MYLOTARG 5 mg inf�zyonluk ��zelti HAZIRLAMADA KULLANILACAK konsantre i�in toz (1 flakon) K�sa �r�n Bilgisi
{ Gemtuzumab }
1. BE�ER� TIBB� �R�N�N ADI
MYLOTARG 5 mg �nf�zyonluk ��zelti Haz�rlamada Kullan�lacak Konsantre ��in Toz
Steril Sitotoksik
2. KAL�TAT�F VE KANT�TAT�F B�LE��M
Etkin madde
Her flakon 5 mg gemtuzumab ozogamisin i�erir.
Suland�rma sonras�nda (bkz. B�l�m 6.6) konsantre ��zelti, 1 mg/mL gemtuzumab ozogamisin i�erir.
Gemtuzumab ozogamisin, sitotoksik madde olan N-asetil-gama-kalikeamisine kovalent olarak ba�lanan, CD33 hedefli monoklonal antikordan (hP67.6; rekombinant insanla�t�r�lm�� imm�noglobulin [Ig] G4, NS0 h�crelerindeki memeli h�cre k�lt�r�nden �retilen kappa antikor) olu�an bir antikor-ila� konjugat�d�r.
Yard�mc� maddeler
Sodyum klor�r 29,2 mg/flakon Monobazik sodyum fosfat monohidrat 0,5 mg/flakon Dibazik sodyum fosfat, susuz 3 mg/flakon
Yard�mc� maddelerin tam listesi i�in, bkz. B�l�m 6.1.
3. FARMAS�T�K FORMU
Konsantre inf�zyon ��zeltisi i�in toz (konsantre i�in toz). Beyaz ile beyaz�ms� aras� kek veya toz.
4. KL�N�K �ZELL�KLER
4.1. Terap�tik endikasyonlar
MYLOTARG, daha �nce tedavi edilmemi�, de novo CD33 pozitif, d���k (akut promyelositik l�semi hari�) ve orta sitogenetik ve molek�ler risk grubunda oldu�u g�sterilmi�, 15 ya� ve �zerinde akut myeloid l�semi (AML) olgular�nda daunorubisin (DNR) ve sitarabin (AraC) tedavisi ile kombine olarak kullan�mda endikedir.
4.2. Pozoloji ve uygulama �ekli
MYLOTARG, kan kanseri tedavisi konusunda deneyim sahibi bir doktorun g�zetimi alt�nda ve tam te�ekk�ll� res�sitasyon olanaklar�n�n haz�r bulundu�u bir ortamda uygulanmal�d�r.
MYLOTARG yaln�zca, yo�un ind�ksiyon kemoterapisi almas� uygun hastalarda kullan�lmal�d�r.
�nf�zyonla ili�kili semptomlar�n d�zeltilmesine yard�mc� olmak i�in, doz uygulanmadan 1 saat �nce, bir kortikosteroid, antihistamin ve asetaminofenle (veya parasetamol) �n ila� uygulamas� �nerilir (bkz. B�l�m 4.4).
T�m�r lizisiyle ili�kili hiper�risemi geli�mesini �nlemeye yard�mc� olmak i�in, hidrasyon, antihiper�risemik veya hiper�riseminin tedavisine y�nelik di�er maddelerin uygulanmas� gibi uygun �nlemler al�nmal�d�r (bkz. B�l�m 4.4).
Pozoloji/uygulama s�kl��� ve s�resi:
�nd�ksiyon
�nerilen MYLOTARG dozu, 1. g�n ila 3. g�nde 30 dakika boyunca inf�zyon yap�lan DNR 60 mg/m/g�n ve 1. g�nden 7. g�ne kadar s�rekli inf�zyonla AraC 200 mg/m/g�n ile kombinasyonlu �ekilde, 1, 4 ve 7. g�nde 2 saatte inf�zyonu yap�lan 3 mg/m/doz (maksimum bir adet 5 mg flakona kadar) �eklindedir.
�kinci bir ind�ksiyon gerekliyse; MYLOTARG, ikinci ind�ksiyon tedavisi s�ras�nda uygulanmamal�d�r. �kinci ind�ksiyon siklusu s�ras�nda yaln�zca DNR ve AraC �u �ekilde �nerilen dozda uygulanmal�d�r: 1. ve 2. g�nlerde DNR 35 mg/m/g�n ve 1. g�n ila 3. g�nde 12 saatte bir AraC
1 g/m.
Konsolidasyon
Transf�zyon olmad���nda periferik kanda 100 × 10/L'ye kadar trombosit say�s�yla 1 × 10 h�cre/L'den fazla mutlak n�trofil say�s� (MNS) ve normosel�ler kemik ili�inde % 5'ten az blast olarak tan�mland��� �ekilde, ind�ksiyonun ard�ndan tam remisyon (CR) ya�ayan hastalar i�in, intraven�z AraC (1. g�n ila 4. g�nde 2 saatte inf�zyonu yap�larak, 12 saatte 1 g/m) ile intraven�z MYLOTARG (1. g�nde bir adet 5 mg flakonluk maksimum doza kadar 2 saatte inf�zyonu yap�lan 3 mg/m/doz) ile kombinasyonlu olarak, 2 adede kadar intraven�z DNR konsolidasyon k�r� (1 g�n [ilk k�r] veya 2 g�n [2. k�r] i�in 60 mg/m) �nerilir.
Tablo 1. Kemoterapi ile kombine olarak uygulanan MYLOTARG i�in doz rejimleri
Tedavi k�r� | MYLOTARG | Daunorubisin | Sitarabin |
�nd�ksiyon | 1, 4 ve 7. g�nde 3 mg/m/doz (maksimum bir adet 5 mg flakona kadar) |
1. g�n ila 3. g�nde 60 mg/m/g�n |
1. g�n ila 7. g�nde 200 mg/m/g�n |
�kinci ind�ksiyon (gerekirse) | MYLOTARG, ikinci ind�ksiyonda uygulanmamal�d�r. |
1. g�n ila 2. g�nde 35 mg/m/g�n |
1. g�n ila 3. g�nde 12 saatte bir 1 g/m |
Konsolidasyon K�r� 1 | 1. g�nde 3 mg/m/doz (maksimum bir adet 5 mg flakona kadar) |
1. g�nde 60 mg/m/g�n |
1. g�n ila 4. g�nde 12 saatte bir 1 g/m |
Konsolidasyon K�r� 2 | 1. g�nde 3 mg/m/doz (maksimum bir adet 5 mg flakona kadar) |
1. g�n ila 2. g�nde 60 mg/m/g�n |
1. g�n ila 4. g�nde 12 saatte bir 1 g/m |
![]()
Doz ve program de�i�iklikleri
Hiperl�kositoz i�in program de�i�iklikleri
Hiperl�kositik (l�kosit say�s� ≥ 30.000/mm) AML'li hastalarda, MYLOTARG uygulanmadan 48 saat �nce �nce periferik akyuvar (WBC) say�s�n� d���rmek amac�yla, l�koferez, oral hidroksi�re veya hiroksi�reli ya da hiroksi�resiz AraC ile sitored�ksiyon yap�lmas� �nerilir.
Kombinasyon tedavisinde MYLOTARG alan de novo hiperl�kositik AML'li olup daha �nce tedavi edilmemi� hastalarda l�kored�ksiyon i�in, hidroksi�reli veya hidroksi�resiz AraC kullan�l�yorsa, a�a��daki modifiye edilmi� program uygulanmal�d�r (Tablo 2):
Tablo 2. Hiperl�kositozun sitarabinle tedavisi i�in program de�i�ikli�i
Tedavi k�r� |
MYLOTARG |
Daunorubisin |
Sitarabin |
Hidroksi�re |
�nd�ksiyon | 3, 6 ve 9. g�nde 3 mg/m/doz (maksimum bir adet 5 mg flakona kadar) |
3. g�n ila 5. g�nde 60 mg/m/g�n |
1. g�n ila 7. g�nde 200 mg/m/g�n |
1.g�n (standart t�bbi uygulamaya g�re) |
Konsolidasyon k�r�ne ili�kin doz tavsiyeleri i�in, bkz. Tablo 1.
Advers ila� reaksiyonlar� i�in doz de�i�ikli�i
Bireysel g�venlilik ve tolere edilebilirli�e g�re MYLOTARG'da doz de�i�ikli�i yap�lmas� tavsiye edilir (bkz. B�l�m 4.4). Baz� advers ila� reaksiyonlar�n�n y�netimi, MYLOTARG dozuna ara verilmesini ya da dozun kal�c� olarak durdurulmas�n� gerektirebilir (bkz. B�l�m 4.4 ve 4.8).
Tablo 3 ve Tablo 4'te s�ras�yla hematolojik olan ve hematolojik olmayan toksisiteler i�in doz de�i�ikli�i k�lavuzlar� g�sterilmektedir.
Tablo 3. Hematolojik toksisiteler i�in doz de�i�iklikleri
Hematolojik toksisiteler | Doz de�i�iklikleri |
Kal�c� trombositopeni (Konsolidasyon k�r�n�n planlanan ba�lang�� tarihinde trombositler < 100.000/mm) |
≥ 50.000/mm de�erine d�zelirse: MYLOTARG yeniden |
Konsolidasyon k�r�n�n ba�lat�lmas� ertelenmelidir.
Konsolidasyon k�r�n�n planlanan ba�lang�� tarihinden sonra trombosit say�s� 14 g�n i�inde ≥ 100.000/mm de�erine d�zelirse: Konsolidasyon tedavisi ba�lat�lmal�d�r (Tablo 1'de a��kland��� gibi).
Konsolidasyon k�r�n�n planlanan ba�lang�� tarihinden sonra trombosit say�s� 14 g�n i�inde < 100.000/mm ve
| uygulanmamal�d�r ve konsolidasyon tedavisi yaln�zca DNR ve AraC'den olu�mal�d�r.
yeniden de�erlendirmek i�in bir K�A yap�lmal�d�r. |
Kal�c� n�tropeni | MYLOTARG uygulanmamal�d�r). |
Trombosit say�s�ndaki d�zelme 14 g�nden fazla s�re boyunca < 50.000/mm de�erinde kal�rsa, konsolidasyon tedavisi yeniden de�erlendirilmeli ve hastalar�n durumunu
N�trofil say�s�, konsolidasyon siklusunun planlanan ba�lang�� tarihinden sonra 14 g�n i�inde (�nceki siklusun ard�ndan olan hematolojik d�zelmeden 14 g�n sonra) 500/mm de�erinin �zerine ��kacak �ekilde d�zelmezse, MYLOTARG kesilmelidir (konsolidasyon sikluslar�nda
K�saltmalar: AML=Akut miyeloid l�semi; AraC=Sitarabin; K�A=Kemik ili�i aspirasyonu, DNR=Daunorubisin.
Tablo 4. Hematolojik olmayan toksisiteler i�in doz de�i�iklikleri
Hematolojik olmayan toksisiteler | Doz de�i�iklikleri |
VOD/SOS | MYLOTARG tedavisi kesilmelidir (bkz. B�l�m 4.4). |
Toplam bilirubin > 2 × ULN ve AST ve/veya ALT > 2,5 × ULN | Her dozdan �nce toplam bilirubin ≤ 2 × ULN de�erine ve AST ile ALT ≤ 2,5 × ULN de�erine d�zelene kadar MYLOTARG ertelenmelidir. S�ral� inf�zyonlar aras�nda 2 g�nden fazla erteleme olursa, planlanan dozun atlanmas� d���n�lmelidir. |
�nf�zyonla ili�kili reaksiyonlar | �nf�zyon durdurulmal� ve semptomlar�n �iddetine g�re uygun t�bbi y�netim uygulanmal�d�r. Hastalar, belirtiler ve semptomlar tamamen ge�inceye kadar izlenmelidir ve inf�zyon devam ettirilebilir. �iddetli ya da ya�am� tehdit edici inf�zyon reaksiyonlar� i�in, tedavinin kal�c� olarak kesilmesi d���n�lmelidir (bkz. B�l�m 4.4). |
Di�er �iddetli veya ya�am� tehdit eden, hematolojik olmayan toksisiteler | Hafiften fazla olmayan bir �iddette d�zelme sa�lanana kadar MYLOTARG ile yap�lan tedavi ertelenmelidir. S�ral� inf�zyonlar aras�nda 2 g�nden fazla erteleme olursa, planlanan dozun atlanmas� d���n�lmelidir. |
K�saltmalar: ALT=Alanin aminotransferaz; AST=Aspartat aminotransferaz; SOS=Sin�zoidal obstr�ksiyon sendromu, ULN=Normalin �st s�n�r�; VOD=Venookl�zif hastal�k.
