OCREVUS 300 mg/ 10ml infüzyonluk çözelti hazırlamak için konsantre (1 flakon) Kısa Ürün Bilgisi
{ Okrelizumab }
1. BEŞERİ TIBBİ ÜRÜNÜN ADI
OCREVUS 300 mg/10 mL infüzyonluk çözelti hazırlamak için konsantre
2. KALİTATİF VE KANTİTATİF BİLEŞİM
Etkin madde
Her flakon, 30 mg/mL konsantrasyonda ocrelizumab çözeltisi içerir. Toplamda her flakonda 10 mL'de 300 mg ocrelizumab bulunmaktadır. Dilüsyon sonrasında elde edilen ilaç konsantrasyonu yaklaşık 1,2 mg/mL' dir.
Ocrelizumab, rekombinant DNA teknolojisiyle Çin Hamsteri Yumurtalığı hücre dizilerinde üretilen bir humanize monoklonal antikorudur.
Yardımcı maddeler
Sodyum asetat trihidrat 21,4 mg Diğer yardımcı maddeler için 6.1'e bakınız.
3. FARMASÖTİK FORMU
Steril
İnfüzyonluk çözelti hazırlamak için konsantre.
Berrak ila hafif opalesan ve renksiz ila açık kahverengi çözelti.
4. KLİNİK ÖZELLİKLER
4.1. Terapötik endikasyonlar
Ataklarla seyreden (RMS) veya primer progresif MS (PPMS) vakalarında endikedir.
4.2. Pozoloji ve uygulama şekli
Tedavi, ciddi infüzyonla ilişkili reaksiyonlar (İİR) gibi şiddetli reaksiyonların yönetiminde uygun tıbbi desteğe erişimi olan, deneyimli bir sağlık uzmanı tarafından başlatılmalı ve takip edilmelidir.
İnfüzyonla ilişkili reaksiyonlar için premedikasyon
İİR'lerin sıklığı ve şiddetini daha da azaltmak amacıyla her ocrelizumab infüzyonundan önce aşağıdaki iki premedikasyon önerilmektedir (bkz. Bölüm 4.4):
Her infüzyondan yaklaşık 30 dakika önce 100 mg intravenöz metilprednisolon veya eşdeğer dozda diğer kortikosteroidlerin uygulanması;
İnfüzyondan yaklaşık 30-60 dakika önce antihistaminik bir ilaç (ör: difenhidramin) uygulanması
İlave olarak, infüzyondan 30-60 dakika önce antipiretikler ile premedikasyon (ör: parasetamol) ayrıca değerlendirilebilir.
Pozoloji/Uygulama sıklığı ve süresi:
Başlangıç dozu:
600 mg başlangıç dozu, iki ayrı intravenöz infüzyon olarak uygulanır; ilk 300 mg infüzyonu 2 hafta sonra ikinci 300 mg infüzyon takip eder (bkz. Tablo 1).
Sonraki dozlar:
Sonraki ocrelizumab dozları 6 ayda bir, tek bir 600 mg intravenöz infüzyon olarak uygulanır (bkz. Tablo 1). İkinci doz, ilk dozun uygulanmasının ardından 6 ay sonra uygulanmalıdır.
Her ocrelizumab dozunun arasında minimum 5 aylık ara olmalıdır.
IIR meydana gelmesi durumunda infüzyon ayarlamaları:
Yaşamı tehdit edici İİR'ler
İnfüzyon sırasında, akut aşırı duyarlılık veya akut solunum güçlüğü sendromu gibi yaşamı tehdit edici veya sakat bırakan bir İİR'nin belirtileri olursa, infüzyon derhal kesilmelidir. Hasta uygun şekilde tedavi görmelidir. Bu hastalarda infüzyon kalıcı olarak kesilmelidir (bkz. Bölüm 4.3).
Şiddetli İİR'ler
Hasta şiddetli bir İİR (ör. dispne) veya kızarma, ateş ve boğaz ağrısı semptomları yaşarsa, infüzyon hemen kesilmeli ve hastaya semptomatik tedavi uygulanmalıdır. İnfüzyon ancak semptomlar düzeldikten sonra tekrar başlatılmalıdır. İnfüzyona yeniden başlandığında infüzyon hızı, reaksiyonun başlangıç zamanında uygulanan infüzyon hızının yarısı olmalıdır. Hasta İİR yaşamazsa, sonraki infüzyonlarda infüzyon hızı ayarlaması gerekli değildir.
Hafif ila orta şiddette İİR'ler
Hasta hafif ila orta şiddette İİR (ör. baş ağrısı) yaşarsa, infüzyon hızı, olay başlangıcında verilen hızın yarısına azaltılmalıdır. Bu azaltılmış hız en az 30 dakika korunmalıdır. Tolere edilirse, infüzyon hızı hastanın başlangıç infüzyon planına göre artırılabilir. Hasta İİR yaşamazsa, sonraki infüzyonlarda infüzyon hızı ayarlaması gerekli değildir.
Tedavi sırasında doz ayarlamaları:
Yukarıdaki doz kesintisi ve yavaşlama örnekleri (hafif ila orta şiddette ve şiddetli İİR'ler için) infüzyon hızındaki bir değişikliğe ve infüzyonun toplam süresinde artmaya neden olur ancak toplam dozu etkilemez. Doz azaltımı önerilmez.
Unutulan veya geç alınan dozlar:
Eğer bir infüzyon unutulursa, mümkün olan en kısa sürede uygulanmalı, planlanan bir sonraki doza kadar beklenmemelidir. Unutulan dozun uygulanmasından 6 ay sonra bir sonraki doz uygulanacak şekilde doz planı yeniden oluşturulmalıdır. Dozları arasında tedavi aralığı minimum 5 ay olmalıdır (bkz. Tablo 1).
Uygulama şekli:
Seyreltme sonrasında, tedavi özel bir infüzyon setiyle IV infüzyon olarak uygulanır. İnfüzyonlar, intravenöz puşe veya bolus olarak uygulanmamalıdır (bkz. Tablo 1).
Hastalar daha önce herhangi ciddi bir infüzyonla ilişkili reaksiyon (İİR) yaşamamışsa, sonraki dozlar için daha kısa (2 saatlik) bir infüzyon süresi uygulanabilir (Tablo 1, Seçenek 2).
Tablo 1: Doz ve Plan
| Uygulanacak ocrelizumab Miktarı | İnfüzyon talimatı | |
İlk doz (600 mg)
İki infüzyona bölünmüş | İnfüzyon 1 | 250 mL'de 300 mg | |
İnfüzyon 2 (2 hafta sonra) | 250 mL'de 300 mg | ||
|
|
| |
|
|
| 30 dakikada bir 30 |
|
|
| mL/saat artırarak |
|
|
| maksimum 180 |
|
|
| mL/saat'e |
|
|
| çıkartılmalıdır |
|
|
| |
|
|
| yaklaşık 2,5 saat |
|
|
| boyunca |
|
|
| uygulanmalıdır |
Sonraki dozlar (600 mg) tek infüzyon 6 ayda bir |
Seçenek 1 | 500 mL'de 600 mg | başlanmalıdır |
| İnfüzyon süresi yaklaşık 3.5 saat |
|
|
|
|
| 30 dakikada bir 40 |
|
|
| mL/saat artırarak |
|
|
| maksimum 200 |
|
|
| mL/saat'e |
|
|
| çıkartılmalıdır |
|
|
| |
|
|
| yaklaşık 3,5 saat |
|
|
| boyunca |
|
|
| uygulanmalıdır |
| Veya | ||
İnfüzyona 30 dakika boyunca 30 mL/sa hızında başlanmalıdır
Sonrasında, hızı her
Her bir infüzyon
İnfüzyona 30 dakika boyunca 40 mL/sa hızında
Sonrasında, hızı her
Her bir infüzyon
| Seçenek 2
İnfüzyon süresi yaklaşık 2 saat | 500 mL'de 600 mg |
|
İnfüzyona ilk 15 dakika için 100 mL/sa hızında başlanmalıdır
Bir sonraki 15 dakika için infüzyon hızı 200 mL/saat'e çıkartılmalıdır
Bir sonraki 30 dk için infüzyon hızını 250 mL/saat'e artırınız
Kalan 60 dakika için infüzyon hızı 300 mL/saat'e çıkartılmalıdır
Her bir infüzyon yaklaşık 2 saat boyunca uygulanmalıdır
Intravenöz infüzyon çözeltileri konsantrenin 9 mg/mL (%0,9) sodyum klorür içeren infüzyon torbası ile dilüe edilerek hazırlanır ve sonuç ocrelizumab konsantrasyonu yaklaşık 1,2 mg/mL'dir. İlave bilgi için Bölüm 6.6'ya bakınız.
