gün boyunca günde 4 kere antimikrobiyal göz damlası uygulanmıştır. Ancak doktorun önerisi ile enjeksiyon öncesinde steril şartların sağlanması halinde antimikrobiyal damla enjeksiyon sonrasında uygulanabilir.
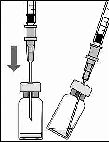
Lucentis’i intravitreal uygulamaya hazırlamak için aşağıdaki talimatları uygulayınız:
1. İlacı çekmeden önce flakonun lastik tıpasının dış kısmı dezenfekte edilmelidir.
2. 5 mikrometrelik filtreli iğneyi (verilmiştir) ile 1ml’lik şırıngaya (verilmiştir) aseptik teknikle takınız. Küt uçlu iğneyi flakon tıpasının ortasına sokarak flakonun tabanına dokununcaya kadar itiniz.
3. Flakondaki tüm sıvıyı flakonu dik pozisyonda tutarak çekiniz, sıvının tamamının çekilmesini kolaylaştırmak için flakonu hafifçe eğiniz.
A.
B.
4. Flakonu boşaltırken iğne ucunun tamamen boşaltmak amacıyla piston çubuğun yeteri kadar geri çekildiğinden emin olun.
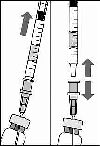
5. Küt uçlu filtreli iğneyi flakon içinde bırakınız ve küt uçlu filtreli iğneyi şırıngadan çıkarınız. Filtreli iğne flakon içeriğinin çekilmesinden sonra atılmalı ve intrevitreal enjeksiyon için kullanılmamalıdır.
C.
6. Enjeksiyon iğnesini (verilmiştir) aseptik ve sıkı bir şekilde şırıngaya takınız.
7. Şırıngadan enjeksiyon iğnesini ayırmadan dikkatlice enjeksiyon iğnesinin başlığını çıkarınız.
Not: Başlığı çıkarırken enjeksiyon iğnesinin sarı göbek kısmından sıkıca tutunuz.
D.
8. Şırıngadaki havayı dikkatle çıkarınız ve dozu şırınga üzerindeki 0.05 ml işaretine ayarlayınız. Şırınga enjeksiyon için hazırdır.
Not: Enjeksiyon iğnesini silmeyiniz. Pistonu geri çekmeyiniz.
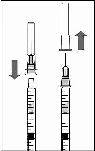
t; | |
0.05 mL | |
Enjeksiyon iğnesi yatay meridyenden kaçınılarak, gözün merkezi hedeflenerek ve gözün aksiyal uzunluğu göz önünde bulundurularak vitreus boşluğuna doğru limbusun 3.5-4.0 mm açığına batırılmalıdır. Sonra 0.05 ml’lik enjeksiyon hacmi verilmelidir.Daha sonraki enjeksiyonlar farklı sklera kadranına enjekte edilmelidir.
Özel popülasyonlara ilişkin ek bilgiler:
Böbrek/Karaciğer yetmezliği: Böbrek yetmezliği olan hastalarda doz ayarlaması gerekmemektedir (Bölüm 5.2 Farmakokinetik özellikler’e bakınız).
Karaciğer yetmezliği olan hastalarda çalışılma yapılmamıştır. Ancak, sistemik maruziyet dikkate değer olmadığı için bu popülasyonda özel önlemler gerekli görülmemiştir.
Pediyatrik popülasyon: 18 yaşın altındaki popülasyonda güvenlilik ve etkinlilik verilerindeki eksiklik nedeniyle LUCENTIS’in çocuk ve ergenlerde kullanımı önerilmemektedir.
4.3. Kontrendikasyonlar
- Etkin maddeye ya da herhangi bir yardımcı maddeye karşı aşırı duyarlılık (bkz. Bölüm 6.1),
- Aktif ya da şüpheli oküler ya da perioküler enfeksiyonlu hastalar,
- Aktif şiddetli göz içi inflamasyonlu hastalarda kullanımı kontrendikedir.
4.4. Özel kullanım uyarılarıve önlemleri LUCENTIS tedavisi sadece intravitreal enjeksiyon ile yapılır
4.8. İstenmeyen etkiler
’e bakınız). LUCENTIS uygulanırken her zaman uygun steril enjeksiyon teknikleri kullanılmalıdır. Ayrıca, bir enfeksiyon oluştuğunda erken tedaviye olanak sağlamak için hastalar enjeksiyonu takip eden hafta sırasında izlenmelidir. Hastalara endoftalmi ya da yukarıda sözü edilen vakaları çağrıştıracak herhangi bir semptomu gecikmeden bildirmeleri öğütlenmelidir.