Uygulama �ekli:
MYLOTARG intraven�z kullan�m i�indir ve uygulanmadan �nce suland�r�lmal� ve seyreltilmelidir (bkz. B�l�m 6.6). 1 mg/mL konsantrasyona suland�r�ld���nda, flakonun ekstrakte edilebilir i�eri�i 4,5 mg'dir (4,5 mL). Suland�r�lm�� ve seyreltilmi� ��zelti, nab�z, tansiyon ve v�cut s�cakl��� da dahil olmak �zere yak�ndan klinik izleme yap�larak, 2 saatlik s�rede inf�zyonla intraven�z yoldan uygulanmal�d�r. MYLOTARG, intraven�z pu�e veya bolus olarak uygulanmamal�d�r (bkz. B�l�m 6.6).
Uygulama �ncesi t�bbi �r�n�n�n suland�r�lmas� ve seyreltilmesi ile ilgili talimatlar i�in bkz. B�l�m
6.6.
�zel pop�lasyonlara ili�kin ek bilgiler:
Karaci�er yetmezli�i:
Toplam bilirubin ≤ 2 × normalin �st s�n�r� (ULN) ve aspartat aminotransferaz (AST)/alanin aminotransferaz (ALT) ≤ 2,5 × ULN ile tan�mlanan karaci�er yetmezli�i bulunan hastalarda ba�lang�� dozunda herhangi bir ayarlama yap�lmas� gerekmez. Her dozdan �nce toplam bilirubin ≤ 2 × ULN
de�erine ve AST ile ALT ≤ 2,5 × ULN de�erine d�zelene kadar MYLOTARG ertelenmelidir (bkz. Tablo 4, B�l�m 4.4 ve 5.2).
B�brek yetmezli�i:
Hafif ila orta d�zeyde b�brek yetmezli�i olan hastalar i�in doz ayarlamas� gerekli de�ildir. MYLOTARG �iddetli b�brek yetmezli�i olan hastalarda ara�t�r�lmam��t�r. MYLOTARG i�in b�brek klirensi s�z konusu de�ildir, �iddetli b�brek yetmezli�i olan hastalardaki farmakokinetik bilinmemektedir (bkz. B�l�m 5.2).
Pediyatrik pop�lasyon:
15 ya��n alt�ndaki hastalarda MYLOTARG'�n g�venlili�i ve etkilili�i belirlenmemi�tir. Mevcut veriler, B�l�m 4.8, 5.1 ve 5.2'de a��klanmaktad�r ancak pozoloji i�in tavsiye verilememektedir.
Geriyatrik pop�lasyon:
Ya�l� hastalarda (≥ 65 ya�) doz ayarlamas� gerekmez (bkz. B�l�m 5.2).
4.3. Kontrendikasyonlar
Etkin madde
bulunan kimselerde kullan�lmamal�d�r.
4.4. �zel kullan�m uyar�lar� ve �nlemleri
Hepatik venookl�zif hastal�k/sin�zoidal obstr�ksiyon sendromu (VOD/SOS) dahil olmak �zere hepatotoksisite
MYLOTARG ile tedavi edilen hastalarda, ya�am� tehdit edici ve bazen �l�mc�l hepatik yetmezlik ve
VOD/SOS'yi i�eren hepatotoksisite rapor edilmi�tir (bkz. B�l�m 4.8).
Potansiyel risk fakt�rlerinin analizine g�re, monoterapi olarak, hematopoetik k�k h�cre naklinden (HKHN) �nce veya sonra MYLOTARG alan yeti�kin hastalar�n ve orta ya da �iddetli karaci�er yetmezli�i olan hastalar�n, VOD geli�tirme riski y�ksektir (bkz. B�l�m 4.8).
VOD/SOS riski nedeniyle, VOD/SOS semptomlar� ve belirtileri yak�ndan izlenmelidir; bunlar, her MYLOTARG dozundan �nce yak�ndan izlenmesi gereken ALT, AST, toplam bilirubin ve alkalen fosfatazdaki art��lar ile hepatomegali (a�r�l� olabilir), h�zl� kilo art��� ve assiti i�erebilir. Yaln�zca toplam bilirubinin izlenmesi, VOD/SOS riski bulunan t�m hastalar�n tan�mlanmas�na yard�mc� olmayabilir. Anormal karaci�er testleri g�r�len hastalar i�in, karaci�er testlerinin ve hepatotoksisiteye ili�kin klinik belirtilerin ve semptomlar�n daha s�k izlenmesi �nerilir. HKHN'ye ge�en hastalar i�in, HKHN sonras� d�nemde, uygun oldu�u �ekilde karaci�er testlerinin yak�ndan takip edilmesi �nerilir. VOD ve HKHN s�resi aras�nda, daha y�ksek MYLOTARG monoterapi dozlar� bak�m�ndan kesin ili�ki saptanmam��t�r, ancak ALFA-0701 �al��mas�nda, son MYLOTARG dozu ve HKHN aras�nda 2 ayl�k bir s�re olmas� �nerilmi�tir.
Hepatik toksisite belirtilerinin veya semptomlar�n�n y�netiminde, MYLOTARG dozuna ara verilmesi veya durdurulmas� gerekebilir (bkz. B�l�m 4.2). VOD/SOS ya�ayan hastalarda MYLOTARG uygulamas� kesilmelidir ve hastalar, standart t�bbi uygulamaya g�re tedavi edilmelidir.
�nf�zyonla ili�kili reaksiyonlar (anaflaksi dahil)
Klinik �al��malarda, anaflaksi de dahil olmak �zere, inf�zyonla ili�kili reaksiyonlar rapor edilmi�tir (bkz. B�l�m 4.8). Pazarlama sonras� deneyimde, �l�mc�l inf�zyon reaksiyonlar� rapor edilmi�tir.
�nf�zyonla ili�kili reaksiyonlar�n belirtileri ve semptomlar�, ate� ve �rperme ile daha az s�kl�kta uygulaman�n ard�ndan ilk 24 saatte ger�ekle�ebilecek hipotansiyon, ta�ikardi ve solunum semptomlar�n� i�erebilir. MYLOTARG inf�zyonu, nab�z, tansiyon ve v�cut s�cakl��� da dahil olmak �zere yak�ndan klinik izleme yap�larak ger�ekle�tirilmelidir. MYLOTARG dozu uygulanmadan 1 saat �nce, kortikosteroid, antihistamin ve asetaminofen (veya parasetamol) ile �n ila� uygulamas� �nerilir (bkz. B�l�m 4.2). �zellikle dispne, bronkospazm veya klinik a��dan anlaml� hipotansiyon olmak �zere, �iddetli reaksiyonlar� olan hastalarda inf�zyona hemen ara verilmelidir. Hastalar, belirtiler ve semptomlar tamamen ortadan kalk�ncaya kadar izlenmelidir. �iddetli solunum semptomlar� veya klinik a��dan anlaml� hipotansiyonu i�erecek �ekilde, anaflaksi belirtileri veya semptomlar� geli�en hastalar i�in, tedavinin durdurulmas� ciddi bir �ekilde g�z �n�nde bulundurulmal�d�r (bkz. B�l�m 4.2).
Miyelos�presyon
Klinik �al��malarda, baz�lar� ya�am� tehdit eden veya �l�mc�l olan n�tropeni, trombositopeni, anemi, l�kopeni, febril n�tropeni, lenfopeni ve pansitopeni bildirilmi�tir (bkz. B�l�m 4.8). N�tropeni ve trombositopeniyle ili�kili komplikasyonlar, s�ras�yla enfeksiyonlar ve kanama/hemarojik reaksiyonlar� i�erebilir. Baz�lar� ya�am� tehdit eden veya �l�mc�l olan enfeksiyonlar ve kanama/hemarojik reaksiyonlar bildirilmi�tir.
Her MYLOTARG dozundan �nce, tam kan say�mlar� izlenmelidir. Tedavi esnas�nda, hastalar enfeksiyon, kanama/hemoraji veya di�er miyelos�presyon etkilerinin belirtileri ve semptomlar� bak�m�ndan izlenmelidir. Tedavi esnas�nda ve sonras�nda rutin klinik ve laboratuvar tarama testleri yap�lmas� belirtilir.
�iddetli enfeksiyon, kanama/hemoraji veya �iddetli n�tropeni veya kal�c� trombositopeniyi i�eren di�er miyelos�presyon etkileri olan hastalar�n tedavisinde, MYLOTARG dozunun ertelenmesi ya da kal�c� durdurulmas� gerekebilir (bkz. B�l�m 4.2).
T�m�r lizis sendromu (TLS)
Klinik �al��malarda TLS rapor edilmi�tir (bkz. B�l�m 4.8). Pazarlama sonras� deneyimde, akut b�brek yetmezli�i komplikasyonuna yol a�an �l�mc�l TLS raporlar� bildirilmi�tir. Hiperl�kositik AML'li hastalarda, TLS ind�kleme riskini azaltmak amac�yla, MYLOTARG uygulanmadan �nce periferik WBC say�s�n� 30.000/mm de�erine d���rmek i�in, hidroksi�re ya da l�koferezle l�kored�ksiyon yap�lmas� g�z �n�nde bulundurulmal�d�r (bkz. B�l�m 4.2).
Hastalar, TLS belirtilerine ve semptomlar�na kar�� izlenmeli ve standart t�bbi uygulamaya g�re tedavi edilmelidir. T�m�r lizisiyle ili�kili hiper�risemi geli�mesini �nlemeye yard�mc� olmak i�in, hidrasyon, antihiper�risemik (�r. allopurinol) veya hiper�riseminin tedavisine y�nelik di�er maddelerin (�r. rasburikaz) uygulanmas� gibi uygun �nlemler al�nmal�d�r.
Advers risk sitogenetikleriyle AML
�yi ve orta riskli sitogenetikleri olan AML hastalar�nda MYLOTARG'�n etkilili�i ortaya konmu�, advers sitogenetikli hastalarda etkinin boyutuna ili�kin bir belirsizlik s�z konusu olmu�tur (bkz. B�l�m 5.1). Yeni tan� de novo AML'li hastalarda daunorubisinle ve sitarabinle kombinasyonlu olarak MYLOTARG ile tedavide, sitogenetik testi sonu�lar� al�nd���nda, MYLOTARG ile tedaviye devam edilmenin hastaya sa�lad��� potansiyel faydan�n risklere g�re daha fazla olup olmad��� de�erlendirilmelidir (bkz. B�l�m 5.1)
Do�um kontrol�
�ocuk do�urma potansiyeli olan kad�nlara veya �ocuk do�urma potansiyeli olan kad�nlar�n partnerlerine, MYLOTARG ile tedavi esnas�nda ve son dozdan sonraki en az 7 ayl�k (kad�nlarda) veya 4 ayl�k (erkeklerde) d�nem boyunca 2 etkin do�um kontrol y�ntemi kullanmalar� konusunda tavsiye verilmelidir (bkz. B�l�m 4.6).
Bu t�bbi �r�n her dozunda 1 mmol (23 mg)'dan daha az sodyum ihtiva eder; yani asl�nda “sodyum
i�ermez”.
Bu t�bbi �r�n ayr�ca sodyum i�eren ��zeltilerle uygulama i�in haz�rlanabilir (bkz. B�l�m 4.2 ve 6.6) ve bu, hastaya uygulanacak t�m kaynaklardan elde edilen toplam sodyum ile ili�kili olarak d���n�lmelidir.
Biyoteknolojik �r�nlerin takip edilebilirli�inin sa�lanmas� i�in uygulanan �r�n�n ticari ismi ve seri
numaras� mutlaka hasta dosyas�na kaydedilmelidir.
4.5. Di�er t�bbi �r�nler ile etkile�imler ve di�er etkile�im �ekilleri
MYLOTARG ile klinik ila� etkile�imi �al��malar� yap�lmam��t�r. �n vitro �al��malardan elde edilen veriler i�in, bkz. B�l�m 5.2.
�zel pop�lasyonlara ili�kin ek bilgiler:
�zel pop�lasyonlara ili�kin herhangi bir etkile�im �al��mas� yap�lmam��t�r.
Pediyatrik pop�lasyon:
Pediyatrik pop�lasyona ili�kin herhangi bir etkile�im �al��mas� yap�lmam��t�r.
4.6. Gebelik ve laktasyon
Genel tavsiye
Gebelik kategorisi: D
�ocuk do�urma potansiyeli bulunan kad�nlar/Do�um kontrol� (Kontrasepsiyon)
�ocuk do�urma potansiyeli olan kad�nlara, MYLOTARG kullan�rken gebe kalmamaya dikkat etmeleri tavsiye edilmelidir.