Tıbbi ürünün uygulanmadan önce seyreltilmesine ilişkin talimatlar için, bkz. Bölüm 6.6.
İnfüzyon sırasında ve infüzyon tamamlandıktan sonra hastalar en az 1 saat gözlenmelidir (bkz. Bölüm 4.4).
Özel popülasyonlara ilişkin ek bilgiler:
Karaciğer yetmezliği:
Karaciğer bozukluğu olan hastalarda ocrelizumabın güvenliliği ve etkililiği üzerinde resmi olarak çalışılmamıştır. Hafif karaciğer yetmezliği olan hastalar klinik çalışmalara dahil edilmiştir. Orta ve ciddi seviyede karaciğer yetmezliği olan hastalarda deneyim mevcut değildir. Ocrelizumab, bir monoklonal antikordur ve atılımı katabolizma yoluyla (karaciğer metabolizmasından ziyade) gerçekleşmekte olup, karaciğer yetmezliği olan hastalar için doz ayarlaması gereksinimi beklenmemektedir (bkz. Bölüm 5.2).
Böbrek yetmezliği:
Böbrek bozukluğu olan hastalarda ocrelizumabın güvenliliği ve etkililiği üzerinde resmi olarak çalışılmamıştır. Hafif böbrek yetmezliği olan hastalar klinik çalışmalara dahil edilmiştir. Orta
ve ciddi seviyede böbrek yetmezliği olan hastalarda deneyim mevcut değildir. Ocrelizumab, bir monoklonal antikordur ve atılımı katabolizma yoluyla (örn. peptitlere ve amino asitlere parçalanma) gerçekleşmekte olup, böbrek yetmezliği olan hastalar için doz ayarlaması gereksinimi beklenmemektedir (bkz. Bölüm 5.2).
Pediyatrik popülasyon:
0 ila 18 yaş arasındaki çocuklarda OCREVUS'un güvenliliği ve etkililiği üzerinde henüz çalışılmamıştır. Bu konu ile ilgili veri mevcut değildir.
Geriyatrik popülasyon:
Mevcut kısıtlı verilere dayanarak (bkz. Bölüm 5.1 ve 5.2) 55 yaşın üzerindeki hastalarda pozoloji ayarlaması gerekmemektedir. Devam eden klinik çalışmalarda yer alan hastalarda, 55 yaş ve üzerine geldiklerinde, her 6 ayda bir 600 mg ocrelizumab uygulanmasına devam edilmektedir.
4.3. Kontrendikasyonlar
OCREVUS aşağıdaki durumlarda kontrendikedir:
Ocrelizumaba veya Bölüm 6.1'de listelenen yardımcı maddelerden herhangi birine karşı aşırı duyarlılığı olan hastalarda
4.4. Özel kullanım uyarıları ve önlemleri
İzlenebilirlik
Biyolojik tıbbi ürünlerin izlenebilirliğini artırmak için, uygulanan ürünün ticari adı ve seri numarası net bir şekilde hasta dosyasına kaydedilmelidir.
İnfüzyonla İlişkili Reaksiyonlar (İİR)
Ocrelizumab, sitokin salınımına ve/veya diğer kimyasal mediyatörlerle ilişkili olabilecek infüzyonla ilişkili reaksiyonlarla ilişkilendirilebilir.
İİR semptomları herhangi bir ocrelizumab infüzyonu sırasında meydana gelebilir ancak sıklıkla ilk infüzyon sırasında rapor edilmiştir. İİR'ler infüzyondan sonra 24 saat içerisinde meydana gelebilir (bkz. Bölüm 4.8). Bu reaksiyonlar, kaşıntı, döküntü, ürtiker, eritem, , boğaz irritasyonu, orofaringeal ağrı, dispne, faringeal veya laringeal ödem, yüzde kızarıklık, hipotansiyon, yüksek ateş, yorgunluk, baş ağrısı, sersemlik hali, bulantı,taşikardi ve anafilaksi şeklinde ortaya çıkabilir.
İnfüzyondan önce:
Şiddetli reaksiyonların yönetimi: İİR, aşırı duyarlılık reaksiyonları ve/veya anafilaktik reaksiyonlar gibi şiddetli reaksiyonların yönetimi için uygun kaynaklar hazır bulundurulmalıdır.
Hipotansiyon: OCREVUS infüzyonları sırasında bir İİR semptomu olarak hipotansiyon görülebilir. Bu nedenle her bir OCREVUS infüzyonundan 12 saat önce ve infüzyon boyunca antihipertansif tedavilerin kesilmesi düşünülmelidir. Konjestif kalp yetmezliği öyküsü (New York Kalp Birliği III ve IV) olan hastalar araştırılmamıştır.
Premedikasyon: İnfüzyon reaksiyonlarının sıklığını ve şiddetini azaltmak için premedikasyon uygulanmalıdır (bkz. Bölüm 4.2 - İnfüzyonla ilişkili reaksiyonlar için premedikasyon).
İnfüzyon esnasında:
Bronkospazm ve astım alevlenmesi gibi şiddetli pulmoner semptomlar görülen hastalarda aşağıdaki önlemler alınmalıdır:
İnfüzyon hemen ve kalıcı olarak durdurulmalıdır
Semptomatik tedavi uygulanmalıdır
4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri
Sitokrom P450 enzimleri, diğer metabolize edici enzimler veya taşıyıcılar üzerinden hiçbir etkileşim beklenmediğinden, etkileşim çalışmaları yürütülmemiştir.
Aşılar
Ocrelizumab tedavisi sonrasında canlı veya canlı atenüe aşılar ile bağışıklamanın güvenliliği henüz çalışılmamıştır.
Ocrelizumab alan hastalarda tetanoz toksoid, 23-valanslı pnömokoksal polisakkarit, keyhole limpet hemosiyanin neoantijen ve mevsimsel influenza aşılarının etkilerine ait veri mevcuttur. (bkz. Bölüm 4.4 ve 5.1)
2 yılı aşkın tedavinin ardından, S. Pnömoni, kabakulak, rubella ve varicella'ya karşı pozitif antikor titresi olan hastaların oranları, başlangıçtaki ile aynıdır.
İmmunosupresif tedaviler:
Relapsların semptomatik tedavisi için kortikosteroidler dışında ocrelizumab ile birlikte diğer immunosupresif tedavilerin kullanılması önerilmez (bkz. Bölüm 4.4).
Özel popülasyonlara ilişkin ek bilgiler:
Ocrelizumab ile herhangi bir farmakokinetik ilaç etkileşimi çalışması yapılmamıştır.
Pediyatrik popülasyon:
Ocrelizumab ile pediyatrik popülasyonda herhangi bir farmakokinetik ilaç etkileşimi çalışması yapılmamıştır.
4.6. Gebelik ve laktasyon
Gebelik kategorisi: C
Çocuk doğurma potansiyeli bulunan kadınlar / Doğum kontrolü (Kontrasepsiyon) Çocuk doğurma potansiyeli bulunan kadınlar, ocrelizumab alırken ve son ocrelizumab infüzyonundan sonra 12 ay boyunca doğum kontrol yöntemi kullanmalıdırlar (bkz. Bölüm 5.1 ve 5.2).
Gebelik dönemi
Ocrelizumabın gebe kadınlarda kullanımına ilişkin yeterli veri mevcut değildir. Hayvanlar üzerinde yapılan araştırmalar üreme toksisitesinin bulunduğunu göstermiştir (bkz. Bölüm 5.3). İnsanlara yönelik potansiyel risk bilinmemektedir.
Ocrelizumabın gebe kadınlarda kullanımı ile ilişkili gelişimsel risk konusundaki veriler yetersizdir. Ocrelizumab bir immünoglobulin G (IgG)'dir. IgG'nin plasenta bariyerini geçtiği bilinmektedir. Gebelikte ocrelizumaba maruz kalan annelerden doğan yenidoğan ve bebeklerde canlı ya da canlı atenüe aşılarla aşılamanın ertelenmesi düşünülmelidir. Ocrelizumaba maruz kalan yenidoğanlarda ve bebeklerde B-hücre sayımı verisi toplanmamıştır ve B-hücre deplesyonun potansiyel devam süresi bilinmemektedir (bkz. Bölüm 4.4).
Gebelik sırasında diğer anti-CD20 antikorlara maruz kalmış olan annelerin bebeklerinde geçici periferik B-hücre deplesyonu ve lenfositopeni rapor edilmiştir.
Hayvan çalışmaları (embriyo-fetal toksisite) teratojenik etkilere işaret etmemektedir. Ancak, uteroda B-hücre deplesyonu tespit edimiştir. Doğum öncesi ve sonrası geliştirme çalışmalarında üreme toksisitesi gözlenmiştir (bkz. Bölüm 5.3).