4.8. İstenmeyen etkiler
’e bakınız). Bu nedenle, hem göz içi basıncı hem de optik sinir başı perfüzyonu izlenmeli ve uygun şekilde tedavi edilmelidir.
Her iki göze aynı anda uygulanan LUCENTIS tedavisinin güvenlilik ve etkinliliği çalışılmamıştır.
VEGF (vasküler endotelyal büyüme faktörü) inhibitörlerinin intravitreal kullanımını takiben arteriyel tromboembolik olayların ortaya çıkması açısından potansiyel risk söz konusudur. Faz 3 çalışmalarda, arteriyel tromboembolik olayların genel sıklığı ranibizumab ile kontrol arasında benzer bulunmuştur. Ranibizumab 0.5 mg ile tedavi edilen hastalarda inme oranları ranibizumab 0.3 mg ya da kontrol ile tedavi edilen hastalara kıyasla sayısal olarak daha yüksek olmuş; ancak farklar istatistiksel olarak anlamlı bulunmamıştır. İnme oranlarındaki farklılık geçici iskemik atak veya inme öyküsü dahil olmak üzere inme için bilinen risk faktörleri olan hastalarda daha yüksek olabilir. Bu yüzden bu hastaların Lucentis tedavisine uygunluğu ve faydanın potansiyel riske göre ağır basıp basmadığı doktorları tarafından dikkatle değerlendirilmelidir.
4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri
Hiçbir klinik etkileşim çalışması yürütülmemiştir.
LUCENTIS intravitreal enjeksiyon, bir faz II klinik çalışmada verteporfin fotodinamik terapiye (PDT) ek olarak kullanılmıştır. 12/105 hastada (%11) ciddi göz içi inflamasyonu gelişmiş, 10/12’sinde LUCENTIS verteporfin/PDT’den 7 gün (± 2) gün sonra uygulandığında oluşmuştur. Ancak, bu çalışmada piyasaya verilmeyecek olan bir liyofilize ranibizumab formülasyonu kullanılmıştır.
Verteporfin/PDT ile LUCENTIS 0.5 mg’ın aynı gün uygulanmasının güvenliğinin değerlendirildiği bir başka çalışmada ilk kombinasyon tedavisini takiben göz içi inflamasyonu insidansı düşük bulunmuştur (2/32 hasta, %6.3). Bu çalışmada piyasaya verilecek olan sıvı formülasyon kullanılmıştır. Böylece sıvı ranibizumab formülasyonu ile verteporfinli PDT’nin aynı gün uygulamasının iyi tolere edildiği gösterilmiştir.
Özel popülasyonlara ilişkin ek bilgiler
Özel popülasyonlara ilişkin hiçbir klinik etkileşim çalışması yürütülmemiştir.
4.6. Gebelik ve laktasyon
Genel tavsiye
Gebelik kategorisi: C
Çocuk doğurma potansiyeli bulunan kad
ı
nlar / Doğum kontrolü (Kontrasepsiyon)
Hayvanlar üzerinde yapılan çalışmalar, gebelik / ve-veya / embriyonal / fetal gelişim / ve-veya / doğum / ve-veya / doğum sonrası gelişim üzerindeki etkiler bakımından yetersizdir (bkz. Kısım 5.3). İnsanlara yönelik potansiyel risk bilinmemektedir.
LUCENTIS gerekli olmadıkça gebelik döneminde kullanılmamalıdır.
Çocuk doğurma potansiyeli olan kadınlar tedavi süresince etkili bir doğum kontrol yöntemi kullanmalıdır.
Gebelik dönemi
LUCENTIS gebe bir kadına tıbbi zorunluluk durumunda risk yarar durumu göz önüne alınarak doktor kararı ile verilmelidir.
Laktasyon dönemi
Ranibizumab’ın insan sütüyle atılıp atılmadığı bilinmemektedir. Ranibizumab’ın süt ile atılımı hayvanlar üzerinde araştırılmamıştır. Emzirmenin durdurulup dur durulmayacağına ya da LUCENTIS tedavisinin durdurulup durdurulmayacağına/tedaviden kaçınılıp kaçınılmayacağına ilişkin karar verilirken, emzirmenin çocuk açısından faydası ve LUCENTIS tedavisinin emziren anne açısından faydası dikkate alınmalıdır.
Üreme yeteneği / Fertilite
Üreme yeteneği üzerine etkileri hakkında çalışma yapılmamıştır.
4.7. Araç ve makine kullanımıüzerindeki etkiler
4.8. İstenmeyen etkiler
’e bakınız). Bu belirtileri yaşayan hastalar bu geçici görme bozuklukları geçene kadar araç ya da makine kullanmamalıdır.