�ocuk do�urma potansiyeli olan kad�nlara veya �ocuk do�urma potansiyeli olan kad�nlar�n partnerlerine, MYLOTARG ile tedavi s�ras�nda ve son dozdan sonraki en az 7 ayl�k (kad�nlarda) veya 4 ayl�k (erkeklerde) d�nem boyunca 2 etkin do�um kontrol y�ntemi kullanmalar� �nerilmelidir.
Gebelik d�nemi
Gebe kad�nlarda gemtuzumab ozogamisin kullan�m�yla ilgili olarak elde edilmi� veri yoktur ya da s�n�rl� say�da veri vard�r. Hayvanlar �zerinde yap�lan �al��malar, �reme toksisitesi bulundu�unu g�stermi�tir (bkz. B�l�m 5.3).
MYLOTARG, annenin elde edece�i potansiyel faydan�n, fet�s�n maruz kalabilece�i riskten daha y�ksek oldu�u durumlar haricinde gebelik s�ras�nda kullan�lmamal�d�r. Gebe kad�nlar ya da gemtuzumab ozogamisin kullan�m� s�ras�nda gebe kalan hastalar ya da gebe kad�nlar�n tedavi g�ren erkek partnerleri, fet�s �zerindeki bu olas� tehlike konusunda uyar�lmal�d�r.
Laktasyon d�nemi
�nsan s�t�nde gemtuzumab ozogamisin veya metabolitlerinin varl���, emzirilen �ocuktaki etkiler veya s�t �retimindeki etkilerle ilgili veri yoktur. Anne s�t� ile beslenen �ocuklarda advers ila� reaksiyonu potansiyeli bulundu�u i�in kad�nlar MYLOTARG tedavisi s�ras�nda ve son dozdan en az 1 ay sonras�na kadar emzirmemelidir (bkz. B�l�m 5.3).
�reme yetene�i/Fertilite
Hastalarda fertilite ile ilgili herhangi bir bilgi bulunmamaktad�r. Klinik d��� bulgulara dayal� olarak, erkek ve kad�n fertilitesi, gemtuzumab ozogamisin tedavisi ile zarar g�rebilir (bkz. B�l�m 5.3). Erkekler ve kad�nlar, tedavi �ncesinde fertilitenin korunmas� ile ilgili tavsiye almal�d�r.
4.7. Ara� ve makine kullan�m� �zerindeki etkiler
MYLOTARG'�n ara� ve makine kullan�m� �zerindeki etkisi orta d�zeydedir. Hastalara, MYLOTARG ile tedavi s�ras�nda yorgunluk, ba� d�nmesi ve ba� a�r�s� g�r�lebilece�i bilgisi verilmelidir (bkz. B�l�m 4.8). Bu nedenle, araba veya makine kullan�l�rken dikkatli olunmas� gerekir.
4.8. �stenmeyen etkiler
G�venlilik profilinin �zeti
MYLOTARG'�n genel g�venlilik profili, ALFA-0701 kombinasyonlu tedavi �al��mas�, monoterapi �al��malar� ve pazarlama sonras� deneyimde, akut miyeloid l�semili hastalardan elde edilen verileri temel almaktad�r. Kombinasyonlu tedavi �al��mas�nda, MYLOTARG'�n g�venlilik profilinin anla��lmas� i�in en �nemli kabul edilen ve se�ilmi� tedavi gerektiren yan etkilerden (TEAE'ler) olu�an g�venlilik verileri, t�m hemoraji derecelerini, t�m VOD derecelerini ve �iddetli enfeksiyonlar� i�ermi�tir. T�m bu TEAE'lerin, advers ila� reaksiyonlar� oldu�u belirlenmi�tir. Toplanan verilerin s�n�rl� olmas� nedeniyle, kombinasyon tedavisi �al��mas�ndan elde edilen laboratuvar verileri ve fraksiyone olmayan rejimin kullan�ld��� monoterapi �al��malar� (201/202/203 �al��malar�), pazarlama sonras� deneyim ve fraksiyone rejimin kullan�ld��� B1761031 monoterapi �al��mas�ndan elde edilen advers ila� reaksiyonlar� bilgisi, advers ila� reaksiyonlar�n�n tam karakterizasyonunun sa�lanmas� amac�yla a�a��da sunulmu�tur.
ALFA-0701 kombinasyonlu tedavi �al��mas�nda, klinik a��dan anlaml� ciddi advers ila� reaksiyonlar VOD/SOS dahil hepatotoksisite (% 3,8), hemoraji (% 9,9), �iddetli enfeksiyon (% 41,2) ve t�m�r lizis sendromudur (% 1,5). Monoterapi �al��malar�nda (201/202/203 �al��malar�), klinik a��dan anlaml� ciddi advers ila� reaksiyonlar�, inf�zyonla ili�kili reaksiyonlar (% 2,5), trombositopeni (% 21,7) ve n�tropenidir (% 34,3). Monoterapi �al��mas� B1761031'de, klinik a��dan anlaml� ciddi advers ila� reaksiyonlar�, enfeksiyon (%30), febril n�tropeni (%22), pireksi (%6), hemoraji (%4), trombositopeni
(%4), anemi (%2), ve ta�ikardidir (%2).
Kombinasyonlu tedavi �al��mas�nda en yayg�n advers ila� reaksiyonlar (> %30) hemoraji ve enfeksiyon olmu�tur. Monoterapi �al��malar�nda (201/202/203 �al��malar�), en yayg�n advers ila� reaksiyonlar� (> %30) pireksi, mide bulant�s�, enfeksiyon, �rperme, hemoraji, kusma, trombositopeni, yorgunluk, ba� a�r�s�, stomatit, ishal, kar�n a�r�s� ve n�tropenidir. Monoterapi �al��mas� B1761031'de en s�k g�r�len advers ila� reaksiyonlar� (> %30), enfeksiyon (%50), febril n�tropeni (%40) ve hemorajidir (%32).
Kombinasyonlu tedavi �al��mas�nda tedavinin kal�c� olarak kesilmesine yol a�an en s�k g�r�len (≥% 1) advers ila� reaksiyonlar�, trombositopeni, VOD, hemoraji ve enfeksiyondur. Monoterapi �al��malar�nda (201/202/203 �al��malar�) tedavinin kal�c� olarak kesilmesine yol a�an en s�k g�r�len (≥% 1) advers ila� reaksiyonlar�, enfeksiyon, hemoraji, �oklu organ yetmezli�i ve VOD'dir.
Monoterapi �al��mas� B1761031'de kal�c� olarak tedavinin kesilmesine yol a�an advers ila� reaksiyonlar�, enfeksiyon ve pireksidir.
�stenmeyen etkiler MedDRA sistemi organ s�n�fland�rmas� baz al�narak a�a��daki kategorilere g�re listelenmi�tir:
�ok yayg�n (≥1/10); yayg�n (≥1/100 ila <1/10); yayg�n olmayan (≥1/1.000 ila <1/100); seyrek (≥1/10.000 ila <1/1.000); �ok seyrek (<1/10.000); bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor) Her s�kl�k grubunda istenmeyen etkiler azalan ciddiyet s�ralamas� ile sunulmaktad�r.
Kombinasyonlu tedavi �al��mas�nda (ALFA-0701) MYLOTARG alan hastalarda se�ilen**
advers ila� reaksiyonlar
Enfeksiyonlar ve enfestasyonlar
�ok yayg�n : Enfeksiyon
Vask�ler hastal�klar
�ok yayg�n : Hemoraji
Hepato-biliyer hastal�klar
Yayg�n : Venookl�zif karaci�er hastal���
Ara�t�rmalar***
�ok yayg�n : Hemoglobin azalmas�, trombosit azalmas�, akyuvar azalmas�, lenfosit (mutlak) azalmas�, n�trofil azalmas�, hiperglisemi, aspartat aminotransferaz (AST) art���, protrombin zaman� art���, aktive parsiyel tromboplastin zaman� art���, alkalen fosfataz art���, alanin aminotransferaz (ALT) art���, kan bilirubin art���, hiper�risemi
![]()
S�kl�k, laboratuvar de�erlerini temel al�r (derece NCI CTCAE v4.03'e g�re).
Monoterapi*** �al��malar�nda MYLOTARG alan hastalarda ve pazarlama sonras�
deneyimdeki advers ila� reaksiyonlar
Enfeksiyonlar ve enfestasyonlar
�ok yayg�n : Enfeksiyon
Kan ve lenf sistemi hastal�klar�
�ok yayg�n : Febril n�tropeni, trombositopeni, n�tropeni, anemi, l�kopeni Yayg�n : Pansitopeni, lenfopeni
Ba����kl�k sistemi hastal�klar�
Yayg�n : �nf�zyonla ili�kili reaksiyon
Metabolizma ve beslenme hastal�klar� �ok yayg�n : Hiperglisemi, i�tah azalmas� Yayg�n : T�m�r lizis sendromu
Sinir sistemi hastal�klar�
�ok yayg�n : Ba� a�r�s�
Kardiyak hastal�klar�
�ok yayg�n : Ta�ikardi
Vask�ler hastal�klar
�ok yayg�n : Hemoraji, hipotansiyon, hipertansiyon
Solunum, g���s bozukluklar� ve mediastinal hastal�klar
�ok yayg�n : Dispne
Bilinmiyor : �nterstisyel pn�moni
Gastrointestinal hastal�klar
�ok yayg�n : Kusma, ishal, kar�n a�r�s�, mide bulant�s�, stomatit, kab�zl�k Yayg�n : Assit, dispepsi, �zofajit
Bilinmiyor : N�tropenik kolit
Hepato-biliyer hastal�klar
�ok yayg�n : Transaminaz art���, bilir�bin d�zeyinde art�� (hiperbilirubinemi)
Yayg�n : Venookl�zif karaci�er hastal���, hepatomegali, sar�l�k, hepatik i�levde anomali,
gama-glutamiltransferaz art���
Yayg�n olmayan: Hepatik yetmezlik, Budd-Chiari sendromu
Deri ve deri alt� doku hastal�klar�
�ok yayg�n : D�k�nt� Yayg�n : Eritem, prurit
B�brek ve idrar yolu hastal�klar�
Bilinmiyor : Hemorajik sistit
Genel bozukluklar ve uygulama b�lgesine ili�kin rahats�zl�klar
�ok yayg�n : Pireksi, �dem, yorgunluk, �rperme Yayg�n : �oklu organ yetmezli�i
Ara�t�rmalar
�ok yayg�n : Kan laktat dehidrojenaz art��� Yayg�n : Kan alkalen fosfat art���
![]()
![]()
ta�ikardi.
kar�n hassasiyeti.
aminotransferaz art���, aspartat aminotransferaz art��� ve karaci�er enzim art���.
hepatik fonksiyon anomalisi.
Se�ilmi� advers reaksiyonlar�n tan�m�
Hepatik VOD/SOS dahil hepatotoksisite
Kombinasyonlu tedavi �al��mas�nda, VOD ve karaci�er laboratuvar anormallikleri hakk�nda veriler toplanm��t�r. Hepatotoksisite advers reaksiyonlar�n�n ilave karakterizasyonu monoterapi �al��malar�ndan sa�lanm��t�r.
Kombinasyonlu tedavi �al��mas�nda (N=131) 6 (% 4,6) hastada tedavi s�ras�nda veya sonras�nda VOD rapor edilmi�tir, bu reaksiyonlar�n 2'si (% 1,5) �l�mc�l olmu�tur. Bu VOD reaksiyonlar�n�n be�i (% 3,8), herhangi bir gemtuzumab ozogamisin dozundan sonra 28 g�n i�inde ger�ekle�mi�tir. Bir VOD olgusu, son gemtuzumab ozogamisin dozundan 28 g�nden fazla s�re sonra ger�ekle�mi�, bu olgular�n 1'i, bir HKHN haz�rlama rejimine ba�land�ktan birka� g�n sonra meydana gelmi�tir. Son gemtuzumab ozogamisin dozundan, VOD ba�lang�c�na kadar olan medyan s�re 9 g�n olmu�tur (aral�k: 2-298 g�n). VOD, kombinasyonlu tedavi �al��mas�n�n kontrol kolunda kemoterapi sonras�nda AML relaps�n�n ard�ndan bir takip tedavisi olarak MYLOTARG alan 2 hastada da rapor edilmi�tir. Bu hastalar�n ikisi de, son gemtuzumab ozogamisin dozundan 28 g�nden uzun s�re sonras�nda VOD deneyimlemi�tir. Bu hastalar�n biri, m�teakip HKHN'den 25 g�n sonra VOD ya�am��t�r.