Anne için olan potansiyel fayda fetüse potansiyel riskten ağır basmadığı sürece gebelikte ocrelizumab uygulamasından kaçınılmalıdır.
Laktasyon dönemi
Ocrelizumabın ve metabolitlerinin insan sütüne karışıp karışmadığı bilinmemektedir. Hayvan çalışmalarında, ocrelizumabın anne sütüyle atılımı gösterilmiştir (bkz. Bölüm 5.3). Yenidoğan ve bebeklerdeki risk göz ardı edilemez. Kadın hastalara tedavi sırasında emzirmeyi kesmeleri önerilmelidir.
Üreme yeteneği/Fertilite
Sinomolgus maymunlarında erkek ve dişi doğurganlığı hakkında yapılan çalışmalara dayanan klinik öncesi veriler, insanlara yönelik özel bir tehlikeye işaret etmemektedir.
4.7. Araç ve makine kullanımı üzerindeki etkiler
OCREVUS'un araç ve makine kullanma yeteneği üzerinde etkisi yoktur yada ihmal edilebilir düzeydedir.
4.8. İstenmeyen etkiler
Güvenlilik profilinin özeti
En önemli ve sık bildirilen advers reaksiyonlar (AİR'ler); İİR'ler (RMS ve PPMS'te sırasıyla %34,3 ve %40,1) , ve enfeksiyonlardır (RMS ve PPMS'te sırasıyla %58,5 ve %72,2) (bkz. Bölüm 4.4). .
Advers olayların tablo halinde listesi
Klinik çalışmalarda bildirilen ve spontan raporlamadan elde edilen advers reaksiyonlar aşağıda Tablo 2'de listelenmiştir. Advers reaksiyonlar MedDRA sistem organ sınıfı ve sıklık kategorilerine göre listelenmiştir. Sıklık kategorileri bu şekilde tanımlanmaktadır: çok yaygın (≥1/10), yaygın (≥1/100 ila <1/10), yaygın olmayan (≥1/1.000 ila <1/100), seyrek (≥1/10.000 ila <1/1.000), çok seyrek (<1/10.000), ve bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). Her bir sıklık grubunun içinde istenmeyen etkiler azalan ciddiyet sırası ile sunulmaktadır.
Tablo 2 OAdvers reaksiyonlar
AİR (MedDRA) Sistem Organ Sınıfı | Çok yaygın | Yaygın | Bilinmiyor |
Enfeksiyonlar ve enfestasyonlar |
Üst solunum yolu enfeksiyonu, nazofarenjit, influenza | Sinüzit, bronşit, oral herpes, gastroenterit, solunum yolu enfeksiyonu, viral enfeksiyon, herpes zoster, konjunktivit, selülit |
|
Kan ve lenf sistemi hastalıkları |
| Nötropeni | Geç nötropeni başlangıcı |
Solunum, göğüs bozuklukları ve mediastinal hastalıklar |
| Öksürük, nezle |
|
Araştırmalar | Kan immunoglobulin M düşüşü | Kan immunoglobulin G düşüşü |
|
Yaralanma, zehirlenme ve prosedürel komplikasyonlar |
İnfüzyon ile ilişkili 1 reaksiyonlar |
|
|

Seçilen advers reaksiyonların açıklaması
İnfüzyonla ilişkili reaksiyonlar
RMS ve PPMS çalışmalarında İİR'lerle ilgili semptomlar belirtilenlerle sınırlı olmamakla birlikte şunları içerir: kaşıntı, döküntü, ürtiker, eritem, yüzde kızarıklık, hipotansiyon, ateş, yorgunluk, baş ağrısı, baş dönmesi, boğazda tahriş, orofaringeal ağrı, dispne, faringeal veya laringeal ödem, bulantı, taşikardi. Kontrollü çalışmalarda ölümcül İİR'ler olmamıştır. Ek olarak, İİR'lerle ilgili semptomlara pazarlama sonrası verilerde anafilaksi dahil edilmiştir.
Aktif kontrollü (RMS) klinik çalışmalarda, interferon beta-1a tedavi grubunda (plasebo infüzyonu) %9,9'luk insidansa kıyasla, ocrelizumab ile tedavi edilen grupta %34,3'lük genel insidansla İİR'ler en yaygın advers reaksiyondur. İİR'lerin insidansı, 1. doz, 1. infüzyon (%27,5) sırasında en yüksek düzeyde olmuş ve zamanla 4. dozda <%10'a düşmüştür. Her iki tedavi grubunda İİR'lerin çoğunluğu hafif ila orta şiddette olmuştur. ocrelizumab ile tedavi edilen hastalar sırasıyla %21,7 ve %10,1'i hafif veya orta, % 2,4'ü şiddetli İİR ve % 0,1 hayatı tehdit eden İİR deneyimlemiştir.
Plasebo kontrollü (PPMS) klinik çalışmada, plasebo grubundaki %25,5'lik insidansa kıyasla, ocrelizumab ile tedavi edilen grupta %40,1'lik genel insidansla İİR'ler en yaygın advers reaksiyondur. İİR'lerin insidansı, 1. doz, 1. infüzyon (%27,4) sırasında en yüksek düzeyde olmuş ve zamanla 4. dozda <%10'a düşmüştür. Her grupta her dozun ilk infüzyonunda o dozun ikinci infüzyonuna kıyasla daha büyük bir hasta oranı İİR'ler yaşamıştır. İİR'lerin çoğunluğu hafif ila orta şiddetli olmuştur. Ocrelizumab ile tedavi edilen hastalar sırasıyla %26,7 ve %11,9'u hafif veya orta şiddette, % 1,4'ü şiddetli İİR deneyimlemiştir. Hayatı tehdit eden İİR görülmemiştir (bkz. Bölüm 4.4).
Sonraki dozlar için alternatif hızlandırılmış infüzyon
Ataklarla seyreden Multipl Sklerozlu (RMS) hastalarda daha kısa (2 saatlik) ocrelizumab infüzyonlarının güvenlilik profilini karakterize etmek için tasarlanmış bir çalışmada (MA30143 Hızlandırılmış İnfüzyon Çalışması), İİR'lerin görülme sıklığı, yoğunluğu ve tipleri 3,5 saat süren infüzyon ile görülen İİR profili ile uyumlu bulunmuştur (bkz Bölüm 5.1). Her iki infüzyon grubunda da ihtiyaç duyulan genel müdahale sayısı düşüktür, ancak 3,5 saatlik infüzyon grubuna kıyasla, kısa (2 saatlik) infüzyon grubunda İİR'leri yönetebilmek için daha fazla müdahale (yavaşlama veya geçici kesintiler) gerekmiştir (sırasıyla% 8,7'ye karşı% 4,8).
Enfeksiyon
Aktif kontrollü RMS çalışmalarında, ocrelizumab alan hastaların %58,5'inde ve interferon beta- 1a alan hastaların %52,5'inde enfeksiyon gözlenmiştir. ocrelizumab alan hastaların %1,3'üne karşılık interferon beta-1a alan hastaların %2,9'unda ciddi enfeksiyon gözlenmiştir. Plasebo kontrollü PPMS çalışmasında ocrelizumab alan hastaların %72,2'sinde ve plasebo alan hastaların %69,9'unda enfeksiyon gözlenmiştir. Ocrelizumab alan hastaların %6,2'sine karşılık plasebo alan hastaların %6,7'sinde ciddi enfeksiyon meydana gelmiştir. Hem RMS hem de PPMS çalışmalarında tüm hastalar açık etiketli fazda ocrelizumaba geçmiştir. RMS'de ciddi enfeksiyon riskinde artış 2. ve 3. yıllar arasında gözlenmiş olup takip eden yıllarda gözlenmemiştir. PPMS'de ise herhangi bir artış gözlenmemiştir.
Solunum yolu enfeksiyonları
Solunum yolu enfeksiyonlarının oranı, interferon beta-1a ve plaseboya kıyasla ocrelizumab ile tedavi edilen hastalarda daha yüksek olmuştur.
RMS klinik çalışmalarında ocrelizumab ile tedavi edilen hastaların %39,9'u ve interferon beta-1a ile tedavi edilen hastaların % 33,2 'ü üst solunum yolları enfeksiyonu; ocrelizumab ile tedavi edilen hastaların %7,5'i ve interferon beta-1a ile tedavi edilen hastaların %5,2'si alt solunum yolları enfeksiyonu deneyimlemiştir. PPMS klinik çalışmasında, ocrelizumab ile tedavi edilen hastaların %48,8'i ve plasebo alan hastaların %42,7'si üst solunum yolları enfeksiyonu; ocrelizumab ile tedavi edilen hastaların %9,9'u ve plasebo alan hastaların %9,2'si alt solunum yolları enfeksiyonu deneyimlemiştir. Ocrelizumab ile tedavi edilen hastaların solunum yolları enfeksiyonu ağırlıklı olarak hafif ila orta olarak raporlanmıştır (%80-90).