Toplam 1315 hasta 24 ay Lucentis kullanacak şekilde ve 440 hasta önerilen 0.5 mg dozla tedavi edilerek üç faz III çalışmada güvenlilik popülasyonunu oluşturmuştur.
4.4. Özel kullanım uyarıları ve önlemleri
’ne bakınız).
4.4. Özel kullanım uyarıları ve önlemleri
’ne bakınız).
| Aşağıda sıralanan advers olaylar, her üç kontrollü faz III çalışmada (FVF2598g-MARINA, FVF2587g-ANCHOR ve FVF3192g-PIER) LUCENTIS 0.5 mg alan hastalarda kontrol tedavisi (plasebo ya da verteporfin/PDT) alanlara göre daha yüksek (en az %3) orandadır. Bu nedenle bunlar advers ilaç reaksiyonları (ADR) olarak kabul edilmiştir. Sebebi enjeksiyon uygulaması ya da tıbbi ürüne bağlı olup olmadığı klinik çalışmalarla saptanamayan yan etkiler aşağıda verilmektedir.
Advers olaylar sistem organ sınıfı ve sıklığa göre şu yaklaşımla sıralanmıştır: çok yaygın (> 1/10), yaygın (> 1/100 ila < 1/10), yaygın olmayan (> 1/1.000 ila < 1/100), seyrek (> 1/10.000 ila < 1/1.000) ve çok seyrek (< 1/10.000).
Enfeksiyon ve enfestasyonlar
Çok yaygın: Nazofarenjit Yaygın: Influenza
Kan ve lenf sistemi hastal
ı
klar
ı
Yaygın: Anemi
Psikiyatrik hastal
ı
klar Yaygın: Anksiyete
Sinir sistemi hastal
ı
klar
ı
Çok yaygın: Baş ağrısı Yaygın: İnme
Göz hastal
ı
klar
ı
Çok yaygın: Göz ağrısı, göz iritasyonu, gözlerde yabancı cisim hissi, göz kanlanması, konjunktiva kanaması, blefarit, göz kuruluğu, görme bozukluğu, vitreusta uçuşan noktalar, göz içi inflamasyonu, vitreus dekolmanı, vitritis, retina kanaması, göz yaşı artması, göz kaşıntısı
Yaygın: Gözde rahatsızlık, konjonktiva hiperemisi, konjuktivit, alerjik konjuktivit, arka kapsülde opaklaşma, retina pigment epitel dekolmanı, retina pigment epitelinin yırtılması, retina dejenerasyonu, retina dekolmanı, retina yırtığı, retinal bozukluklar, görme keskinliğinde azalma, vitreal kanama, vitreal bozukluklar, uveit, iritis, iridosiklitis, katarakt, subkapsüler katarakt, punktat keratit, kornea abrazyonu, ön kamarada flare, bulanık görme, enjeksiyon yerinde kanama, göz kanaması, göz akıntısı, fotopsi, fotofobi, göz kapağı ödemi, göz kapağında ağrı,
Yaygın olmayan: göz kapağı irritasyonu, keratopati, korneal stria, kornea ödemi, hipopion, endoftalmi, körlük, enjeksiyon yerinde ağrı, enjeksiyon yerinde rahatsızlık, gözde anormallik hissi, iris adezyonu, korneal deposit, hifemi.
Solunum, göğüs hastal
ı
klar
ı
ve mediastinal hastal
ı
klar
Yaygın: Öksürme
Gastrointestinal hastal
ı
klar
Yaygın: Bulantı
Deri ve deri alt
ı
doku hastal
ı
klar
ı
Yaygın: Alerjik reaksiyonlar (kızarıklık, ürtiker, pruritus, eritema)
Kas-iskelet bozukluklar, bağ doku ve kemik hastal
ı
klar
ı
Çok yaygın: Eklem ağrısı
Araşt
ı
rmalar
Çok yaygın: Göz içi basıncının yükselmesi
4. 9 Doz aş
ı
m
ı
ve tedavisi
Klinik çalışmalarda ve pazarlama sonrası verilerde kazayla ortaya çıkan doz aşımı vakaları bildirilmiştir. Rapor edilen bu vakalarla ilişkilendirilebilen en sık görülen advers etkiler göz içi basıncının yükselmesi ve gözde ağrı hissidir. Eğer bir doz aşımı oluşursa, ilgili hekim tarafından gerekli görülmesi durumunda intraoküler basınç takip edilmeli ve tedavi edilmelidir.