Monoterapi �al��mas� B1761031'de hi�bir hasta i�in VOD bildirilmemi�tir. Ancak, 1 (%2) hastada VOD ile uyumlu semptomlar (asit ve hiperbilirubinemi) ile �l�mc�l kapiller ka��� sendromu g�r�lm��t�r. Derece 3 hepatotoksisite olgular� aras�nda gama-glutamiltransferaz art��� (%4), alanin aminotransferaz art��� (%2), aspartat aminotransferaz art��� (%2), hipoalb�minemi (%2) ve transaminazlarda art�� (%2) yer alm��t�r. Hi�bir hastada Derece 4 veya Derece 5 hepatotoksisite g�r�lmemi�tir.
Potansiyel risk fakt�rlerinin bir analizine g�re, monoterapi olarak fraksiyone olmayan MYLOTARG alan yeti�kin hastalar aras�nda, gemtuzumab ozogamisine maruz kalmadan �nce bir HKHN alan hastalarda VOD geli�me olas�l���, gemtuzumab ozogamisinle tedaviden �nce HKHN almam�� olan hastalara g�re 2,6 kat fazla olmu�tur (% 95 g�ven aral��� [GA]: 1,448-4,769); gemtuzumab ozogamisinle tedavinin ard�ndan bir HKHN alan hastalarda VOD geli�me olas�l���, gemtuzumab ozogamisinle tedavinin ard�ndan HKHN almayan hastalara g�re 2,9 kat fazla olmu�tur (% 95 GA: 1,502-5,636) ve ba�lang��ta orta seviye/�iddetli karaci�er yetmezli�i olan hastalarda VOD geli�me olas�l���, ba�lang��ta orta seviye/�iddetli karaci�er yetmezli�i olmayan hastalara g�re 8,7 kat fazla olmu�tur (% 95 GA: 1,879-39,862).
Hastalar, B�l�m 4.4'te �nerildi�i gibi hepatotoksisite i�in izlenmelidir. Hepatik toksisite belirtilerinin veya semptomlar�n�n y�netiminde, MYLOTARG dozuna ara verilmesi veya dozun kesilmesi gerekebilir (bkz. B�l�m 4.2).
Miyelos�presyon
Daha �nce tedavi edilmemi� de novo AML'li olan ve kemoterapiyle birlikte fraksiyone gemtuzumab ozogamisin dozlar�yla tedavi edilen hastalar �zerinde yap�lan kombinasyonlu tedavi �al��mas�nda, l�kosit, n�trofil ve trombositlerdeki 3/4. derece azalmalar s�ras�yla 131 (% 100), 124 (% 96,1) ve 131 (% 100) hastada g�zlenmi�tir.
�nd�ksiyon faz�nda, trombosit say�lar�nda 50.000/mm ve 100.000/mm de�erlerine kadar d�zelme s�ras�yla 109 (% 83,2) ve 99 (% 75,6) hastada g�r�lm��t�r. 50.000/mm ve 100.000/mm de�erlerine kadarki trombosit say�lar�ndaki d�zelmesi i�in medyan s�reler s�ras�yla 34 ve 35 g�n olmu�tur. Birinci konsolidasyon faz�nda, 50.000/mm ve 100.000/mm trombosit say�lar�na d�zelme s�ras�yla 92 (% 94,8) ve 71 (% 73,2) hastada g�r�lm��t�r. Bu d�zelmelerin medyan s�releri 50.000/mm ve 100.000/mm trombosit de�erleri i�in s�ras�yla 32 ve 35 g�n olmu�tur. �kinci konsolidasyon faz�nda, 50.000/mm ve 100.000/mm trombosit say�lar�na d�zelme s�ras�yla 80 (% 97,6) ve 70 (% 85,4) hastada g�r�lm��t�r. Bu d�zelmelerin medyan s�releri 50.000/mm ve 100.000/mm trombosit de�erleri i�in s�ras�yla 36,5 ve 43 g�n olmu�tur.
Yan�t veren hastalar i�in tedaviye ba�land�ktan sonra trombosit say�lar� 45 g�n boyunca <50.000/mm de�erinde kalan trombositopeni (CR ve tamamlanmam�� trombosit d�zelmesi [CRp]), 22 (% 20,4) hastada meydana gelmi�tir. Kal�c� trombositopenisi olan hasta say�s� tedavi fazlar� aras�nda benzer olmu�tur (ind�ksiyon faz�nda 8 [% 7,4] hasta, birinci konsolidasyon faz�nda 8 [% 8,5] hasta ve ikinci konsolidasyon faz�nda 10 [% 13,2] hasta).
�nd�ksiyon faz�nda, 500/mm ve 1.000/mm MNS de�erlerine kadar n�trofil toparlanmas� s�ras�yla 121 (% 92,4) ve 118 (% 90,1) hastada g�r�lm��t�r. 500/mm ve 1.000/mm MNS de�erlerine kadar n�trofil toparlanmas� i�in medyan s�re 25 g�n olarak belirlenmi�tir. Tedavinin birinci konsolidasyon faz�nda, 94 (% 96,9) hastan�n n�trofil say�lar�ndaki toparlanma 500/mm ve 91 (% 94) hastan�n n�trofil say�lar�ndaki toparlanma 1000/mm olarak belirlenmi�tir. 500/mm ve 1000/mm MNS de�erlerine kadar n�trofil toparlanmas� i�in medyan s�reler s�ras�yla 21 ve 25 g�n olarak
belirlenmi�tir. Tedavinin ikinci konsolidasyon faz�nda, 80 (% 97,6) hastan�n n�trofil say�lar�ndaki toparlanma 500/mm ve 79 (% 96,3) hastan�n n�trofil say�lar�ndaki toparlanma 1.000/mm olarak belirlenmi�tir. 500/mm ve 1.000/mm MNS de�erlerine kadar n�trofil toparlanmas� i�in medyan s�reler s�ras�yla 22 ve 27 g�n olarak belirlenmi�tir.
Kombinasyonlu tedavi �al��mas�nda, de novo AML'li olan ve kemoterapiyle kombine fraksiyone gemtuzumab ozogamisin dozlar�yla tedavi edilen hastalar�n (N=131) 102'si (% 77,9), t�m tedaviyle ili�kili �iddetli (derece ≥3) enfeksiyonlar deneyimlemi�tir. Septik �ok nedeniyle tedaviye ba�l� �l�m 1 (% 0,8) hastada rapor edilmi�tir. �l�mc�l �iddetli enfeksiyon, MYLOTARG kolundaki 2 (% 1,53) hastada ve kontrol kolundaki 4 (% 2,92) hastada rapor edilmi�tir.
Kombinasyonlu tedavi �al��mas�nda (N=131), t�m derecelerde ve 3/4. derece kanama/hemorajik reaksiyon, s�ras�yla 118 (% 90,1) ve 27 (% 20,6) hastada rapor edilmi�tir. En s�k g�r�len 3. derece kanama/hemorajik reaksiyon, hematemez (% 3,1), hemoptizi (% 3,1) ve hemat�ri (% 2,3) olmu�tur.
4. derece kanama/hemorajik reaksiyonlar, 4 (% 3,1) hastada rapor edilmi�tir (gastrointestinal hemoraji, hemoraji ve pulmoner alveolar hemoraji [2 hasta]). �l�mc�l kanama/hemorajik reaksiyonlar 3 (% 2,3) hastada rapor edilmi�tir (serebral hematom, intrakraniyal hematom ve subdural hematom).
Monoterapi �al��mas� B1761031'de (N=50), 10 (%20) hastada Derece 3/4 enfeksiyonlar rapor edilmi�tir. En s�k (≥%5) bildirilen Derece 3/4 enfeksiyonlar, her biri 3 (%6) hastada g�r�lm�� olan sepsis ve pn�monidir. Alt� (6) (%12) hastada Derece 5 enfeksiyon g�r�lm��t�r (4 [%8] hastada sepsis,
4 [%8] hastada atipik pn�moni ve 1 hastada [%2] COVID-19 pn�monisi). T�m derecelerde
kanama/hemorajik olgular 16 (%32) hastada rapor edilmi�tir. 2 (%4) hastada Derece 3/4 hemorajik olgu meydana gelmi�tir (her biri 1 hastada olmak �zere; mide kanamas� Derece 3 ve travmatik intrakraniyal kanama Derece 4). �l�mc�l kanama/hemorajik olgu bildirilmemi�tir.
�iddetli enfeksiyon, kanama/hemoraji veya �iddetli n�tropeni veya kal�c� trombositopeni dahil miyelos�presyonun di�er etkileri g�r�len hastalar�n y�netiminde, MYLOTARG dozunun ertelenmesi veya kal�c� olarak kesilmesi gerekebilir (bkz. B�l�m 4.2 ve 4.4).
�mm�nojenite
T�m terap�tik proteinlerle oldu�u gibi, imm�nojenite potansiyeli vard�r.
Monoterapi �al��mas� B1761031'de relaps veya refrakter CD-33-pozitif AML'li 50 yeti�kin hastada, MYLOTARG'a kar�� anti-ila� antikoru (ADA), elektrokemil�minesans (ECL) y�ntemi kullan�larak de�erlendirilmi�tir. ADA numuneleri pozitif ��kan hastalar i�in, MYLOTARG'a kar�� n�tralize edici antikoru (Nab) �l�mek amac�yla h�cre bazl� bir test geli�tirilmi�tir.
ADA ve NAb insidans� s�ras�yla 6 (%12) ve 1 (%2) dir. ADA varl���n�n, toplam hP67.6 antikorunun veya konjuge kalikeamisinin farmakokineti�i �zerinde istatistiksel olarak anlaml� veya klinik olarak bir etkisi olmam��t�r. Hastalar�n hi�birinde ADA ile ili�kili anafilaksi, a��r� duyarl�l�k veya di�er klinik sekeller g�r�lmemi�tir. ADA varl���n�n herhangi bir potansiyel g�venlilik durumu ile do�rudan bir ili�kisi oldu�una dair hi�bir kan�t yoktur.
ADA'lar�n saptanmas�, y�ksek d�zeyde, miktar tayininin duyarl�l���na ve �zg�ll���ne ba�l�d�r. Miktar tayinindeki antikor pozitifli�inin insidans�, miktar tayini y�ntemi, dola��mdaki gemtuzumab ozogamisin konsantrasyonlar�, numunenin i�lenmesi, numune alma zamanlamas�, e�lik eden tedaviler ve altta yatan hastal�k gibi �e�itli fakt�rlerden etkilenebilir. Bu nedenlerle, gemtuzumab ozogamisine
kar�� antikor geli�me s�kl���n�n, di�er �r�nlere kar�� antikor geli�me s�kl��� ile kar��la�t�r�lmas� yan�lt�c� olabilir.
Pediyatrik pop�lasyon
Daha �nce tedavi edilmemi� AML
MYLOTARG'�n daha �nce tedavi edilmemi� AML'li �ocuklarda ve 15 ya��n alt�ndaki adolesanlardaki g�venlili�i ve etkilili�i belirlenmemi�tir (bkz. B�l�m 4.2).
0 ila 29 ya� aral���nda de novo AML'li yeni tan� konmu� 1.063 �ocuk (hastalar�n % 93,7'si 18 ya� alt�) ve gen� eri�kinde (hastalar�n % 6,3'�), yo�un birinci basamak tedaviyle kombine olarak verilen gemtuzumab ozogamisin ile yap�lan tamamlanm�� AAML0531 Faz 3 randomize pediyatrik �al��mada (bkz. B�l�m 5.1), gemtuzumab ozogamisinin g�venlilik profili, de novo AML'li yeti�kin hastalarda a��r kemoterapiyle kombine �ekilde verilen gemtuzumab ozogamisin �zerinde yap�lan di�er �al��malarda g�zlenen profile benzer olmu�tur. Ancak, AAML0531 �al��mas�nda ikinci gemtuzumab ozogamisin dozundan sonra ikinci konsolidasyon d�neminde, gemtuzumab ozogamisin kolunda n�trofil toparlanma s�resi uzam�� olan (> 59 g�n) hasta oran�, kar��la�t�rma koluna k�yasla daha y�ksek oldu�u i�in (% 21'e kar��l�k % 11,5) ve remisyonda daha fazla hasta hayat�n� kaybetti�i i�in (% 5,5'ye kar��l�k % 2,8), pediyatrik hastalarda kullan�lacak optimum gemtuzumab ozogamisin dozu belirlenmemi�tir.