Herpes
Aktif kontrollü (RMS) klinik çalışmalarda, herpes enfeksiyonları, ocrelizumab ile tedavi edilen hastalarda interferon beta-1a ile tedavi edilen hastalara kıyasla daha sık bildirilmiştir: herpes zoster (%2,1 ile %1), herpes simpleks (% 0,7 ile % 0,1) ve oral herpes (%3 ile %2,2), genital herpes (%0,1 ile %0) ve herpes virüs enfeksiyonu (%0,1 ile %0). Tüm enfeksiyonlar, bir tane Derece 3 hariç, hafif ila orta şiddetli olmuş ve hastalar standart tedavi uygulanmasıyla iyileşmişlerdir.
Plasebo kontrollü (PPMS) klinik çalışmada, oral herpesi olan hastalar ocrelizumab tedavi kolunda, plaseboya göre daha sık rapor edilmiştir (%2,7'ye karşı %0,8).
Laboratuvar anormallikleri İmmunoglobulinler
Ocrelizumab tedavisi, ağırlıklı olarak IgM'deki azalmaya bağlı olarak çalışmaların kontrollü döneminde toplam immunoglobulinlerde azalmayla sonuçlanmıştır. Klinik çalışma verileri, IgG'de sürekli azalma (ve IgM ve IgA için daha az) ile ciddi enfeksiyonlar arasında bir ilişki
olduğunu göstermiştir.
Lenfositler
RMS'de, interferon beta-1a ile tedavi edilen hastaların %32,6'sına karşılık ocrelizumab ile tedavi edilen hastaların %20,7'sinde lenfositin NAS'ın (normalin alt sınırı) altına düştüğü gözlenmiştir. PPMS'de plaseboyla tedavi edilen hastaların %11,7'sine karşılık ocrelizumab ile tedavi edilen hastaların %26,3'ünde lenfositlerin NAS'ın altına düştüğü gözlenmiştir.
Ocrelizumab ile tedavi edilen hastalarda bildirilen bu düşüşlerin çoğunun şiddet olarak Derece
1 (<NSA– 800 hücre/mm) ve Derece 2 (500-800 hücre/mm) olduğu görülmüştür. Ocrelizumab grubundaki hastaların yaklaşık %1'inde Derece 3 lenfopeni (200-500 hücre/mm) olduğu görülmüştür. Hastaların hiçbirinde Derece 4 lenfopeni (<200 hücre/mm) bildirilmemiştir.
Ocrelizumabla tedavi edilen hastalarda doğrulanmış total lenfosit sayımı düşüş epizotları esnasında ciddi enfeksiyonların oranında artış gözlenmiştir. Ciddi enfeksiyon sayısının, kesin sonuçlara varmak için çok düşük olduğu görülmüştür.
Nötrofil
Aktif kontrollü (RMS) tedavi döneminde, interferon beta-1a ile tedavi edilen hastaların %40,9'una kıyasla, ocrelizumab ile tedavi edilen hastaların %14,7'sinde nötrofillerin NSA'ın altına düştüğü gözlenmiştir. Plasebo kontrollü (PPMS) klinik çalışmada, nötrofil seviyelerinde azalma görülen ocrelizumab hastalarının oranı, plasebo hastalarına (%12,9) kıyasla daha yüksek (%10,0) olmuştur. Plasebo grubundaki hastaların %1,3'üne karşılık ocrelizumab grubunda daha yüksek oranda hastada (%4,3) yada ≥Derece 2 nötropeni görülmüş; plasebo grubundaki hastaların %0'ına karşılık ocrelizumab grubundaki hastaların yaklaşık %1'inde Derece 4 nötropeni görülmüştür.
Nötrofil düşüşlerinin çoğunun geçici nitelikte (sadece ocrelizumab ile tedavi edilen belirli bir hastada bir defa gözlenmiştir) ve şiddet olarak Derece 1 ve 2 (sırasıyla <NAS ve 1500 hücre/mm arasında ve 1000-1500 hücre/mm) olduğu belirlenmiştir. Genel olarak, ocrelizumab grubundaki hastaların yaklaşık %1'inde Derece 3 veya 4 nötropeni vardı. Derece 3 (500 – 1000 hücre/mm) nötropenili bir hasta ve Derece 4(<500 hücre/mm) nötropenili bir hasta granülosit-koloni uyarıcı faktörle spesifik tedaviye ihtiyaç duymuş ve epizot sonrasında ocrelizumab tedavisine devam etmiştir. Nötropeni, ocrelizumab uygulamasından birkaç ay sonra ortaya çıkabilir (bkz. Bölüm 4.4).
Diğer
2000 mg ocrelizumab alan başka bir hasta ise en son infüzyondan 12 hafta sonra uygulanan manyetik rezonans görüntülemesinin (MRG) ardından etiyolojisi bilinmeyen sistemik enflamatuvar yanıt sendromu (SEYS) nedeniyle ölmüştür; MRG için uygulanan gadolinyumlu kontrast maddeye karşı gelişen anafilaktoid reaksiyon SEYS'nin ortaya çıkmasına katkıda bulunmuş olabilir.
Şüpheli advers reaksiyonların raporlanması
Ruhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar/risk dengesinin sürekli olarak izlenmesine olanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu Türkiye Farmakovijilans Merkezi'ne (TÜFAM) bildirmeleri gerekmektedir (www.titck.gov.tr: e-posta: tufam@titck.gov.tr, tel: 0 800 314 00 08; faks: 0 312 218 35 99).
4.9. Doz aşımı ve tedavisi
Onaylı intravenöz ocrelizumab dozundan daha yüksek dozlarda klinik çalışma deneyimi sınırlıdır. MS hastalarında bugüne kadar test edilen en yüksek doz, 2 hafta arayla iki 1000 mg intravenöz infüzyon olarak uygulanan 2000 mg'dır (RRMS'de Faz II doz bulma çalışması). Advers reaksiyonları, merkezi klinik çalışmalarda güvenlilik profiliyle tutarlı olmuştur.
Doz aşımı olması durumunda spesifik bir antidot yoktur; infüzyon hemen durdurulmalı ve hasta İİR'ler açısından gözlenmelidir (bkz. Bölüm 4.4).
5. FARMAKOLOJİK ÖZELLİKLER
5.1. Farmakodinamik özellikler
Farmakoterapötik grup: Antineoplastik ve İmmünomodülatör Ajanlar, İmmünosupresanlar, Selektif İmmünosupresanlar
ATC kodu: L04AA36
Etki mekanizması
Ocrelizumab, CD20 eksprese eden B hücrelerini seçici olarak hedefleyen bir rekombinant hümanize monoklonal antikordur.
CD20, ön-B hücreleri, olgun ve bellek B hücrelerinde bulunan ama lenfoid kök hücreleri ve plazma hücrelerinde eksprese edilmeyen bir hücre yüzeyi antijenidir.
Ocrelizumabın MS'te terapötik klinik etkilerini gösterdiği kesin mekanizmalar tam olarak açıklanmamıştır ama CD20 eksprese eden B hücrelerinin sayısı ve fonksiyonunda azalmayla immunomodülasyona neden olduğu varsayılır. Hücre yüzeyindeki bağlanmanın ardından, ocrelizumab, antikor bağımlı sellüler fagositoz (ADCP), antikor bağımlı sellüler sitotoksisite (ADCC), kompleman bağımlı sitotoksisite (CDC) ve apoptoz aracılığıyla CD20 eksprese eden B hücrelerini seçici olarak tüketir. B hücresi rekonstitüsyon kapasitesi ve önceden var olan humoral bağışıklık korunur. Ayrıca doğuştan bağışıklık ve toplam T hücresi sayıları etkilenmez.
Farmakodinamik etkiler
Ocrelizumab ile tedavi, beklenen bir farmakolojik etki olarak, tedaviden 14 gün sonra (değerlendirmenin ilk zaman noktası) kanda CD19+ B hücrelerinin hızlı tükenmesine yol açar. Bu, tedavi dönemi boyunca sürdürülmüştür. Ocrelizumabın varlığı miktar tayiniyle CD20'nin tanınmasını engellediği için, B hücre sayımları için CD19 kullanılır.
Faz III çalışmalarda, her ocrelizumab dozunun arasında, hastaların %5'ine kadarı en az bir zaman noktasında B hücresi deplesyonu (> normalin alt sınırı (NAS) veya başlangıç) sergilemiştir. B hücresi tükenmesinin derecesi ve süresi, PPMS ve RMS çalışmalarında tutarlı olmuştur.