5. FARMAKOLOJİK ÖZELLİKLER
5.1. Farmakodinamik özellikler
Farmakoterapötik grubu: Antineovaskülarizasyon ilaçları ATC kodu: S01LA04
Ranibizumab insan vasküler endotelyal büyüme faktörü A’ya (VEGF-A) hedeflenmiş bir insanlara uyarlanmış rekombinan monoklonal antikor parçasıdır. VEGF-A izoformlarına (VEGF110, VEGF121 ve VEGF165) yüksek afiniteyle bağlanarak VEGF-A’nın reseptörleri VEGFR-1 ve VEGFR-2’ye bağlanmasını önlemektedir. VEGF-A’nın reseptörlerine bağlanması, yaşa bağlı makula dejenerasyonunun neovasküler biçiminin ilerlemesine katkıda bulunduğu düşünülen endotel hücresi proliferasyonu ve neovaskülarizasyonunun yanı sıra damar geçirgenliği artışına yol açmaktadır.
LUCENTIS’in klinik güvenlilik ve etkinliği randomize, çift-kör, plasebo ya da aktif kontrollü üç çalışmada neovasküler YBMD’li hastalarda değerlendirilmiştir. Bu çalışmalara toplam 1323 hasta (879 aktif ve 444 kontrol) alınmıştır.
FVF2598g (MARINA) çalışmasında minimal klasik ya da klasik komponenti olmayan okült KNV’li hastalara aylık 0.3 ya da 0.5 mg LUCENTIS veya plasebo intravitreal enjeksiyonlar verilmiştir. Bu çalışmaya toplam 716 hasta (plasebo 238, LUCENTIS 0.3 mg 238, LUCENTIS 0.5 mg 240) alınmıştır. 24. ayın sonuna kadar veriler mevcuttur.
FVF2587g (ANCHOR) çalışmasında baskın klasik KNV lezyonlu hastalara aylık LUCENTIS 0.3 mg intravitreal enjeksiyonlar ve plasebo PDT aylık LUCENTIS 0.5 mg intravitreal enjeksiyonlar ve plasebo PDT ya da plasebo intravitreal enjeksiyonlar ve aktif verteporfin/PDT verilmiştir. Plasebo ya da aktif verteporfin/PDT ilk LUCENTIS enjeksiyonu ile birlikte ve florescein anjiyografisi inatçı ya da yineleyen damar sızıntısını gösterdiği taktirde ondan sonra üç ayda bir verilmiştir. Bu çalışmaya toplam 423 hasta (plasebo 143, LUCENTIS 0.3 mg 140, LUCENTIS 0.5 mg 140) alınmıştır. 24. ayın sonuna kadar veriler mevcuttur.
Her iki çalışmada birincil etkinlik sonlanım noktası görme keskinliğinde 12. ayda kontrole göre < 15 harf kaybetmiş şeklinde tanımlanan görme keskinliğini koruyan hastaların oranıdır. LUCENTIS ile tedavi edilen hastaların hemen hepsi (yaklaşık %95) görme keskinliğini korumuştur. LUCENTIS ile tedavi edilen hastaların %34-40’ı görme keskinliğinde 12 ayda > 15 harf kazanma şeklinde tanımlanan klinik açıdan anlamlı bir düzelme ile karşılaşmıştır. Lezyon boyutu sonuçları anlamlı şekilde etkilememiştir. Ayrıntılı sonuçlar aşağıdaki tablolarda sunulmaktadır.
Tablo 5-1 FVF2598g (MARINA) çal
ı
şmas
ı
nda 12. ve 24. aydaki sonuçlar
Sonuç ölçümü | Ay | Plasebo (n=238) | LUCENTIS 0.5 mg (n=240) |
Görme keskinliğinde < 15 harf kaybı (%) (Görmenin idamesi) | 12. | %62 | %95 |
24. | %53 | %90 | |
Görme keskinliğinde > 15 harf kazanma (%)a | 12. | %5 | %34 |
24. | %4 | %33 | |
Görme keskinliğinde ortalama değişiklik (harf) (Std. Sapma)a | 12. | -10.5 (16.6) | +7.2 (14.4) |
24. | -14.9 (18.7) | +6.6 (16.5) | |
a p<0.01. Tablo 5-2 FVF2587g (ANCHOR) çal | |||
Sonuç ölçümü | Ay | Verteporfin/PDT (n=143) | LUCENTIS 0.5 mg (n=140) |
Görme keskinliğinde < 15 harf kaybı (%)a (Görmenin idamesi) | 12. | %64 | %96 |
24. | %66 | %90 | |
Görme keskinliğinde > 15 harf kazanma (%) Görme keskinliğinde ortalama değişiklik (harf) (Std. Sapma)a | 12. | %6 | %40 |
24. | %6 | %41 | |
12. | -9.5 (16.4) | +11.3 (14.6) | |
24. | -9.8 (17.6) | +10.7 (16.5) |
a p<0.01.