Relaps veya refrakter AML
Relaps veya refrakter AML'li pediyatrik hastalarda MYLOTARG'�n g�venlili�i ve etkilili�i belirlenmemi�tir (bkz. B�l�m 4.1 ve 4.2).
Pediyatrik hastalarda MYLOTARG'� de�erlendiren �al��malar�n sistematik bir literat�r taramas�nda g�zlenen g�venlilik sonu�lar� (bkz. B�l�m 5.1) Tablo 5'te sunulmu�tur.
Tablo 5. MYLOTARG alan relaps veya refrakter AML'li pediyatrik hastalarda sistematik bir literat�r taramas�ndan elde edilen g�venlilik sonu�lar�
| ||||||||||||
| ||||||||||||
| ||||||||||||
| ||||||||||||
![]()
��pheli advers reaksiyonlar�n raporlanmas�
Ruhsatland�rma sonras� ��pheli ila� advers reaksiyonlar�n�n raporlanmas� b�y�k �nem ta��maktad�r. Raporlama yap�lmas�, ilac�n yarar/risk dengesinin s�rekli olarak izlenmesine olanak sa�lar. Sa�l�k mesle�i mensuplar�n�n herhangi bir ��pheli advers reaksiyonu T�rkiye Farmakovijilans Merkezi (T�FAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e- posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks: 0 312 218 35 99)
4.9. Doz a��m� ve tedavisi
Klinik deneyimde, MYLOTARG ile doz a��m� vakalar� rapor edilmemi�tir. Yeti�kinlerde 9 mg/m'den y�ksek tekil dozlar test edilmemi�tir. MYLOTARG doz a��m� tedavisi, genel destekleyici �nlemlerden olu�mal�d�r.
5. FARMAKOLOJ�K �ZELL�KLER
5.1. Farmakodinamik �zellikler
Farmakoterap�tik grup: Antineoplastik ajanlar, monoklonal antikorlar ve antikor ila� konjugatlar�, di�er monoklonal antikorlar ve antikor ila� konjugatlar�
ATC kodu: L01FX02.
Etki mekanizmas�
Gemtuzumab ozogamisin, CD33-hedefli bir antikor ila� konjugat� (ADC)'dir. Gemtuzumab, �zellikle insan CD33'�n� tan�yan bir h�manize imm�noglobulin s�n�f G alt tip 4 (IgG4) antikorudur. Antikor b�l�m�, miyeloid l�semik blastlar�n veya miyelomonositik seri immat�r normal h�crelerin y�zeyinde bulunan, fakat normal hematopoetik k�k h�crelerinin y�zeyinde bulunmayan, siyalik aside ba��ml� bir adhezyon proteini olan CD33 antijenine �zel olarak ba�lan�r. K���k molek�l, N-asetil-gama- kalikeamisin, sitotoksik yar� sentetik bir do�al �r�nd�r. N-asetil-gama-kalikeamisin, bir AcBut (4-(4- asetilfenoksi) b�tanoik asit) ba�lay�c� arac�l���yla antikora kovalent olarak ba�lan�r. Klinik olmayan veriler gemtuzumab ozogamisinin antikanser aktivitesinin; ADC'nin CD33- ekprese eden kanser h�crelerine ba�lanmas�n�n, ADC-CD33 kompleksinin h�cre i�ine al�nmas� sonras� ba�lay�c�n�n hidrolitik yolla par�alanmas�yla N-asetil-gama-kalikeamisin dimetilhidrazidin h�cre i�inde serbest kalmas� yoluyla oldu�unu g�stermektedir. N-asetil-gama-kalikeamisin dimetilhidrazidin aktivasyonu, �ift sarmall� DNA k�r�lmalar�na neden olur ve sonras�nda h�cre d�ng�s�n� durdurma ve apoptotik h�cre �l�m�n� tetikler.
Kalikeamisinin l�semik blast h�crelerine en y�ksek d�zeydeiletilmesi i�in, CD33 antijenik b�lgelerinin y�ksek bir y�zdesinin sat�rasyonunun gerekece�i kabul edilmektedir. �e�itli monoterapi �al��malarda, relaps ya da refrakter AML'li hastalarda MYLOTARG dozu sonras�ndaki CD33 sat�rasyonu �l��lm��t�r. T�m �al��malarda, 2 mg/m² ve �st� d�zeylerdeki MYLOTARG dozunun ard�ndanen y�ksek de�ere yak�n periferik CD33 sat�rasyonu g�zlenmi�tir, bu da d���k bir gemtuzumab ozogamisin dozunun mevcut t�m CD33 b�lgelerini ba�lamak i�in yeterli oldu�unu g�stermektedir.
Klinik etkililik ve g�venlilik
De novo AML'li olup daha �nce tedavi edilmeyen hastalar �zerindeki ALFA-0701 �al��mas�
Daunorubisin ve sitarabinden (DA) olu�an standart bir kemoterapi ind�ksiyonuna MYLOTARG eklenmesinin, tek ba��na DA ile kar��la�t�r�ld��� �ok merkezli, randomize, a��k etiketli bir Faz 3 �al��mas�nda, MYLOTARG'�n etkilili�i ve g�venlili�i de�erlendirilmi�tir. Uygun hastalar, de novo AML'li olup daha �nce tedavi edilmeyen 50 ve 70 ya� aras�ndaki ki�iler olmu�tur (ALFA-0701 �al��mas�). Akut promiyelositik l�semili hastalar (APL, AML3) ve miyelodisplastik sendrom kaynakl� AML hastalar� (MDS) ya da ikincil AML'li hastalar �al��man�n d���nda tutulmu�tur.
Birincil sonlan�m noktas�, olays�z sa�kal�m (EFS) olmu�tur. �kincil sonlan�m noktalar� CR ve CRp oranlar�n�, relapss�z sa�kal�m� (RFS), genel sa�kal�m� (OS) ve MYLOTARG dahil edildi�inde veya edilmedi�ine kombinasyon DA'n�n g�venlili�i olmu�tur.
Bu �al��mada, 135'i 3+7 DA ile birlikte fraksiyone 3 mg/m × 3 doz MYLOTARG ve 136's� tek ba��na 3+7 DA ind�ksiyon tedavisiyle, toplamda 271 hasta randomize edilmi�tir (bkz. B�l�m 4.2). Randomizasyon koluna bak�lmaks�z�n, DA olarak uygulanan ancak MYLOTARG i�ermeyen ikinci bir ind�ksiyon k�r�ne izin verilmi�tir. Bu iki koldan birinde yer alan, ikinci ind�ksiyon tedavisi k�r�n� almayan ve ind�ksiyonun ard�ndan bir CR elde etmeyen hastalar, idarubisin, AraC ve gran�losit koloni stim�le edici fakt�rden (G-CSF) olu�an bir kurtarma k�r� alabilmi�tir.
CR veya CRp'li hastalar, ilk randomizasyonlar�na g�re MYLOTARG dahil edilerek veya edilmeyerek DNR ve AraC'yi i�eren 2 tedavi k�r�yle konsolidasyon tedavisi alm��t�r. Remisyona giren hastalar, allojenik transplantasyon i�in de uygun olmu�tur. Son MYLOTARG dozu ve transplantasyon aras�nda en az 2 ayl�k bir s�re olmas� �nerilmi�tir.
Genel olarak, hastalar�n medyan ya�� 62 olmu� (aral�k 50 ila 70 ya�) ve �o�u hastan�n (% 87,8) Do�u Kooperatif Onkoloji Grubu (Eastern Cooperative Oncology Group) performans durumu (ECOG PS) ba�lang��ta 0 ila 1 olmu�tur. Tedavi kollar� aras�nda ba�lang��taki �zellikler dengeli olmu�tur, bunun tek istisnas� olan cinsiyette, kaydolan erkek y�zdesi tek ba��na DA koluna (% 44,1) k�yasla MYLOTARG kolunda (% 54,8) daha y�ksek olmu�tur. Amerikan Ulusal Geli�mi� Kanser A�� (National Comprehensive Cancer Network) (NCNN) ve European LeukaemiaNet (ELN) 2010 risk s�n�fland�rmalar�yla, toplamda hastalar�n s�ras�yla % 59'unda ve % 65,3'�nde iyi/orta riskli hastal�k belgelenmi�tir. Yerel laboratuvar sonu�lar�ndan uyumlu hale getirilen ak�m sitometrisiyle, AML blastlar� �zerindeki CD33 ekspresyonu toplamda 194/271 (% 71,6) hastada belirlenmi�tir. Az say�da hastada (% 13,7) CD33 ekspresyonu d���k bulunmu�tur (blastlar�n % 30'undan az�).
�al��mada, de novo AML'li olup daha �nce tedavi edilmemi� hastalar i�in standart ind�ksiyon kemoterapisine fraksiyone dozlarda eklenen MYLOTARG'�n (3 mg/m × 3 doz) EFS'de istatistiksel a��dan �nemli ve klinik a��dan anlaml� iyile�meye yol a�t��� ortaya konarak, birincil sonlan�m noktas�na ula��lm��t�r. Medyan EFS, MYLOTARG kolunda 17,3 ay (% 95 GA: 13,4-30) ve tek ba��na DA kolunda 9,5 ay (% 95 GA: 8,1-12) olmu�tur; tehlike oran� (HR) 0,562 (% 95 GA: 0,415-0,762); log-rank testiyle 2-y�nl� p=0,0002. ALFA-0701 �al��mas�nda elde edilen etkililik verileri Tablo 6'da �zetlenmektedir ve EFS i�in Kaplan-Meier grafi�i �ekil 1'de sunulmaktad�r.
Tablo 6. ALFA 0701 �al��mas�ndan etkililik sonu�lar� (mITT pop�lasyonu)
| MYLOTARG + daunorubisin + sitarabin |
Daunorubisin + sitarabin |
Olays�z sa�kal�m (Ara�t�rmac� incelemesi) | N=135 | N=136 |
Olay say�s�, n (%) | 73 (54,1) | 102 (75) |
Medyan EFS (ay olarak) [% 95 GA] | 17,3 [13,4-30] | 9,5 [8,1-12] |
2 y�ll�k EFS olas�l��� [% 95 GA] | 42,1 [32,9-51] | 18,2 [11,1-26,7] |
3 y�ll�k EFS olas�l��� [% 95 GA] | 39,8 [30,2-49,3] | 13,6 [5,8-24,8] |
Tehlike oran� [% 95 GA] | 0,562 [0,415, 0,762] |
|
p-de�eri | 0,0002 |
|
Relapss�z sa�kal�m (Ara�t�rmac� incelemesi) |
N=110 |
N=100 |
Olay say�s�, n (%) | 49 (44,5) | 66 (66) |
Medyan RFS (ay olarak) [% 95 GA] | 28 [16,3-NE] | 11,4 [10-14,4] |
Tehlike oran� [% 95 GA] | 0,526 [0,362-0,764] |
|
p-de�eri | 0,0006 |
|
Genel sa�kal�m | N=135 | N=136 |
�l�m say�s�, n (%) | 80 (59,3) | 88 (64,7) |
Medyan OS (ay olarak) [% 95 GA] | 27,5 [21,4-45,6] | 21,8 [15,5-27,4] |
Tehlike oran� [% 95 GA] | 0,807 [0,596-1,093] |
|
p-de�eri | 0,1646 |
|
Yan�t oran� (Ara�t�rmac� incelemesi) | N=135 | N=136 |
Genel yan�t % [% 95 GA] | 81,5 [73,89-87,64] | 73,5 [65,28-80,72] |
CR | 70,4 | 69,9 |
CRp | 11,1 | 3,7 |
Risk fark� [% 95 GA] | 7,95[-3,79-19,85] |
|
p-de�eri | 0,1457 |
|
EFS'nin ana tan�m�na dayan�larak: Ara�t�rmac� de�erlendirmesiyle belirlenen olay tarihleri (ind�ksiyon ba�ar�s�zl���, relaps ya da �l�m).
mITT pop�lasyonu, tedaviye ba�lanmadan �nce onam�n geri �ekilmedi�i takdirde randomize edilen ve ilk randomizasyon koluna g�re analiz edilen t�m hastalar� i�ermi�tir.
K�saltmalar: CR=Tam remisyon; CRp=Tamamlanmam�� trombosit d�zelmesiyle tam remisyon; GA=G�ven aral���; EFS=Olays�z sa�kal�m; mITT=Modifiye ITT; n=Say�; N=Say�; NE=Tahmin edilemez; OS=Genel sa�kal�m; RFS=Relapss�z sa�kal�m.