Son Ocrelizumab infüzyonundan sonraki en uzun takip süresi (Faz II WA21493, N=51), B hücresi çoğalmasına kadar geçen medyan sürenin (hangisinin daha önce olduğuna bağlı olarak başlangıca veya LLN'ye geri dönmüştür) 72 hafta (27 - 175 haftalık aralık) olduğunu göstermektedir. Tüm hastaların yüzde doksanında, son infüzyondan yaklaşık iki buçuk yıl sonra B hücreleri LLN veya başlangıca göre çoğalmıştır.
Klinik etkililik ve güvenlilik
Ataklarla seyreden multipl skleroz formları (RMS)
Ocrelizumabın etkililiği ve güvenliliği, ataklarla seyreden MS formları olan hastalarda tasarımları aynı olan iki randomize, çift kör, çift plasebolu, aktif komparatör kontrollü klinik çalışmada (WA21092 ve WA21093) değerlendirilmiştir (2010 McDonald kriterlerine uygun olarak). Çalışma tasarımı ve çalışma popülasyonunun başlangıç özellikleri Tablo 3'te özetlenmektedir.
Demografi ve başlangıç özellikleri açısından her iki tedavi grubu iyi dengelenmiştir. Ocrelizumab alan hastalara (Grup A) 6 ayda bir 600 mg (1. Doz 2 hafta arayla 2 x 300 mg intravenöz infüzyonlar halinde) verilmiş ve sonraki dozlar tek bir 600 mg intravenöz infüzyon olarak uygulanmıştır. Grup B'deki hastalara haftada 3 kez subkütan (S.C.) enjeksiyonla İnterferon beta-1a 44 mcg verilmiştir.
Tablo 3 Çalışma Tasarımı ve Demografik Özellikler
| Çalışma 1 | Çalışma 2 | ||
Çalışma adı | WA21092 (OPERA I) (n=821) | WA21093 (OPERA II) | ||
Çalışma tasarımı | ||||
Çalışma popülasyonu | Ataklarla seyreden MS formları görülen hastalar | |||
Taramadaki hastalık öyküsü | İki yıl içinde en az iki relaps veya bir yıl içinde bir relaps; EDSS 0 ve 5,5 (5,5 dahil) arasında, | |||
Çalışma süresi | 2 yıl | |||
Tedavi grupları | Grup A: 600 mg ocrelizumab Grup B: 44 mcg SC interferon beta-1a (IFN) | |||
Başlangıçtaki özellikler | Ocrelizumab 600 mg (n=410) | IFN 44 mcg (n=411) | Ocrelizumab 600 mg (n=417) | IFN 44 mcg (n=418) |
Medyan yaş (yıl) | 37,1 | 36,9 | 37,2 | 37,4 |
Dahil edilen yaş aralığı (yıl) | 18-56 | 18-55 | 18-55 | 18-55 |
Cinsiyet dağılımı (%erkek/%kadın) | 34,1/65,9 | 33,8/66,2 | 35/65 | 33/67 |
Tanıdan bu yana geçen ortalama/medyan hastalık süresi (yıl) | 3,82/1,53 | 3,71/1,.57 | 4,15/2,1 | 4,13/1,84 |
Daha önce DMT almamış hastaların %'si** | 73,4 | 71 | 72,7 | 74,9 |
Geçen yıldaki ortalama relaps sayısı | 1,31 | 1,33 | 1,32 | 1,34 |
T1 lezyonunda Gd tutulumlu hastaların oranı | 42,5 | 38,1 | 39,0 | 41,4 |
Ortalama EDSS* | 2,82 | 2,71 | 2,73 | 2,79 |
* Genişletilmiş Yeti Yitimi Durumu Ölçeği
**Randomizasyondan önceki 2 sene içinde bir hastalık modifiye edici tedavi (DMT) ile tedavi edilmemiş hastalar
Temel klinik ve MRG etkililiği sonuçları Tablo 4 ve Şekil 1'de sunulmaktadır.
Bu çalışmaların sonuçları, interferon beta-1a 44mcg SC'ye kıyasla ocrelizumabın relapsları, MRG ile ölçülen klinik ve subklinik hastalık aktivitesi ve hastalık progresyonunu önemli ölçüde baskıladığını göstermektedir.
Tablo 4 WA21092 ve WA21093 Çalışmalarından Temel Klinik ve MRG Sonlanım Noktaları (RMS)
Sonlanım Noktaları | Çalışma 1: WA21092 (OPERA I) | Çalışma 2: WA21093 | |||
Ocrelizumab 600 mg (n=410) | IFN 44 mcg (n=411) | Ocrelizumab 600 mg (n=417) | IFN 44 mcg (n=418) | ||
Klinik Sonlanım Noktaları |
| ||||
Yıllık Relaps Oranı (primer sonlanım noktası) Rölatif Azalma | 0-156 | 0,292 | 0,155 | 0,290 | |
%46 (p<0,0001) | %47 (p<0,0001) | ||||
12 Haftalık Doğrulanmış Yeti Yitimi Progresyonu Görülen Hasta Oranı Riskte Azalma (Birleştirilmiş Analiz) Riskte Azalma (Ayrı Ayrı Çalışmalar) | %9,8 Ocrelizumab - %15,2 IFN %40 (p=0,0006) | ||||
%43 (p=0,0139) | %37 (p=0,0169) | ||||
24 Haftalık Doğrulanmış Yeti Yitimi Progresyonu Görülen Hasta Oranı (CDP) Riskte Azalma (Birleştirilmiş Analiz) Riskte Azalma (Ayrı Ayrı Çalışmalar) | %7,6 Ocrelizumab - %12,0 IFN %40 (p=0,0025) | ||||
%43 (p=0,0278) | %37 (p=0,0370) | ||||
En az 12 haftalık Doğrulanmış Yeti Yitimi İyileşmesi görülen hasta oranı(Birleştirilmiş) Rölatif Artış (Birleştirilmiş Analiz) Rölatif Artış (Ayrı Ayrı Çalışmalar) | %20,7 Ocrelizumab - %15,6 IFN | ||||
%33 (p=0,0194) | |||||
%61 (p=0,0106) | %14 (p=0,4019) | ||||
96 haftada relaps gözlenmeyen hastaların oranı | %80,4 | %66,7 | %78,9 | %64,3 | |
(p=0,0001) | (p<0,0001) | ||||
Hastalık Aktivitesi Kanıtı Görülmeyen (NEDA) Hasta Oranı Rölatif Artış | %48 | %29 | %48 | %25 | |
%64 (p<0,0001) | %89 (p<0,0001) | ||||
MRG Sonlanım Noktaları |
| ||||
MRG taraması başına ortalama Gd tutulumlu T1 lezyon sayısı Rölatif Azalma | 0,016 | 0,286 | 0,021 | 0,416 | |
%94 (p<0,0001) | %95 (p<0,0001) | ||||
MRG taraması başına yeni ve/veya büyüyen T2 hiperintens lezyonların ortalama sayısı Rölatif Azalma | 0,323 | 1,413 | 0,325 | 1,904 | |
%77 (p<0,0001) | %83 (p<0,0001) | ||||
|
|
|
|
| |
|
| ||||
24. hafta ila 96. hafta arasında beyin hacmindeki değişiklik oranı Beyin hacmi kaybında rölatif azalma | -0,572 | -0,741 | -0,638 | -0,750 | |
%22,8 (p=0,0042) | %14,9 (p=0,0900) | ||||
tahminleri
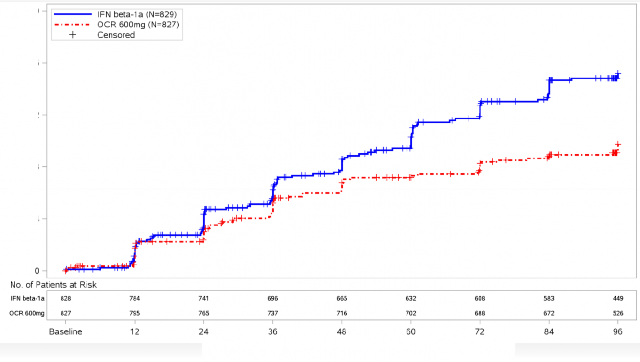
Şekil 1: En Az 12 Hafta Boyunca Sürdürülen Doğrulanmış Yeti Yitimi Progresyonunun Başlangıcına Kadar Geçen Süre ve Çift-Kör Tedavi Dönemi Boyunca Meydana Gelen İlk Nörolojik Kötüleşme Olayına İlişkin Kaplan-Meier Grafiği* (Birleştirilmiş ITT popülasyonu)*
*OPERA I ve II'nin önceden belirlenmiş birleştirilmiş analizi
En az 12 hafta sürdürülen CDP'ye kadar geçen sürenin önceden belirlenen havuzlanmış analizlerinin sonuçları (interferon beta-1a'ya kıyasla ocrelizumab için %40 risk azaltımı, (p=0,0006), en az 24 hafta sürdürülen sonuçlarla (interferon beta-1a'ya kıyasla ocrelizumab için %40 risk azaltımı, p=0,0025) son derece tutarlı olmuştur.