Şekil 5-1 FVF2598g (MARINA) ve FVF2587g (ANCHOR) çal
ı
şmas
ı
nda başlang
ı
çtan 24. aya kadar görme keskinliğinde ortalama değişiklik
Çalışma FVF2598g
15 1D 5 0
-5 1D -15
+h E \
-e ° Ö
JŞ
■ S -
>M M
Ü I
’M <2
S
QJ
> +21 5
-u-o-o-o -14.9
8 10 12 14 16 1B 20 22 24
O
O
0 2 4
Ay
Çalışma FVF2587g
15 İD 5 0
-5 -1D -15 "
0
al la
-e
o
+ 11.3 -v
3 £
JŞ
> +20.a
a 3
’M ’<2
0 >M
O
* * +
-9.5 j
o O
2 4 6 8 10 12
Ay
ANCHOR
MARINA
LUCENTIS 0.5 mg (n=240) Plasebo (n=238)
-■- LUCENTIS 0.5 mg (n=139)
Verteporfin PDT (n=143)
LUCENTIS ile tedavi edilen gruptaki hastalarda ortalama olarak en az düzeyde gözlenebilen KNV lezyonu büyümesi gözlenmiştir. 12. ayda KNV lezyonu toplam alanı kontrol kollarındaki 2.3-2.6 DA’ya karşı LUCENTIS’te 0.1-0.3 DA bulunmuştur. Her iki çalışmada elde edilen bulgular, devam eden ranibizumab tedavisinin, tedavinin ilk yılında en iyi düzeltilmiş görme keskinliğinde (BCVA) >15 harf kayıp olan hastalarda da faydalı olabileceğini göstermiştir.
Çalışma FVF3192g (PIER) neovasküler YBMD’li hastalarda (klasik KNV bileşeni bulunan ve bulunmayan) LUCENTIS’in güv enlilik ve etkinliğini değerlendirmek için tasarlanmış randomize, çift-kör, plasebo kontrollü iki yıllık bir çalışmadır. 12. ayın sonuna kadar veriler mevcuttur. Hastalar ilk 3 ay, ayda bir LUCENTIS 0.3 mg ya da 0.5 mg veya plasebo intravitreal enjeksiyonları ve takiben her üç ayda bir 1 doz almıştır. Bu çalışmaya toplam 184 hasta (plasebo 63, LUCENTIS 0.3 mg 60, LUCENTIS 0.5 mg 61) alınmış, 171 ’i (%93) 12 aylık çalışmayı tamamlamıştır. PIER’de LUCENTIS ile tedavi edilen hastalar 0. günden 12. aya kadar planlanan ortalama 6 tedavinin hepsini almıştır.
PIER’de etkinlik sonlanım noktası görme keskinliğinde başlangıca göre 12. aydaki ortalama değişikliktir (Şekil 2’ye Bakınız). Ortalama görme keskinliğindeki ilk 3 aylık artıştan sonra (aylık dozu takiben) üç ayda bir 1 doz verilen LUCENTIS hastalarında görme keskinliği 12. ayda başlangıç değerine dönmüştür. PIER’de LUCENTIS ile tedavi edilen hastaların hemen hepsi (%90) 12. ayda görme keskinliğini korumuştur.
Ay
LUCENTIS 0.5 mg (n=240) Plasebo (n=238)
Şekil 5-2 FVF3192g (PIER) çal
ı
şmas
ı
nda başlang
ı
çtan 12. aya kadar görme keskinliğinde ortalama değişiklik: ITT popülasyonu
Çalışma FVF3192g
10

0 2 4 6 8 10 12
32 hastanın 9 ay boyunca izlendiği, aynı gün uygulanan verteporfin PDT ve LUCENTIS 0.5 mg’ın güvenliliğinin değerlendirildiği açık bir çalışmadan (PROTECT) elde edilen veriler, başlangıç tedavisinin ardından intraoküler inflamasyon insidansının % 6.3 olduğunu göstermektedir (32 hastanın 2’si).