Sa�kal�m Olas�l���
�ekil 1. ALFA-0701 �al��mas�ndan ara�t�rmac� de�erlendirmesiyle Kaplan-Meier olays�z sa�kal�m grafi�i (mITT pop�lasyonu)
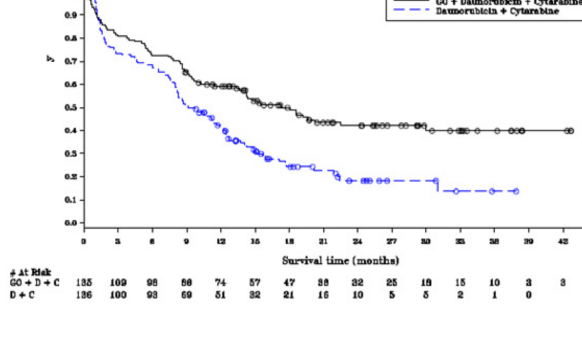
Notlar: Daireler, sans�rlenen g�zlemleri belirtmektedir.
D + C, Daunorubisin + Sitarabin'i ifade etmektedir.
Sa�kal�m s�resi (Ay)
- GO + Daunorubisin + Sitarabin
- Daunorubisin + Sitarabin
K�saltmalar: C=Sitarabin; D=Daunorubisin; GO=Gemtuzumab ozogamisin; mITT= Modifiye ITT.
Advers risk sitogenetikleriyle AML'de kullan�m
ALFA-0701 �al��mas�ndaki alt grup analizlerinde, standart kombine kemoterapiye MYLOTARG eklenmesiyle, advers risk sitogenetikleri olan hasta alt grubunda EFS'de iyile�me olmam��t�r (HR 1,11; % 95 GA: 0,63-1,95). Sitogenetik risk s�n�fland�rmas� ve sitogenetik/molek�ler risk s�n�fland�rmas�yla analiz edilen EFS ve OS, a�a��da Tablo 7'da ve 8'de sunulmaktad�r.
Tablo 7. ALFA-0701 �al��mas�ndan AML risk s�n�fland�rmalar�yla ara�t�rmac� de�erlendirmesinden olays�z sa�kal�m (mITT Pop�lasyonu)
| MYLOTARG + daunorubisin + sitarabin | Daunorubisin + sitarabin |
Sitogenetik (iyi/orta), N | 94 | 95 |
Olay say�s�, n (%) | 44 (46,8) | 68 (71,6) |
Medyan EFS (ay olarak) [% 95 GA] | 22,5 [15,5-NE] | 11,6 [8,3-13,7] |
Tehlike oran� [% 95 GA] | 0,460 [0,313-0,676] |
|
p-de�eri | < 0,0001 |
|
Sitogenetik (advers), N | 27 | 30 |
Olay say�s�, n (%) | 23 (85,2) | 26 (86,7) |
Medyan EFS (ay olarak) [% 95 GA] | 4,5 [1,1-7,4] | 2,8 [1,6-8,7] |
Tehlike oran� [% 95 GA] | 1,111 [0,633-1,949] |
|
p-de�eri | 0,7151 |
|
ELN (iyi/orta), N | 86 | 91 |
Olay say�s�, n (%) | 40 (46,5) | 63 (69,2) |
Medyan EFS (ay olarak) [% 95 CI] | 22,5 [15,5-NE] | 12,2 [8,5-14,3] |
Tehlike oran� [% 95 GA] | 0,485 [0,325-0,724] |
|
p-de�eri | 0,0003 |
|
ELN (zay�f/advers), N | 37 | 36 |
Olay say�s�, n (%) | 27 (73) | 32 (88,9) |
Medyan EFS (ay olarak) [% 95 GA] | 7,4 [3,7-14,3] | 4 [1,7-8,6] |
Tehlike oran� [% 95 GA] | 0,720 [0,430-1,205] |
|
p-de�eri | 0,2091 |
|
ALFA-0701 �al��mas�, MYLOTARG'�n alt gruplardaki faydas�n� gelece�e y�nelik olarak de�erlendirmek i�in tasarlanmam��t�r; analiz yaln�zca tan�mlay�c� ama�larla sunulmu�tur.
EFS'nin ana tan�m�na dayan�larak: Ara�t�rmac� de�erlendirmesiyle belirlenen olay tarihleri (ind�ksiyon ba�ar�s�zl���, relaps ya da �l�m).
mITT pop�lasyonu, tedaviye ba�lanmadan �nce onam�n geri �ekilmedi�i takdirde randomize edilen ve ilk randomizasyon koluna g�re analiz edilen t�m hastalar� i�ermi�tir.
K�saltmalar: AML=Akut miyeloid l�semi; GA=G�ven aral���; EFS=Olays�z sa�kal�m; ELN=European LeukaemiaNet; mITT= Modifiye ITT; n=Say�; N=Say�; NE=Tahmin edilemez.
Tablo 8. ALFA-0701 �al��mas�ndan AML risk s�n�fland�rmalar�yla genel sa�kal�m (mITT Pop�lasyonu)
| MYLOTARG + daunorubisin + sitarabin | Daunorubisin + sitarabin |
Sitogenetik (iyi/orta), N | 94 | 95 |
�l�m say�s�, n (% ) | 51 (54,3) | 57 (60) |
Medyan OS (ay olarak) [% 95 GA]a | 38,6 [24,4-NE] | 26 [18,9-39,7] |
Tehlike oran� [% 95 GA]b | 0,747 [0,511-1,091] |
|
p-de�eric | 0,1288 |
|
Sitogenetik (advers), N | 27 | 30 |
�l�m say�s�, n (% ) | 24 (88,9) | 24 (80) |
Medyan OS (ay olarak) [% 95 GA]a | 12 [4,2-14,2] | 13,5 [9,4-27,3] |
Tehlike oran� [% 95 GA]b | 1,553 [0,878-2,748] |
|
p-de�eric | 0,1267 |
|
ELN (iyi/orta), N | 86 | 91 |
�l�m say�s�, n (% ) | 44 (51,2) | 53 (58,2) |
Medyan OS (ay olarak) [% 95 GA]a | 45,6 [25,5-NE] | 26,9 [19,3-46,5] |
Tehlike oran� [% 95 GA]b | 0,730 [0,489-1,089] |
|
p-de�eric | 0,1216 |
|
ELN (zay�f/advers), N | 37 | 36 |
�l�m say�s�, n (% ) | 31 (83,8) | 29 (80,6) |
Medyan OS (ay olarak) [% 95 GA]a | 13,2 [7-18,5] | 13,5 [10,8-19,8] |
Tehlike oran� [% 95 GA]b | 1,124 [0,677-1,867] |
|
p-de�eric | 0,6487 |
|
ALFA-0701 �al��mas�, MYLOTARG'�n alt gruplardaki faydas�n� gelece�e y�nelik olarak de�erlendirmek i�in tasarlanmam��t�r; analiz yaln�zca tan�mlay�c� ama�larla sunulmu�tur.
mITT pop�lasyonu, tedaviye ba�lanmadan �nce onam�n geri �ekilmedi�i takdirde randomize edilen ve ilk randomizasyon koluna g�re analiz edilen t�m hastalar� i�ermi�tir.
K�saltmalar: AML=Akut miyeloid l�semi; GA=G�ven aral���; ELN=European LeukaemiaNet; mITT=Modifiye ITT; n=Say�; N=Say�; NE=Tahmin edilemez; OS=Genel sa�kal�m.
Pediyatrik pop�lasyon
Daha �nce tedavi edilmemi� AML
1.063 adet yeni tan� konmu� AML'li �ocuk (hastalar�n % 93,7'si < 18 ya�) ve gen� yeti�kinde (hastalar�n % 6,3'�); ortalama ya� 8,9 (aral�k: 0-29 ya�), tek ba��na standart kemoterapi veya MYLOTARG ile kombine kemoterapinin de�erlendirildi�i randomize bir �al��mada (COG AAML0531) de novo AML'li hastalar, tek ba��na standart 5 k�rl�k kemoterapiye veya ayn� kemoterapiye ind�ksiyonun 1. k�r�nde bir kez ve yo�un doz uygulaman�n 2. k�r�nde bir kez verilen 2 MYLOTARG dozu eklenerek (3 mg/m/doz) randomize edilmi�tir. �al��mada, yo�un kemoterapiye MYLOTARG eklenmesinin, de novo AML'de, daha d���k bir relaps riski sa�lamas� sayesinde EFS'yi iyile�tirdi�i (3 y�l: % 50,6'ya kar��l�k % 44; HR 0,838; % 95 GA: 0,706-0,995; p=0,0431) ortaya konmu�tur ve MYLOTARG kolunda daha uzun OS e�ilimi g�r�lm��t�r ancak bu, istatistiksel a��dan anlaml� olmam��t�r (3 y�l: % 72,4'e kar��l�k % 67,6; HR 0,904; % 95 GA: 0,721-1,133; p=0,3799). Ancak, d���k riskli AML'li hastalarda y�ksek toksisite (remisyon sonras� toksisite ile ili�kili mortalite) g�zlendi�i saptanm��t�r ve bu, yo�un doz uygulaman�n 2. k�r�nde gemtuzumab ozogamisin ald�ktan sonra ger�ekle�en uzun s�reli n�tropeniye atfedilmi�tir (bkz. B�l�m 4.2 ve 4.8). Genel olarak MYLOTARG kolundaki hastalar�n 29'u (% 5,5'i) ve kar��la�t�rma kolundaki 15 (% 2,8) hasta remisyon s�ras�nda �lm��t�r. Bu nedenle, pediyatrik hastalar i�in optimum gemtuzumab ozogamisin dozu belirlenmemi�tir (bkz. B�l�m 4.2).
Relaps veya refrakter AML
Relaps veya refrakter AML'li pediyatrik hastalarda MYLOTARG'� de�erlendirmek i�in MYLOTARG'� monoterapi (tek veya fraksiyone doz) veya kombinasyon olarak alan 454 hastay� kapsayan; yay�nlanm�� 16 makaleye ek olarak US Expanded Access Study �al��malar�n�n sistematik bir literat�r taramas� yap�lm��t�r (bkz. B�l�m 4.8). Bu �al��malardaki hasta say�s� 5-105 aras�nda olup medyan �al��ma b�y�kl��� 15 hastad�r. Genel en k���k ve en b�y�k ya� aral��� 0 ile 22,3 ya� aras�nda olup tedavi s�ras�nda genel medyan ya� 8,7 olarak belirlenmi�tir.
�o�u �al��ma, insani ama�l� ilaca erken eri�im program� kapsam�nda (% 70,6) yap�lm��t�r. MYLOTARG, �al��malar�n % 47,1'inde monoterapi, % 23,5'inde kombinasyon ve % 29.4'�nde monoterapi ve kombinasyon olarak verilmi�tir. MYLOTARG'�n toplam dozu 1,8 mg/m ile 9 mg/m aras�nda de�i�mi�tir. MYLOTARG kombinasyon olarak verildi�inde, 9 �al��man�n 8'inde sitarabin i�eren bir tedavi rejimi kullan�lm��t�r. �al��malar�n % 23,5'inde hastalar�n b�y�k �o�unlu�u fraksiyone (1, 4, 7. g�nlerinde 3 mg/m) MYLOTARG dozlar� al�rken; �al��malar�n % 35,3'�nde 3 mg/m'den y�ksek dozlar verilmi�tir. MYLOTARG �o�u �al��mada ind�ksiyon tedavisi olarak verilmi�tir (% 82,4).
MYLOTARG monoterapisi ile yan�t oran� (CR/CRp/CRi; �al��malar aras�nda a��rl�kl� ortalama) fraksiyone dozlama (1 �al��ma) ile % 33,3 ve fraksiyone olmayan dozlama (9 �al��ma) ile % 24,3 olmu�tur. Kombinasyon tedavisinde; yan�t oran�, fraksiyone olmayan MYLOTARG (3 �al��ma) ile % 49 ve fraksiyone MYLOTARG (2 �al��ma) ile % 38,8 olmu�tur.
MYLOTARG i�in bilinen advers olaylar olan miyelosupresyon, enfeksiyonlar, genel VOD ve HKHN sonras� VOD ve �l�m hakk�ndaki g�venlilik bilgileri (bkz. B�l�m 4.8 ve Tablo 5) literat�rden elde edilmi�tir.
Bu analizin s�n�rlamalar�, baz� �al��malar�n k���k �rneklem b�y�kl�kleri, �al��malar�n heterojenli�i ve bu ortamda kontrol verilerinin eksikli�ini i�erir.