Çalışmalarda aktif hastalığı olan hastalar dahil edilmiştir. Bu hastalarda, klinik ve radyolojik açıdan aktif, tedavi naif ve önceki tedavilerine yetersiz cevap geliştirmiş hastalar dahildir. Çok aktif ve aktif hastalık durumu da dahil olmak üzere farklı başlangıç seviyelerinde hastalık aktiviteleri olan hasta popülasyonu analizleri, ocrelizumab etkililiğinin ARR'de ve CDP ile 12 haftada genel popülasyon ile aynı olduğunu ortaya koymuştur.
Primer progresif multipl skleroz (PPMS)
Ocrelizumabın etkililik ve güvenliliği ayrıca ana dahil etme kriterlerine (18-55 yaş (bu yaşlar dahil), taramada 3– 6,5 puan arası EDSS, MS semptomlarının ortaya çıkışından itibaren geçen hastalık süresi: taramadaki EDSS puanı ≤5olan hastalarda <10 yıl ve EDSS puanı <5olan hastalarda <15 yıl) göre hastalığın erken evresinde olan primer progresif MS'li hastalara yönelik randomize, çift-kör, plasebo-kontrollü bir çalışmada (Çalışma WA25046) değerlendirilmiştir. Hastalık aktivitesi bakımından, progresif MS'te dahi enflamatuvar etkinliğe özgü özellikler görüntülemeyle ilişkili olabilir (T1 Gd-kontrast tutan lezyonlar ve/veya aktif (yeni veya büyüyen) T2 lezyonları). Bütün hastalarda enflamatuvar etkinliği doğrulamak için MRG bulguları kullanılmalıdır. 55 yaşın üzerindeki hastalar incelenmemiştir. Çalışma tasarımı ve çalışma popülasyonunun başlangıç özellikleri Tablo 5'te sunulmuştur.
Demografik özellikler ve başlangıç özelliklerinin iki tedavi grubu arasında dengeli olduğu görülmüştür. Kraniyal MRG T1 Gd-kontrast tutan lezyonlar veya T2 lezyonları olarak enflamatuvar etkinliğe özgü görüntüleme özellikleri göstermiştir.
Faz III PPMS çalışması sırasında, hastalar her 6 ayda bir 600 mg ocrelizumab dozunu, iki hafta ara ile uygulanan 2 adet 300 mg'lık infüzyonlar şeklinde almıştır. RMS'de 600 mg infüzyonlar ve PPMS'de 300 mg x 2 infüzyonlar uyumluPK/PD profilleri göstermiştir. İnfüzyon başına İİR profilleri tek seferde 600 mg veya iki hafta arayla 300 mg doz uygulanmasından bağımsız olarak benzerdir (bkz. Bölüm 4.8 ve 5.2). Ancak 2x300 mg doz rejimi ile toplamdaki infüzyon sayısı daha yüksek olduğunda, toplam İİR sayısı daha yüksektir. Bu nedenle, Doz 1'den sonra toplam infüzyon sayısını düşürmek için (metilprednisolon ve antihistaminik ile eş zamanlı uygulanması sırasında) ocrelizumabın 600 mg'lık tek bir infüzyon olarak uygulanması tavsiye edilir (bkz. Bölüm 4.2).
Tablo 5 WA25046 Çalışmasının çalışma tasarımı, demografik ve başlangıç özellikleri
Çalışma adı | WA25046 Çalışması ORATORIO (n=732) | |
Çalışma tasarımı | ||
Çalışma popülasyonu | Primer progresif MS formu görülen hastalar | |
Çalışma süresi | Olaya dayalı (Minimum 120 hafta ve 253 doğrulanmış yeti yitimi progresyonu olayı) (Medyan takip süresi: Ocrelizumab 3 yıl, Plasebo 2,8 yıl | |
Taramadaki hastalık öyküsü | Yaş 18-55, EDSS 3 ila 6,5 | |
Tedavi grupları | Grup A: 600 mg Ocrelizumab Grup B: Plasebo, 2:1 randomizasyon | |
Başlangıçtaki özellikler | 600 mg Ocrelizumab (n=488) | Plasebo (n=244) |
Ortalama Yaş (yıl) | 44,7 | 44,4 |
Dahil edilen yaş aralığı | 20-56 | 18-56 |
Cinsiyet dağılımı (%erkek/%kadın) | 51,4/48,6 | 49,2/50,8 |
PPMS tanısından bu yana geçen ortalama/medyan hastalık süresi (yıl) | 2,9/1,6 | 2,8/1,3 |
Ortalama EDSS | 4,7 | 4,7 |
Temel klinik ve MRG etkililiği sonuçları Tablo 6 ve Şekil 2'de sunulmaktadır.
Bu çalışmanın sonuçları, ocrelizumabın plaseboya kıyasla hastalık progresyonunu önemli ölçüde geciktirdiğini ve yürüme hızındaki kötüleşmeyi azalttığını göstermektedir.
Tablo 6 WA25046 Çalışmasından (PPMS) Temel Klinik ve MRG Sonlanım Noktaları
| Çalışma 3 | |
Sonlanım Noktaları | WA25046 (ORATORIO) | |
Ocrelizumab 600 mg (n=488) | Plasebo (n=244) | |
Klinik Sonlanım Noktaları | ||
Primer etkililik sonlanım noktası 12 Haftalık Doğrulanmış Yeti Yitimi Progresyonu Görülen Hasta Oranı (primer sonlanım noktası) Riskte azalma | %30,2 | %34 |
%24 (p=0,0321) | ||
24 Haftalık Doğrulanmış Yeti Yitimi Progresyonu Görülen Hasta Oranı Riskte azalma | %28,3 | %32,7 |
%25 (p=0,0365) | ||
Başlangıca kıyasla 120. Haftada Süreli 25 Adım Yürüme Testindeki Değişiklik Oranı Yürüme süresindeki progresyon oranında rölatif azalma | 38,9 | 55,1 |
%29,4 (p=0,0404) | ||
MRG Sonlanım Noktaları | ||
Başlangıca kıyasla 120. Haftada T2 hiperintens lezyon hacmindeki değişiklik oranı | -3,4 | 7,4 |
(p< 0,0001) | ||
24. hafta ila 120. hafta arasında beyin hacmindeki değişiklik oranı Beyin hacmi kayıp oranında rölatif azalma | -0,902 | -1,093 |
%17,5 (p=0,0206) | ||
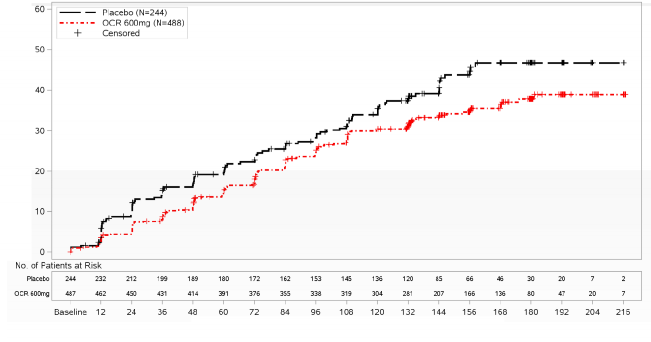
CDP riskinde %24 azalma HR (%95 GA): 0,76 (0,59,
0,98); p=0,0321
Şekil 2: Çift Kör Tedavi Döneminde Meydana Gelen İlk Nörolojik Kötüleşme Olayıyla En Az 12 Hafta Sürdürülen Doğrulanmış Sakatlık Progresyonunun Başlangıcına Kadar Geçen Sürenin Kaplan-Meier Grafiği (ITT Popülasyonu)*
* Bu analizdeki tüm hastalar en az 120 hafta boyunca takip edilmiştir. Birincil analiz değerlendirilen tüm olaylara dayanmaktadır.
Birincil sonlanım noktasına yönelik önceden tanımlanmış, yeterli güce sahip olmayan alt grup analizi, genç veya başlangıçta T1 Gd-kontrast tutan lezyonları olan hastaların yaşlı veya T1 Gd-kontrast tutan lezyonları olmayan hastalara kıyasla tedaviden daha fazla fayda sağladığını göstermektedir (≤ 45 yaş: HR 0,64 [0,45, 0,92], >45 yaş: HR 0,88 [0,62, 1,26]; başlangıçta T1 Gd-kontrast tutan lezyonları olan hastalar: HR 0,65 [0,4-1,06], T1 Gd-kontrast tutan lezyonları olmayan hastalar: HR 0,84 [0,62-1,13]).