MARINA’da 12. ayda LUCENTIS ile tedavi edilen hastalar NEI VFQ-25 ile ölçüldüğü gibi ortalamada yakını görme, uzağı görme ve görmeye spesifik bağımlılıkla ilgili aktivite yetilerinde istatistiksel ve klinik açıdan anlamlı bir iyileşme bildirirken plasebo uygulanan hastalar bu aktivite yetilerinde azalma olduğunu bildirmiştir. Yakın aktiviteler ölçeğinde 0.5 mg LUCENTIS ile tedavi edilen hastalar +10.4 puanlık artış bildirirken, plasebo uygulanan hastalar 2.6 puanlık azalma bildirmiştir (p<0.01). Uzak aktiviteler ölçeğinde LUCENTIS ile tedavi edilen hastalar +7.4 puanlık artış bildirirken plasebo uygulanan hastalar 5.9 puanlık azalma bildirmiştir (p<0.01). Görmeye spesifik bağımlılık ölçeğinde LUCENTIS ile tedavi edilen hastalar +6.8 puanlık artış bildirirken plasebo uygulanan hastalar 4.7 puanlık azalma bildirmiştir (p<0.01).
12. ayda bu üç VFQ-25 alt ölçeğinin her birindeki başlangıçtan olan artış LUCENTIS ile tedavi edilen hastalarda 24. ayda korunurken plasebo enjeksiyon grubundaki başlangıçtan olan ortalama değişiklik bu alt ölçeklerin her birinde 12. aydan 24. aya kadar daha da azalmıştır. Bu nedenle, LUCENTIS’in plasebo ile kontrol grubuna göre 24. aydaki tedavi yararı 12. aydakinden yüksektir.
ANCHOR’da 12. ayda LUCENTIS ile tedavi edilen hastalar verteporfin/PDT alan hastalara göre yakını görme, uzağı görme ve görmeye spesifik bağımlılıkla ilgili aktivite yetilerinde istatistiksel ve klinik açıdan anlamlı bir iyileşme bildirmiştir. Yakın aktiviteler ölçeğinde 0.5 mg LUCENTIS ile tedavi edilen hastalar +9.1 puanlık artış bildirirken verteporfin/PDT uygulanan hastalar +3.7 puanlık artış bildirmiştir (p<0.01). Uzak aktiviteler ölçeğinde LUCENTIS ile tedavi edilen hastalar +9.3 puanlık artış bildirirken verteporfin/PDT uygulanan hastalar +1.7 puanlık artış bildirmiştir (p<0.01). Görmeye spesifik bağımlılık ölçeğinde LUCENTIS ile tedavi edilen hastalar +8.9 puanlık artış bildirirken verteporfin/PDT uygulanan hastalar 1.4 puanlık azalma bildirmiştir (p<0.01). Verteporfin PDT grubunda, görmeye özgü bağımlılık alt ölçek skorlarında 12. ayda başlangıca göre ortalama azalma 24. ayda da korunurken, yakın ve uzak aktivitelerde alt ölçek skorlarında 12. ayda başlangıca göre gözlenen ortalama iyileşme 24. ayda kaybolmuştur. Her bir tedavi grubunda 12. ve 24. aylar arasındaki bu değişiklikler, ranibizumabın görmeye özgü bağımlılık alt ölçeğinde tedaviye faydası 24. ayda 12. aya kıyasla daha az olmakla birlikte, ranibizumabın verteporfin PDT’ye göre tedavi faydasını artırmasını ya da korumasını sağlamış, (p-değerleri 0.0023 ile 0.0006 arasında değişmektedir).
Çalışma FVF3689g (SAILOR) YBMD’ye sekonder KNV ile önceden tedavi edilmiş ve hiç tedavi denenmemiş gönüllülerdeki bir yıllık, çok merkezli, tek kör, faz IIIb çalışmadır. Çalışmanın asıl amacı 12 ay tedavi edilen gönüllülerde gözle ilgili olan ve olmayan ciddi yan etkilerin sıklığı hakkında fikir edinmek olmuştur. İki bin üç yüz yetmiş sekiz hasta birbirini takip eden üç ay zarfında her ay bir kez 0.3 mg veya 0.5 mg intravitreal ranibizumab enjeksiyonu ve bunu takiben gerektiğinde ayda birden fazla olmamak üzere gerektiği şekilde tedavi uygulanmak üzere 1:1 oranda randomize edilmiştir.
Genel olarak, gözle ilgili olan ve olmayan yan etkilerin sıklığında her iki doz grubu arasında dengesizlik gözlenmemiştir. 0.5 mg grubunda 0.3 mg grubuyla karşılaştırıldığında inme oranlarında istatistiksel olarak anlamlı olmayan yükselme eğilimi vardı. Tüm inme oranları için ilgili %95 CI değerleri geniş olarak saptanmıştır (0.3 mg grubu için %0.3 ila %1.3, 0.5 mg grubu için %0.7 ila %2.0). Her iki doz grubunda da inme sayısı az olmuştur ve tedavi grupları arasında inme oranlarında gerçek bir farklılık olduğu sonucunu çıkarmak (ya da bu durumu olasılık dışı bırakmak) için yeterli delil saptanmamıştır. İnme oranlarındaki farklılık geçici iskemik atak veya inme öyküsü dahil olmak üzere inme için bilinen risk faktörleri olan hastalarda daha yüksek olabilir.