Kardiyak elektrofizyoloji
MYLOTARG'�n d�zeltilmi� QT aral��� �zerindeki etkisi, B1761031 monoterapi �al��mas�nda, relaps veya refrakter CD-33-pozitif AML'si olan 50 yeti�kin hastada de�erlendirilmi�tir. Terap�tik plazma konsantrasyonlar�nda ba�lang�ca g�re en b�y�k ortalama QTcF aral��� de�i�ikli�i 5,1 milisaniye olmu�tur (%90 GA: 2,15, 8,06 milisaniye). Ba�lang��tan itibaren maksimum QTcF art��� > 60 milisaniye ve QTcF > 480 milisaniye olan hasta bulunmamaktad�r. Ayn� hastada atriyal fibrilasyon (Derece 3) ve supraventrik�ler ta�ikardi (Derece 3) olay� meydana gelmi�tir. Derece 4 veya Derece 5 kardiyak iletim yan etkileri bildirilmemi�tir.
Konsantrasyon-QTc aral��� analizine dayal� olarak, toplam hP67.6 antikoru i�in ba�lang�ca g�re QTcF'de beklenen medyan de�i�iklik, g�zlemlenen ortalama plazma Cde�erinde 0,842 milisaniye (%95 GA: -1,93, 3,51 milisaniye) olmu�tur. Konjuge olmayan kalikeamisin i�in, QTcF'de ba�lang��tan itibaren beklenen medyan de�i�im, MYLOTARG'�n �nerilen doz rejiminde uygulanmas�n�n ard�ndan g�zlemlenen yakla��k plazma Cde�erinde 0,602 milisaniye (%95 GA:- 2,17, 2,72 milisaniye) olmu�tur.
5.2. Farmakokinetik �zellikler
Genel �zelliklerGemtuzumab ozogamisin, sitotoksik ajan N-asetil-gama-kalikeamisine kovalent olarak ba�lanan CD33 hedefli monoklonal antikordan (hP67.6) olu�an bir antikor-ila� konjugat�d�r (ADC). Gemtuzumab ozogamisinin farmakokineti�i (PK), antikorun (hP67.6) PK �zellikleriyle birlikte, konjuge ve konjuge olmayan kalikeamisin t�revleri �l��lerek a��klan�r..
Emilim:
Klinik PK verileri, MYLOTARG'�n bir monoterapi doz rejimini (1, 4, 7. g�nlerde 3 mg/m ila bir 5 mg'l�k flakon) takiben toplanm��t�r. Konjuge kalikeamisin ve toplam hP67.6 antikoru i�in �oklu dozlar� takiben geometrik ortalama AUCve Cile �l��len maruz kalmalar, s�ras�yla 461.500 pg∙sa/mL ve 11.740 pg/mL ve 26.820 ng∙sa/mL ve 585,6 ng/mLdir. Konjuge olmayan kalikeamisin i�in PK verileri, plazmadaki istikrars�zl�k nedeniyle sunulmam��t�r.
Da��l�m
�n vitro, N-asetil-gama-kalikeamisin dimetil hidrazidin insan plazmas� proteinlerine ba�lanma oran� yakla��k % 97'dir. �n vitro, N-asetil-gama-kalikeamisin dimetil hidrazid, P-glikoproteinin (P-gp) bir substrat�d�r. Hastalarda, hP67.6 antikorunun (V1 [13 L] ve V2 [6,91 L] toplam�) toplam da��l�m hacmi yakla��k 20 L olarak saptanm��t�r.
Biyotransformasyon
Gemtuzumab ozogamisinin birincil metabolik yola��n�n, N-asetil-gama-kalikeamisin dimetil hidrazidin hidrolitik sal�m� oldu�u �ng�r�lmektedir. �n vitro �al��malarda, N-asetil-gama- kalikeamisin dimetil hidrazidin, esas olarak dis�lf�r k�sm�n�n enzimatik olmayan indirgenmesi yoluyla yayg�n olarak metabolize oldu�u g�sterilmi�tir. Sonu�ta ortaya ��kan metabolitlerin aktivitesinin (sitotoksisite), �nemli �l��de azalm�� olaca�� beklenmektedir.
Di�er t�bbi �r�nlerle etkile�imler
Di�er t�bbi �r�nlerin gemtuzumab ozogamisin �zerindeki etkisi
�n vitro, N-asetil-gama-kalikeamisin dimetil hidrazid a��rl�kl� olarak, enzimatik olmayan indirgeme yolu ile metabolize edilir. Bu nedenle, gemtuzumab ozogamisinin sitokrom P450 (CYP) veya �ridin difosfat glukuronosiltransferaz (UGT) ila� metabolizma enzim inhibit�rleri ya da ind�kleyicileriyle birlikte uygulanmas�n�n, N-asetil-gama-kalikeamisin dimetil hidrazide maruziyeti de�i�tirmesi olas� de�ildir.
Pop�lasyon farmakokinetik (PK) analizlerine dayan�larak, gemtuzumab ozogamisinin hidroksi�re, DNR ve AraC ile kombinasyonunun, hP67.6 PK's�nda ya da konjuge olmayan kalikeamisinde klinik a��dan anlaml� de�i�ikliklere neden olmayaca�� tahmin edilmektedir.
Gemtuzumab ozogamisinin di�er t�bbi �r�nler �zerindeki etkisi CYP substratlar� �zerindeki etki
�n vitro, N-asetil-gama-kalikeamisin dimetil hidrazid ve gemtuzumab ozogamisinin, klinik a��dan anlaml� konsantrasyonlarda, CYP1A2, CYP2A6 (yaln�zca gemtuzumab ozogamisin kullan�larak test edilmi�tir), CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 ve CYP3A4/5'in aktivitelerini engelleme potansiyeli d���k bulunmu�tur. �n vitro, N-asetil-gama-kalikeamisin dimetil hidrazid ve gemtuzumab ozogamisinin, klinik a��dan anlaml� konsantrasyonlarda, CYP1A2, CYP2B6 ve CYP3A4 aktivitelerini ba�latma (ind�kleme) potansiyeli d���k bulunmu�tur.
UGT substratlar� �zerindeki etki
�n vitro, N-asetil-gama-kalikeamisin dimetil hidrazidin, klinik a��dan anlaml� konsantrasyonlarda, UGT1A1, UGT1A4, UGT1A6, UGT1A9 ve UGT2B7 aktivitelerini engelleme potansiyeli d���kt�r.
�la� ta��y�c� substratlar �zerindeki etki
�n vitro, N-asetil-gama-kalikeamisin dimetil hidrazidin, klinik a��dan anlaml� konsantrasyonlarda, P-gp, meme kanserine diren� proteini (BCRP), safra tuzu atma pompas� (BSEP), �oklu ila� direnci ili�kili protein (MRP) 2, �oklu ila� ve toksin d��a at�m proteini (MATE)1 ve MATE2K, organik anyon ta��y�c� (OAT)1 ve OAT3, organik katyon (OCT)1 ve OCT2, ve organik anyon ta��y�c� polipeptid (OATP)1B1 ve OATP1B3 aktivitelerini engelleme potansiyeli d���k olmu�tur.
Birlikte uygulanan kemoterap�tikler �zerindeki etki
Pop�lasyon farmakokinetik (PK) analizlerine dayan�larak, gemtuzumab ozogamisinin DNR ve AraC ile kombinasyonunun, bu maddelerin PK's�nda klinik a��dan anlaml� de�i�ikliklere neden olmayaca�� tahmin edilmektedir.
Eliminasyon
Gemtuzumab ozogamisin farmakokineti�i, do�rusal ve zamana ba�l� klerens bile�enlerine sahip 2 b�lmeli bir modelle iyi karakterize edilmi�tir. MYLOTARG'�n bir monoterapi dozlama rejimini (1, 4, 7. g�nlerde 3 mg/m ila bir 5 mg'l�k flakon) takiben relaps veya refrakter AML'si olan 50 hastada, toplam hP67.6 antikorunun klerensi 0,288 L/saat olmu�tur ve terminal eliminasyon yar� �mr�n�n (t½) 96,6 saat oldu�u tahmin edilmi�tir.
Spesifik g�n�ll� veya hasta gruplar�ndaki farmakokinetik
Ya�, �rk ve cinsiyet
Pop�lasyon PK analizine dayal� olarak, ya��n, �rk�n ve cinsiyetin gemtuzumab ozogamisin e�ilimini kayda de�er oranda etkilemedi�i tespit edilmi�tir.
Karaci�er yetmezli�i
Karaci�er yetmezli�i bulunan hastalarda gemtuzumab ozogamisin i�in resmi PK �al��malar� ger�ekle�tirilmemi�tir.
Pop�lasyon PK analizine dayan�larak, gemtuzumab ozogamisin klirensinin (hP67.6 antikoru ve konjuge olmayan kalikeamisin), Ulusal Kanser Enstit�s� Organ Disfonksiyonu �al��ma Grubu (National Cancer Institute Organ Dysfunction Working Group - NCI ODWG) taraf�ndan tan�mland��� �ekilde, hafif karaci�er yetmezli�i durumundan etkilenmesi beklenmemektedir. Analiz, �u NCI ODWG yetmezlik durumu kategorilerinde 405 hasta ile yap�lm��t�r: Hafif (B1, n=58 ve B2, n=19), orta (C, n=6) ve normal hepatik fonksiyon (n=322) (bkz. B�l�m 4.2).
B�brek yetmezli�i
B�brek yetmezli�i bulunan hastalarda gemtuzumab ozogamisin i�in resmi PK �al��malar� ger�ekle�tirilmemi�tir.
406 hasta �zerinde yap�lan bir pop�lasyon PK analizine dayan�larak, hafif d�zeyde b�brek yetmezli�i bulunan hastalarda (kreatinin klirensi [CL60-89 mL/dk; n=149) veya orta d�zeyde b�brek yetmezli�i bulunan hastalarda (CL30-59 mL/dk; n=47) gemtuzumab ozogamisin klirensi, normal b�brek fonksiyonuna sahip hastalarla benzer (CL≥ 90 mL/dk; n=209) olmu�tur. Gemtuzumab ozogamisinin PK's�, �iddetli b�brek yetmezli�i olan hastalarda ara�t�r�lmam��t�r.
Pediyatrik pop�lasyon
Pop�lasyon modellemesinin sonu�lar�, gemtuzumab ozogamisinin PK davran���n�n (hP67.6 antikoru ve konjuge olmayan kalikeamisin), 9 mg/m doz uygulama rejiminin ard�ndan yeti�kin ve pediyatrik AML hastalar� aras�nda benzer oldu�unu g�stermi�tir.
5.3. Klinik �ncesi g�venlilik verileri
Tekrarl� doz toksisitesi
En �nemli toksik etkiler, karaci�er, kemik ili�i ve lenfoid organlar, hematoloji parametreleri (d���k RBC k�tle ve WBC say�lar�, a��rl�kl� olarak lenfositler), b�brek, g�z ve erkek ile di�i �reme organlar�nda meydana gelmi�tir. S��anlarda karaci�er, b�brek ve erkek �reme organlar� ve maymunlarda lenfoid dokular �zerindeki etkiler (EAA'e g�re 3 mg/m de�erindeki ���nc� insan dozundan sonra insan klinik maruziyetinin, s��anlar i�in yakla��k 18 kat� ve maymunlar i�in 36 kat�) geri d�n��l� olmam��t�r. 12 haftal�k �al��mada maymunlarda di�i �reme organlar� ve g�z �zerindeki etkiler advers etkilerdir (EAA'e g�re, 3 mg/m de�erindeki ���nc� insan dozundan sonra insan klinik maruziyetinin s�ras�yla yakla��k 193 ve 322 kat�). Hayvanlarda g�r�len geri d�n��s�z bulgular�n insanlar �zerindeki ge�erlili�i belirsizdir. Hayvanlarda, MYLOTARG uyguland�ktan sonra sinir sistemine etkileri g�zlenmemi�tir. S��anlarda di�er antikor-kalikeamisin konjugatlar�yla, s��anlarda sinir sistemi de�i�iklikleri belirlenmi�tir.
Genotoksisite
Gemtuzumab ozogamisinin klastojenik oldu�u saptanm��t�r. Bu, kalikeamisinin ve di�er enedin antit�m�r antibiyotiklerinin bilinen DNA k�r�lmalar�n� ind�kleme etkisi ile tutarl� bir sonu�tur. N-asetil-gama-kalikeamisin DMH'nin (sal�nan sitotoksin) mutajenik ve klastojenik oldu�u saptanm��t�r.