Ayrıca, post-hoc analizler başlangıçta T1 Gd-kontrast tutan lezyonları olan genç hastalarda tedavinin daha iyi bir etki gösterdiğini ortaya koymuştur (≤ 45 yaş: HR 0,52 [0,27-1]; ≤ 46 yaş [WA25046 çalışmasındaki medyan yaş]; HR 0,48 [0,25-0,92]; <51 yaş: HR 0,53 [0,31-0,89]).
Açık Etiket Uzantısına (AEU) devam etmeden önce veya çalışma tedavisinden ayrılana kadar ek olarak yaklaşık 9 aylık kontrollü takip ve çift kör tedavi içeren Genişletilmiş Kontrollü Dönemde (GKD) post-hoc analizler gerçekleştirilmiştir. 24 haftalık Doğrulanmış Engellilik Progresyonunun (CDP) EDSS≥7 olan hastaların oranı (EDSS≥7'nin 24W-CDP'si, tekerlekli sandalyeye kadar geçen süre), tedavinin 144. haftasında plasebo grubunda %9,1 iken, ocrelizumab grubunda %4,8 olmuş ve bu oran GKD sırasında tekerlekli sandalyeye geçiş süresinin %47 risk azalmasına (HR 0,53, [0,31, 0,92]) neden olmuştur. Bu sonuçlar doğaları gereği keşif amaçlı olduğundan ve körleme kaldırıldıktan sonra veriler içerdiğinden, dikkatle yorumlanmalıdır.
Hızlandırılmış infüzyon çalışması
Hızlandırılmış (2 saatlik) ocrelizumab infüzyonunun güvenliliği, diğer hastalık modifiye edici tedavilere naif olan Ataklarla Seyreden Multipl Sklerozlu (RMS) hastalarda MA30143 (Ensemble) çalışmasının alt kolu olan prospektif, çok merkezli, randomize, çift kör, kontrollü,
paralel kollu bir çalışma ile değerlendirilmiştir. İlk doz 14 gün arayla iki 300 mg infüzyon (toplam 600 mg) olarak uygulanmıştır. Hastalar ikinci dozlarından sonra (Doz 2 ila 6) 1: 1 oranında ya ocrelizumabın 24 haftada bir uygulanan yaklaşık 3.5 saat boyunca infüze edilmiş konvansiyonel infüzyon grubuna ya da ocrelizumabın 24 haftada bir uygulanan yaklaşık 2 saat süren daha kısa infüzyon süresi grubuna randomize edilmiştir. Randomizasyon, bölgeye ve hastaların ilk randomize edildiği doza göre tabakalandırılmıştır.
Birincil sonlanım noktası, ilk randomize infüzyonu sırasında yada takip eden 24 saat içinde İİR meydana gelen hastaların oranıdır. Primer analiz 580 hasta randomize edildiğinde yapılmıştır. İlk randomize infüzyon sırasında yada takiben 24 saat içinde İİR meydana gelen hastaların oranı, hızlandırılmış infüzyonda % 24,6 iken, konvansiyonel infüzyon grubunda % 23,1 olmuştur. Tabakalı grup farkı benzerdir. Genel olarak, tüm randomize dozlarda, İİR'lerin çoğunluğu hafif veya orta düzeydeydi ve her iki grupta birer İİR olmak üzere sadece iki İİR şiddetli olmuştur. Hayatı tehdit eden, ölümcül veya ciddi İİR'lergörülmemiştir.
İmmunojenisite
MS çalışmalarındaki (WA21092, WA21093 ve WA25046) hastalar birçok zaman noktasında (başlangıç ve çalışma süresi boyunca tedavi sonrası 6 ayda bir) anti-ilaç antikorları (AİA'lar) açısından test edilmiştir. Ocrelizumab ile tedavi edilen 1311 hastadan 12'si (~%1) tedaviyle ortaya çıkan AİA'lar açısından pozitif sonuç vermiş, bunların 2'si nötralize edici antikorlar açısından pozitif sonuç vermiştir. Ocrelizumab ile ilişkili düşük AİA insidansı düşünülürse, tedaviyle ortaya çıkan AİA'ların güvenlilik ve etkililik üzerindeki etkisi değerlendirilemez.
İmmunizasyonlar
RMS hastalarında gerçekleştirilen açık etiketli, randomize bir çalışmada (N=102), aşı uygulamasından sonra 8. haftada tetanoz aşısına pozitif yanıt veren hastaların oranı, kontrol grubunda %54,5'e kıyasla ocrelizumab grubunda %23,9 olarak bulunmuştur (interferon-beta haricinde hastalığı modifiye edici herhangi bir tedavi mevcut değildir). 8. haftada anti-tetanoz toksoidine spesifik antikor titresi geometrik ortalaması sırasıyla 3,74 ve 9,81 IU/mL olarak tespit edilmiştir. Aşı uygulamasından sonra 4. haftada 23-PPV'de bulunan ≥5 serotipe verilen pozitif yanıt oranı, kontrol grubunda %100 ve ocrelizumab grubunda %71,6'dır. Ocrelizumab ile tedavi edilen hastalarda 23-PPV'den 4 hafta sonra uygulanan rapel aşısı (pekiştirme dozu/13-PCV), 23-PPV ile ortak 12 serotipe verilen yanıtı belirgin bir şekilde arttırmamıştır. Beş influenza suşuna karşı seroprotektif titreleri bulunan hastaların yüzdesi, ocrelizumab ile tedavi edilen hastalarda ve kontrol grubunda sırasıyla, aşılama öncesinde %20,0-60,0 ve %16,7-43,8 ve aşılamadan 4 hafta sonra %55,6-80,0 ve %75,0-97,0 aralıklarında
bulunmuştur. (bkz. Bölüm 4.4 ve 4.5)
5.2. Farmakokinetik özellikler
Genel özellikler
Emilim:
Ocrelizumab intravenöz infüzyon olarak uygulanır. Diğer uygulama yollarıyla çalışma gerçekleştirilmemiştir.
Dağılım:
Merkezi dağıtım hacminin popülasyon farmakokinetiği tahmini 2,78 L olmuştur. Periferal hacim ve kompartmanlar arası klerens 2,68 L ve 0,294 L/gün olarak tahmin edilmiştir.
Biyotransformasyon:
Antikorlar ağırlıklı olarak katabolizmayla (ör: peptid ve amino asitlere yıkılma)
uzaklaştırıldığından, ocrelizumabın metabolizması üzerinde doğrudan çalışılmamıştır. Eliminasyon:
Sabit klerens 0,17 L/gün tahmin edilirken, başlangıçtaki zamana bağımlı klerens 0,0489 L/gün olarak tahmin edilmiş ve 33 haftalık yarı ömürle birlikte düşmüştür. Ocrelizumabın terminal eliminasyon yarı ömrü 26 gün olmuştur.
Doğrusallık/doğrusal olmayan durum:
MS çalışmalarında ocrelizumabın farmakokinetiği, zamana bağlı klerens ve bir IgG1 monoklonal antikor için tipik PK parametreleri sergileyen, iki bölümlü bir modelle açıklanmıştır.
Genel maruziyet (24 haftalık dozlama aralıklarında EAA), aynı dozun uygulandığı düşünülürse bekleneceği üzere, PPMS çalışmalarında 2 x 300 mg ve RMS çalışmalarında 1 x 600 mg'da aynı olmuştur. 600 mg ocrelizumabın 4. dozundan sonra eğri altı alan (EAA) 3510 mcg/mL•gün ve ortalama maksimum konsantrasyon (C) RMS'de (600 mg infüzyon) 212 mcg/mL, PPMS'de (300 mg infüzyonlar) 141 mcg/mL olmuştur.
Özel popülasyonlara ilişkin ek bilgiler:
Pediyatrik popülasyon:
18 yaşının altındaki çocuklarda ocrelizumabın farmakokinetiğini araştırmak amacıyla çalışma yürütülmemiştir.
Geriyatrik popülasyon:
55 yaş ve üzerindeki hastalarda ocrelizumabın farmakokinetiğini araştırmak amacıyla çalışma yürütülmemiştir.
Böbrek yetmezliği :
Resmi farmakokinetik çalışma yürütülmemiştir. Hafif böbrek yetmezliği olan hastalar klinik çalışmalara dahil edilmiş ve bu hastalarda ocrelizumabın farmakokinetiğinde değişiklik gözlenmemiştir. Ciddi veya orta seviyede böbrek hasarı olan hastalara ait farmakokinetik veriler mevcut değildir.