5.2. Farmakokinetik özellikler
Genel özellikler
LUCENTIS, neovasküler (yaş tip) yaşa bağlı maküla dejenerasyonu (YBMD) tedavisinde kullanılan, steril, koruyucu içermeyen, berrak, renksiz ile soluk sarı arasında sulu enjektabl çözeltidir.
Emilim:
Neovasküler YBMD’li hastalara aylık intravitreal LUCENTIS uygulamasını takiben serum ranibizumab konsantrasyonları, genellikle VEGF’nin biyolojik aktivitesini %50 inhibe etmesi için gerekli ranibizumab konsantrasyonlarının altındaki en yüksek değerlerden (Cmax) düşüktür (in vitro hücre proliferasyonu tayininde değerlendirildiği gibi 11-27 ng/ml). Cmax 0.05 ila 1.0 mg/göz doz aralığında dozla orantılıdır.
Aylık LUCENTIS 0.5 mg/göz intravitreal uygulaması üzerine dozdan yaklaşık 1 gün sonra ulaşılan serum ranibizumab Cmax’unun genellikle 0.79 ila 2.90 ng/ml aralığında ve Cmin’unun genellikle 0.07 ila 0.49 ng/ml aralığında olduğu tahmin edilmektedir. Serum ranibizumab maruziyetinin vitreal ranibizumab maruziy etinden yaklaşık 90,000 kat düşük olduğu tahmin edilmektedir.
Dağılım:
Intravitreal yolla uygulandığından dağılım bilgisi mevcut değildir. Biy otransformasy on:
Intravitreal yolla uygulandığından biyotransformasyon bilgisi mevcut değildir. Eliminasyon:
Popülasyon farmakokinetik analizine ve 0.5 mg dozla tedavi edilen hastalarda ranibizumabın serumdan kaybolmasına göre, ranibizumabın vitreusta ortalama eliminasyon yarı ömrü yaklaşık 10 gündür.
Doğrusallık / doğrusal olmayan durum:
Intravitreal yolla uygulandığından bu bilgi mevcut değildir.
Hastalardaki karakteristik özellikler
Böbrek/Karaciğer yetmezliği: Böbrek yetmezliği olan hastalarda LUCENTIS’in farmakokinetiğini incelemek için hiçbir klinik çalışma yürütülmemiştir. Bir popülasyon farmakokinetik analizinde %68 hastada (136/200) böbrek yetmezliği (%46.5 hafif [50-80 ml/dakika], %20 orta [30-50 ml/dakika] ve %1.5 şiddetli [< 30 ml/dakika]) bulunmuştur. Sistemik klerens, hafif düşük olup klinik açıdan anlamlı değildir.
Karaciğer yetmezliği olan hastalarda LUCENTIS’in farmakokinetiğini incelemek için hiçbir klinik çalışma yürütülmemiştir.
Pediyatrik hastalar: 18 yaş altındaki hastalarda yeterli farmakokinetik veri mevcut değildir.
5.3. Klinik öncesi güvenlilik verileri
Sinomolgus maymunlarına 26 haftaya kadar iki haftada bir 0.25 mg/göz ve 2.0 mg/göz arasındaki dozlarda uygulanan iki taraflı intravitreal ranibizumab doza bağlı oküler etkilerle sonuçlanmıştır.
İntraoküler enjeksiyondan 2 gün sonra tepe noktasına ulaşan ön kamarada flare ve hücrelerde doza bağlı artışlar saptanmıştır. İnflamatuar yanıtın şiddeti genellikle takip eden enjeksiyonlarla ya da iyileşme sırasında azalmıştır. Arka bölmede doza bağlı azalma eğiliminde olan ve genellikle tedavi süresinin sonuna kadar süren vitreus hücre infiltrasyonu ve uçuşan noktalar saptanmıştır. 26 haftalık çalışmada vitreus inflamasyonunun şiddeti enjeksiyon sayısıyla artmıştır. Ancak bunun geri dönüşümlü olduğu ve iyileşmeden sonra ortadan kalktığı gözlenmiştir. Arka kamara inflamasyonunun yapısı ve zamanlaması klinik açıdan önemli olmayabilen bir immün aracılı antikor yanıtını düşündürmektedir. Bazı hayvanlarda nispeten uzun olan şiddetli inflamasyon döneminden sonra, lensteki değişikliklerin şiddetli inflamasyona ikincil olduğunu gösteren katarakt oluşumu gözlenmiştir. İntravitreal enjeksiyonları takiben dozdan bağımsız olarak doz sonrası göz içi basıncında geçici bir yükselme gözlenmiştir.