Karsinojenisite
Gemtuzumab ozogamisin ile resmi karsinojenisite �al��malar� yap�lmam��t�r. Toksisite �al��malar�nda, insanda ���nc� 3 mg/m dozu uyguland�ktan sonra EAA'e g�re insan�n klinik olarak maruz kald��� ilac�n yakla��k 54 kat�na maruziyette s��anlar�n karaci�erlerinde prenoeplastik lezyonlar (minimum ile hafif aras� oval h�cre hiperplazisi) meydana gelmi�tir. Maymunlarda ise, insanda ���nc� 3 mg/m dozu uyguland�ktan sonra EAA'e g�re insan�n klinik olarak maruz kald��� ilac�n yakla��k 115 kat�na kadar maruziyette, preneoplastik ya da neoplastik lezyon g�zlenmemi�tir. Hayvanlarda g�r�len bu geri d�n��s�z bulgular�n insanlar �zerindeki ge�erlili�i belirsizdir.
Reprod�ktif toksisite
Bir di�i s��an fertilite �al��mas�nda, maternal toksisite varl���nda (insanda ���nc� 3 mg/m dozu uyguland�ktan sonra EAA'e g�re insan�n klinik olarak maruz kald��� ilac�n yakla��k 9,7 kat�) corpus luteum say�s�nda hafif bir azalma ve artm�� embriyoletalite g�zlenmi�tir. 12 haftal�k �al��mada, di�i maymunlar�n �reme sistemi �zerinde etkiler g�zlenmi�tir (over, ovid�kt, uterus ve serviks atrofisi; 3 mg/m de�erindeki ���nc� dozdan sonraki insan klinik maruziyetinin yakla��k 193 kat�).
Bir erkek fertilite �al��mas�nda, erkek �remesi �zerindeki etkiler, d���k spermatogoni ve spermatositler, testik�ler spermatidlerde ve epididimal spermde azalmalar, spermatidlerde n�kleus bo�lu�u olu�umunu ve/veya dev h�cre g�r�lmesini i�ermi�tir. Ek bulgular, testisler, epididimler, meme bezi ve fertilite �zerindeki etkiler �eklindedir. Erkek s��anlar, doz uygulanmayan 9 haftal�k s�reden sonra tekrar �iftle�tirildi�inde testislerinde, sperm ve fertilite �zerindeki etkiler daha k�t� olmu�, ancak d��m�� olan spermatosit ve spermatogoni say�lar�nda k�smi toparlanma olmu�tur. Erkek s��anlar�n �reme organlar� �zerindeki etkiler, geri d�n��s�z veya k�smen geri d�n��l� olmu�tur (bkz. B�l�m 4.6). Maymunlarda erkek �remesi �zerine etkiler (testisler, epididimler, meni keseleri), 3 mg/m de�erindeki ���nc� dozun ard�ndan insan klinik maruziyetinin yakla��k 66 kat�nda g�zlenmi�tir.
Bir embriyo-fetal toksisite �al��mas�nda, daha d���k fetal v�cut a��rl���, daha y�ksek fetal e�ri kaburga s�kl��� ve daha d���k fetal iskelet osifikasyon s�kl��� g�zlenmi�tir. Y�ksek embriyoletalite ve fetal morfolojik anormallikler aras�nda parmaklarda malformasyonlar, aort ark� yoklu�u, �st ekstremitelerdeki uzun kemiklerde anomaliler, bi�imsiz skapula, vertebral centrum yoklu�u ve sternum kemiklerinde erime g�r�lm��t�r. Maternal toksisite varl���nda y�ksek embriyoletalite de g�zlenmi�tir. Embriyo-fetal etkiler ile ili�kilendirilen en d���k doz, insanda ���nc� 3 mg/m dozu uyguland�ktan sonra EAA'e g�re insan�n klinik olarak maruz kald��� ilac�n yakla��k 9,7 kat� ile ili�kili bulunmu�tur (bkz. B�l�m 4.6).
6. FARMAS�T�K �ZELL�KLER
6.1. Yard�mc� maddelerin listesi
Dekstran 40 S�kroz Sodyum klor�r
Monobazik sodyum fosfat monohidrat Dibazik sodyum fosfat, susuz
6.2. Ge�imsizlikler
�la�lar�n birbirleriyle ge�imlilik �al��malar�n�n bulunmad��� durumlarda, bu t�bbi �r�n, di�er t�bbi �r�nler ile kar��t�r�lmamal�d�r.
6.3. Raf �mr�
A��lmam�� flakon 60 ay.
Suland�r�lm�� ve seyreltilmi� ��zelti
Suland�r�lm�� ve seyreltilmi� MYLOTARG ��zeltileri ���ktan korunmal� ve hemen kullan�lmal�d�r. Suland�r�lm�� veya seyreltilmi� ��zeltiyi dondurmay�n�z.
�r�n hemen kullan�lam�yorsa;
Suland�rman�n ard�ndan, orijinal flakonda 16 saate kadar buzdolab�nda (2°C-8°C) veya oda s�cakl���nda (30°C'nin alt�nda) 3 saate kadar saklanabilir.
Seyreltilmi� ��zelti, buzdolab�nda (2°C-8°C) 18 saate kadar ve oda s�cakl���nda (30C'nin alt�nda) 6 saate kadar saklanabilir. Bu s�re, oda s�cakl���nda (30C'nin alt�nda), seyreltilmi� ��zeltinin haz�rlanmas�, gerekirse dengelenmesi ve hastaya uygulanmas� i�in gereken s�reyi i�erir. Seyreltilmi� ��zeltinin haz�rlanmas�ndan uygulamaya kadar olan s�re 24 saati a�mamal�d�r.
6.4. Saklamaya y�nelik �zel tedbirler
Buzdolab�nda (2°C-8°C) saklay�n. Dondurmay�n.
I��ktan korumak i�in flakonu orijinal kutusunun i�inde saklay�n. Suland�rma ve seyreltme sonras� saklama ko�ullar� i�in bkz. B�l�m 6.3.
6.5. Ambalaj�n niteli�i ve i�eri�i
5 mg gemtuzumab ozogamisin i�eren, b�til kau�uk t�pal� ve ge�me kapakl� s�k��t�rmal� ba�l�kl� amber
renkli Tip 1 cam flakon.
Her kutuda 1 flakon bulunur.
6.6. Be�eri t�bbi �r�nden arta kalan maddelerin imhas� ve di�er �zel �nlemler
Suland�rma ve seyreltme prosed�rleri i�in uygun aseptik tekni�i kullan�n. MYLOTARG, ����a kar�� hassast�r ve suland�rma, seyreltme ve uygulama esnas�nda mor�tesi ����a kar�� korunmal�d�r.
Suland�rma
Gerekli MYLOTARG dozunu (mg) hesaplay�n.
Suland�rmadan �nce, flakonun yakla��k 5 dakika oda s�cakl���na (30°C'nin alt�nda) ula�mas�n� bekleyin. Tek kullan�ml�k 1 mg/mL gemtuzumab ozogamisin ��zeltisi elde etmek i�in, her bir 5 mg'l�k flakonu 5 mL enjeksiyonluk su ile suland�r�n.
��z�nmeye yard�mc� olmak i�in flakonu nazik�e �evirin. �alkalamay�n.
Suland�r�lm�� ��zeltiyi partik�l ve renk bozuklu�u a��s�ndan inceleyin. Suland�r�lm�� ��zelti, k���k beyaz ile beyaz�ms�, opak ile yar� saydam aras� ve amorf ile lif benzeri aras� partik�ller i�erebilir.
MYLOTARG, bakteriyostatik koruyucu i�ermez.
Suland�r�lm�� ��zelti hemen kullan�lam�yorsa, orijinal flakonda 16 saate kadar buzdolab�nda (2°C-8°C) veya oda s�cakl���nda (30°C'nin alt�nda) 3 saate kadar saklanabilir. I��ktan koruyun ve dondurmay�n.
Seyreltme
Hastan�n v�cut y�zey alan�na g�re uygun dozu elde etmek �zere gerekli suland�r�lm�� ��zelti hacmini hesaplay�n. Bir enjekt�r kullanarak bu miktarda �r�n� flakondan �ekin. MYLOTARG flakonlar�, fazla dolum olmadan 5 mg t�bbi �r�n i�erir. Y�nlendirildi�i gibi 1 mg/mL konsantrasyona suland�r�ld���nda, flakonun ekstrakte edilebilir i�eri�i 4,5 mg'dir (4,5 mL). I��ktan koruyun. Flakonda kalan kullan�lmam�� suland�r�lm�� ��zeltiyi at�n.
Dozlar, a�a��daki y�nergelere g�re 0,075 mg/mL ila 0,234 mg/mL aras�nda bir konsantrasyon
elde edilmek �zere kar��t�r�lmal�d�r:
3,9 mg'den d���k dozlar, enjekt�rle uygulanmak �zere haz�rlanmal�d�r. Suland�r�lm�� MYLOTARG ��zeltisini, 9 mg/L (% 0,9) enjeksiyon i�in sodyum klor�r i�eren bir enjekt�re ekleyerek 0,075 mg/mL ila 0,234 mg/mL aras�nda son konsantrasyon elde edin. I��ktan koruyun.
3,9 mg veya �st� dozlar, 9 mg/mL'lik (% 0,9) uygun sodyum klor�r hacimli enjeksiyonluk ��zelti i�eren bir enjekt�r ya da IV torbas� i�inde seyreltilerek, 0,075 mg/mL ila 0,234 mg/mL aras�nda son konsantrasyon elde edilmelidir. I��ktan koruyun.
 A��r� Alkol Kullan�m�, Alkolizm
Alkol ba��ml�l���, alkol kullan�m� ve alkol sorunlar� aras�ndaki fark� a��klamak g��t�r.
�rne�in, ge�mi�te alkol kullanm�� olan bir kimsenin mutlaka alkol ba��ml�s� olmas�
gerekmez.
A��r� Alkol Kullan�m�, Alkolizm
Alkol ba��ml�l���, alkol kullan�m� ve alkol sorunlar� aras�ndaki fark� a��klamak g��t�r.
�rne�in, ge�mi�te alkol kullanm�� olan bir kimsenin mutlaka alkol ba��ml�s� olmas�
gerekmez. |
 Belso�uklu�u, Chlamydia ve Frengi
Belso�uklu�u, bakterilerin sebep oldu�u bir enfeksiyondur. Cinsel ili�ki
yoluyla bula��r ve d�lyata�� boynunda, idrar yollar�nda, an�ste, makatta ve
bo�azda enfeksyona sebep olabilir.
Belso�uklu�u, Chlamydia ve Frengi
Belso�uklu�u, bakterilerin sebep oldu�u bir enfeksiyondur. Cinsel ili�ki
yoluyla bula��r ve d�lyata�� boynunda, idrar yollar�nda, an�ste, makatta ve
bo�azda enfeksyona sebep olabilir. |
 |
Diyabet Hastal��� Diyabet, ins�lin hormonu ile ilgili problemlerden kaynaklanan bir hastal�kt�r. |
 |
Ast�m Ast�ml� ki�ilerin akci�erlerindeki hava borular� (bron�lar) hassast�r. Bu ki�iler belirli tetikleyici fakt�rlere maruz kald�klar�nda, hava borular� nefes almalar�n� g��le�tirecek �ekilde daral�r. |
 |
Ruh ve Ak�l Sa�l���m�z� Geli�tirmek �yi ak�l ve ruh sa�l��� sahip olmaktan ziyade, yapt���n�z �eylerdir. Ak�l ve ruhsal olarak sa�l�kl� olmak i�in kendinize de�er vermeli ve kendinizi kabul etmelisiniz. |
�LA� GENEL B�LG�LER�
Pfizer �la�lar� Ltd.�ti.
| Sat�� Fiyat� | 213112.88 TL [ 17 Dec 2024 ] |
| �nceki Sat�� Fiyat� | 206868.78 TL [ 2 Dec 2024 ] |
| Original / Jenerik | Original �la� |
| Re�ete Durumu | Normal Re�eteli bir ila�d�r. |
| Barkodu | 8681308271099 |
| Etkin Madde | Gemtuzumab |
| ATC Kodu | L01XC05 |
| Birim Miktar | 5 |
| Birim Cinsi | MG |
| Ambalaj Miktar� | 1 |
| Antineoplastik ve �mm�nomod�lat�r Ajanlar > Di�er Kanser �la�lar� |
| �thal ( ref. �lke : Abd ) ve Be�eri bir ila�d�r. |