Karaciğer yetmezliği :
Resmi farmakokinetik çalışma yürütülmemiştir. Hafif karaciğer yetmezliği olan hastalar klinik çalışmalara dahil edilmiş ve bu hastalarda OCREVUS'un farmakokinetiğinde değişiklik gözlenmemiştir. Ciddi veya orta seviyede karaciğer hasarı olan hastalara ait farmakokinetik veriler mevcut değildir.
5.3. Klinik öncesi güvenlilik verileri
Klinik dışı veriler; güvenlilik farmakolojisi, tekrarlı doz toksisitesi ve embriyo-fötal gelişime yönelik konvansiyonel çalışmalara dayalı olarak insanlar için özel bir risk olmadığını göstermektedir. Ocrelizumabla karsinojenisite veya mutajenisite çalışmaları yapılmamıştır.
Sinomolgus maymunları üzerinde gerçekleştirilen iki pre- ve post-natal gelişim çalışmalarında gestasyonun 20. gününden en azından doğumda kadar ocrelizumab uygulanması; glomerülopati, kemik iliğinde lenfoid folikül oluşumu, lenfoplazmasitik renal enflamasyon ve yavrunun testis ağırlığında azalmayla ilişkilendirilmiştir. Bu çalışmalarda uygulanan maternal dozlar, klinik ortamda beklenenden 4,5 ila 21 kat daha yüksek maksimum ortalama serum konsantrasyonlarına (C) neden olmuştur.
Çalışmada beş ölümcül vaka görülmüş (5/24) olup, biri prematüre doğum nedeniyle güçsüzlükle birlikte fırsatçı enfeksiyona, diğeri aktif enfeksiyonlu (mastit) bir anne hayvanın yavrusunun serebellumunu içeren bir enfektif meningoensefaliteye ve diğer üçü sarılık ve hepatik hasar kanıtı, şüphelenilen viral etiyoloji, muhtemelen bir poliomavirüse atfedilmiştir. Bu beş doğrulanmış veya şüphelenilen neonatal enfeksiyonun seyri B hücre tükenmesinden potansiyel olarak etkilenmiş olabilir. Ocrelizumab maruz kalmış anne hayvanların yenidoğan yavrularında post natal fazda B hücresi popülasyonlarının tükendiği fark edilmiştir. Emzirme döneminde sütte ölçülebilir ocrelizumab düzeyleri saptanmıştır (serum düzeylerinde kararlı durumun yaklaşık %0,2'si).
6. FARMASÖTİK ÖZELLİKLER
6.1. Yardımcı maddelerin listesi
Sodyum asetat trihidrat (E262)
Glasiyal asetik asit α,α-Trehaloz sihidrat Polisorbat 20 (E432) Enjeksiyonluk su
6.2. Geçimsizlikler
Bu tıbbi ürün ile polivinil klorür (PVC) veya polyolefin (PO) poşetler ve intravenöz uygulaması setleri arasında uyumsuzluk gözlenmemiştir.
Bu tıbbi ürün, Bölüm 6.6'da bahsedilenlerden başka tıbbi ürünlerle karıştırılmamalıdır.
6.3. Raf ömrü
Açılmamış flakon
24 ay
Seyreltilmiş intravenöz infüzyonluk çözelti
Kullanım sırasındaki kimyasal ve fiziksel stabilitesi, 2-8°C'de 24 saat ve oda sıcaklığında 8 saat süreyle gösterilmiştir.
Mikrobiyolojik açıdan, hazırlanan infüzyon hemen kullanılmalıdır. Hemen kullanılmazsa, kullanım sırasındaki saklama süreleri ve kullanım öncesi koşullar kullanıcının sorumluluğundadır ve seyreltme işlemi kontrol altında ve valide edilmiş aseptik koşullarda yapılmadığı takdirde, bu süre normalde 2-8°C'de 24 saati, oda sıcaklığında 8 saati geçmemelidir.
Bir intravenöz infüzyonun aynı gün tamamlanamaması durumunda, kalan çözelti atılmalıdır.
6.4. Saklamaya yönelik özel tedbirler
Buzdolabında saklayınız (2ºC – 8 ºC). Dondurmayınız.Işıktan korumak için flakonları karton kutusunda saklayınız.
Tıbbi ürünün seyreltmeden sonraki saklama koşulları için, bkz. Bölüm 6.3.
6.5. Ambalajın niteliği ve içeriği
Kutuda, flororeçine ile lamine butil kauçuk tıpalı, ALU mühürlü, plastik geçme kapaklı, 10 mL konsantre içeren 15mL'lik renksiz tip I cam flakon.
1 veya 2 flakonluk ambalajlarda bulunmaktadır.
6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler
Seyreltme talimatları
Ürün bir sağlık uzmanı tarafından aseptik teknik kullanılarak hazırlanmalıdır. Flakonu çalkalamayınız. Seyreltilmiş infüzyon çözeltisini hazırlamak için steril bir iğne ve şırınga kullanılmalıdır.
Ürün koruyucu madde içermez ve sadece tek kullanım içindir.
Rengi bozulmuşsa veya yabancı partikül madde içeriyorsa, konsantreyi kullanmayınız.
Tıbbi ürünü uygulama öncesinde seyreltilmelidir. İntravenöz uygulama için çözeltiler, tıbbi ürünün izotonik sodyum klorür 9 mg/mL (%0,9) enjeksiyon için çözelti (300 mg /250 mL veya 600 mg/500 mL) içeren bir infüzyon torbasında seyreltilerek yaklaşık 1,2 mg/mL ocrelizumab konsantrasyonu elde edilmesiyle hazırlanır.
Seyreltilmiş çözelti, 0,2 veya 0,22 mikron genişliğinde filtreli bir infüzyon seti kullanılarak uygulanmalıdır.
İntravenöz infüzyon başlatılmadan önce, infüzyon torbasının içeriği oda sıcaklığında olmalıdır. İmha
Kullanılmamış olan ürünler ya da atık materyaller, “Tıbbi Atıkların Kontrolü Yönetmeliği'' ve “Ambalaj ve Ambalaj Atıklarının Kontrolü Yönetmeliğiâ€ne uygun olarak imha edilmelidir.
 Şizofrenlik
Şizofrenliğin psikiatrik teşhisi hakkında çok fazla anlaşmazlık vardır. Bu
sayfadaki bilgiler, şizofrenliğin teşhisi, nedenleri ve tedavisi hakkındaki faklı teoriler
hakkında bilgi verecektir.
Şizofrenlik
Şizofrenliğin psikiatrik teşhisi hakkında çok fazla anlaşmazlık vardır. Bu
sayfadaki bilgiler, şizofrenliğin teşhisi, nedenleri ve tedavisi hakkındaki faklı teoriler
hakkında bilgi verecektir. |
 Travma Sonrası Bunalımı
Travmatik bir olay, günlük olağan olayların dışında olan ve kişiyi derinden
rahatsız eden bir olaydır.Birçok olay böyle bir etki gösterebilir.
Travma Sonrası Bunalımı
Travmatik bir olay, günlük olağan olayların dışında olan ve kişiyi derinden
rahatsız eden bir olaydır.Birçok olay böyle bir etki gösterebilir. |
 |
Tiroid Kanseri En sık görülen tiroid kanseri türü olan papiller tiroid kanseri, tüm tiroid kanserlerinin yaklaşık %70'ini oluşturur. |
 |
Mide Kanseri Mide kanseri genellikle mideyi tümüyle kaplayan ve mukus üretmekle görevli hücrelerde başlar. Bu kanser tipine adenokarsinom denir. |
 |
Depresyonu Anlamak Depresyon farklı kişileri farklı biçimlerde etkiler. Duygusal veya fiziksel olmak üzere geniş alanda belirtilere sebep olabilir.Depresyona neler sebep olur? |
İLAÇ GENEL BİLGİLERİ
Roche Müstahzarları Sanayi A.Ş.
| Geri Ödeme Kodu | A16804 |
| Satış Fiyatı | 118602.86 TL [ 24 Mar 2025 ] |
| Önceki Satış Fiyatı | 118602.86 TL [ 17 Mar 2025 ] |
| Original / Jenerik | Original İlaç |
| Reçete Durumu | Normal Reçeteli bir ilaçdır. |
| Barkodu | 8699505761978 |
| Etkin Madde | Okrelizumab |
| ATC Kodu | L04AA36 |
| Birim Miktar | 300 |
| Birim Cinsi | MG |
| Ambalaj Miktarı | 1 |
| Antineoplastik ve İmmünomodülatör Ajanlar > İmmünsupresif Ajanlar |
| İthal ( ref. ülke : Isvicre ) ve Beşeri bir ilaçdır. |