Mikroskopik göz değişiklikleri inflamasyonla ilişkili olmuş, dejeneratif süreç göstermemiştir. Bazı gözlerin optik diskinde granülomatöz inflamatuar değişiklikler kaydedilmiştir. İyileşme dönemi sırasında bu arka bölme değişiklikleri azalmış ve bazı vakalarda kaybolmuştur.
İntravitreal uygulamayı takiben hiçbir sistemik toksisite belirtisi saptanmamıştır. Uygulama yapılan hayvanların bir alt kümesinde ranibizumaba karşı serum ve vitreus antikorları bulunmuştur.
Hiçbir karsinojeniklik, mutajeniklik, üreme ve gelişim toksisitesi verisi bulunmamaktadır.
6. FARMASÖTİK ÖZELLİKLER
6.1. Yardımcımaddelerin listesi
6.2. Geçimsizlikler
Geçimlilik araştırmaları bulunmadığından, bu tıbbi ürün başka tıbbi ürünlerle karıştırılmamalıdır.
6.3. Raf ömrü
6.4. Saklamaya yönelik özel tedbirler
Buzdolabında (2-8°C) saklayınız.
6.5. Ambalajın niteliği ve içeriği
Klorobutil kauçuk tapalı bir cam flakon (renksiz tip I cam) içinde 0.3 ml enjeksiyonluk LUCENTIS çözeltisi. Bir ambalaj; bir flakon, şişe içeriğinin çekilmesi için bir filtreli iğne, intravitreal enjeksiyon için bir iğne, şişe içeriğinin çekilip intravitreal enjeksiyonu için bir şırınga içermektedir.
6.6. Beşeri tıbbi üründen arta kalan maddelerin imhasıve diğer özel önlemler
Flakonlar sadece tek kullanım içindir (Bölüm 4.2 Pozoloji ve uygulama şekli’ne bakınız).
Geçerli olduğu takdirde kullanılmamış olan ürünler ya da atık materyaller "Tıbbi Atıkların Kontrolü Yönetmeliği" ve "Ambalaj ve Ambalaj Atıklarının Kontrolü Yönetmelik"lerine uygun olarak imha edilmelidir.
 Diyabet Hastalığı
Diyabet, insülin hormonu ile ilgili problemlerden kaynaklanan bir hastalıktır.
Diyabet Hastalığı
Diyabet, insülin hormonu ile ilgili problemlerden kaynaklanan bir hastalıktır. |
 Gıda Alerjisi
Her yıl milyonlarca insan yiyeceklere alerji gösteriyor.
Gıda Alerjisi
Her yıl milyonlarca insan yiyeceklere alerji gösteriyor. |
|
Diyabet Hastalığı Diyabet, insülin hormonu ile ilgili problemlerden kaynaklanan bir hastalıktır. |
 |
İnme İnme, beynin hasar görmesinin sonucudur. Bu hasar, beynin bir kısmındaki ya bir kanama ya da akut kan eksikliği nedeniyle o kısmın geçici ya da kalıcı olarak işlevini yapamamasına yol açar. |
 |
Yüksek Tansiyon Hipertansiyon sürekli anormal derecede yüksek olan kan basıncıdır. Tansiyon atardamarlarınızdaki kanın basıncıdır. |
İLAÇ GENEL BİLGİLERİ
Novartis Sağlık,Gıda ve Tarım Ürünleri San. Tic. A.Ş.
| Geri Ödeme Kodu | A10722 |
| Satış Fiyatı | 10592.36 TL [ 2 May 2025 ] |
| Önceki Satış Fiyatı | 10592.36 TL [ 25 Apr 2025 ] |
| Original / Jenerik | Original İlaç |
| Reçete Durumu | Normal Reçeteli bir ilaçdır. |
| Barkodu | 8699504770254 |
| Etkin Madde | Ranibizumab |
| ATC Kodu | S01LA04 |
| Birim Miktar | 10 |
| Birim Cinsi | MG/ML |
| Ambalaj Miktarı | 1 |
| Duyu Organları > Oküler Damar Bozukluğu Ajanları > Ranibizumab |
| İthal ( ref. ülke : Isvicre ) ve Beşeri bir ilaçdır. |