SERONDIR 10 MG+20 MG+30 mg film kapl� tablet tedaviYE BASLAMA paketi K�sa �r�n Bilgisi
{ Apremilast }
1. BE�ER� TIBB� �R�N�N ADI
SEROND�R 10mg + 20mg + 30mg film kapl� tablet tedaviye ba�lama paketi
2. KAL�TAT�F VE KANT�TAT�F B�LE��M
Etkin madde
SEROND�R tedaviye ba�lama paketi i�eri�inde bulunan,
Apremilast 10 mg film kapl� tablet, 10 mg apremilast i�erir. Apremilast 20 mg film kapl� tablet, 20 mg apremilast i�erir. Apremilast 30 mg film kapl� tablet, 30 mg apremilast i�erir.
Yard�mc� maddeler
Apremilast 10 mg film kapl� tablet, 57 mg laktoz monohidrat (s���r kaynakl�) i�erir. Apremilast 20 mg film kapl� tablet, 114 mg laktoz monohidrat (s���r kaynakl�) i�erir. Apremilast 30 mg film kapl� tablet, 171 mg laktoz monohidrat (s���r kaynakl�) i�erir.
Yard�mc� maddeler i�in b�l�m 6.1'e bak�n�z.
3. FARMAS�T�K FORMU
Apremilast 10 mg film kapl� tablet: Pembe renkli, elmas �ekilli, bir y�z�nde “10” yaz�l� film kapl� tabletler.
Apremilast 20 mg film kapl� tablet: Kahverengi renkli, elmas �ekilli, bir y�z�nde “20” yaz�l� film kapl� tabletler.
Apremilast 30 mg film kapl� tablet: Bej renkli, elmas �ekilli, bir y�z�nde “30” yaz�l� film kapl� tabletler.
4. KL�N�K �ZELL�KLER
4.1. Terap�tik endikasyonlar
Ps�riatik artrit
Apremilast, tek ba��na veya Hastal��� Modifiye Edici Antiromatizmal �la�larla (DMARD'lar) kombinasyon halinde �nceki DMARD tedavisine yetersiz yan�t veren veya tolere edemeyen yeti�kin hastalarda aktif ps�riatik artritin (PsA) tedavisinde endikedir (b�l�m 5.1'e bak�n�z).
Ps�riazis
Apremilast siklosporin, metotreksat veya psoralen ve ultraviyole-A ����� (PUVA) dahil olmak �zere di�er sistemik tedavilere yan�t vermeyen, s�z konusu tedavilere kar�� kontrendikasyonu veya intolerans� bulunan yeti�kin hastalarda orta ve �iddetli kronik plak tipi ps�riazisin tedavisinde endikedir.
Pozoloji/uygulama s�kl��� ve s�resi:
Tedavi, ps�riazis veya ps�riatik artrit tan� ve tedavisinde deneyimli bir hekim taraf�ndan ba�lat�lmal� ve kontrol edilmelidir.
Apremilast�n �nerilen dozu yemek k�s�tlamas� olmaks�z�n yakla��k 12 saat arayla sabah ve ak�am olmak �zere g�nde iki kez oral yoldan al�nan 30 mg'd�r. A�a��daki Tablo 1'de g�sterildi�i �ekilde bir ba�lang�� titrasyon program� gerekmektedir. Ba�lang�� titrasyonundan sonra ba�ka bir titrasyon gerekli de�ildir.
Tablo 1: Doz titrasyonu program�
1. G�n | 2. G�n | 3. G�n | 4. G�n | 5. G�n | 6. G�n ve sonras� | |||||
Sabah | Sabah | Ak�am | Sabah | Ak�am | Sabah | Ak�am | Sabah | Ak�am | Sabah | Ak�am |
10 mg | 10 mg | 10 mg | 10 mg | 20 mg | 20 mg | 20 mg | 20 mg | 30 mg | 30 mg | 30 mg |
Hastalar bir dozu atlarsa, bir sonraki doz m�mk�n olan en k�sa zamanda al�nmal�d�r. Bir sonraki dozlar� i�in zaman yakla�m��sa atlanan doz al�nmamal� ve sonraki doz normal zaman�nda al�nmal�d�r.
Pivotal �al��malar s�ras�nda en fazla iyile�me tedavinin ilk 24 haftas� i�inde g�zlenmi�tir. Bir hasta 24 haftadan sonra terap�tik fayda kan�t� g�stermezse tedavi yeniden de�erlendirilmelidir. Hastan�n tedaviye yan�t� d�zenli olarak de�erlendirilmelidir.
4.2. Pozoloji ve uygulama �ekli
SEROND�R a��zdan kullan�m i�indir.
SEROND�R film kapl� tabletler a� veya tok karn�na b�t�n olarak yutulmal�d�r.
�zel pop�lasyonlara ili�kin ek bilgiler:
B�brek yetmezli�i:
Hafif ve orta �iddette b�brek yetmezli�i olan hastalarda herhangi bir doz ayarlamas�na gerek yoktur. Apremilast dozu ciddi b�brek yetmezli�i (Cockcroft-Gault denklemi ile hesaplanan dakikada 30 mL'den az kreatinin klirensi) olan hastalarda g�nde bir kez 30 mg'a azalt�lmal�d�r. Bu grup hastalarda ba�lang�� doz titrasyonu i�in apremilast�n sadece Tablo 1'de listelenen SABAH program� kullan�larak titre edilmesi ve AK�AM dozlar�n�n atlanmas� �nerilir (b�l�m 5.2'ye bak�n�z).
Karaci�er yetmezli�i:
Karaci�er yetmezli�i olan hastalar i�in herhangi bir doz ayarlamas�na gerek yoktur (b�l�m 5.2'ye bak�n�z).
Pediyatrik pop�lasyon:
0-18 ya� aras�ndaki �ocuklarda apremilast�n g�venlili�i ve etkilili�i belirlenmemi�tir. Herhangi bir veri mevcut de�ildir.
Geriyatrik pop�lasyon:
Bu hasta pop�lasyonu i�in herhangi bir doz ayarlamas�na gerek yoktur (b�l�m 4.8 ve 5.2'ye bak�n�z).
4.3. Kontrendikasyonlar
Etkin madde
Gebelikte (b�l�m 4.6'ya bak�n�z) kontrendikedir.
4.4. �zel kullan�m uyar�lar� ve �nlemleri
�shal, Bulant� ve KusmaPazarlama sonras� apremilast kullan�m� ile ili�kili �iddetli ishal, bulant� ve kusma raporlar� al�nm��t�r. �o�u olay tedavinin ilk birka� haftas� i�inde meydana gelmi�tir. Baz� durumlarda hastalar hastaneye yat�r�lm��t�r. 65 ya� veya �zerindeki hastalar komplikasyonlar a��s�ndan daha y�ksek risk ta��yabilir. Hastalarda �iddetli ishal, bulant� veya kusma geli�irse apremilast ile tedavinin kesilmesi gerekebilir.
Psikiyatrik bozukluklar
Apremilast kullan�m�, uykusuzluk ve depresyon gibi psikiyatrik bozukluklar�n artma riski ile ili�kilidir. Depresyon �yk�s� olan veya olmayan hastalarda intihar da dahil olmak �zere intihar d���ncesi ve davran��� g�zlemlenmi�tir (b�l�m 4.8'e bak�n�z). Hastalar �ncesine ait veya mevcut psikiyatrik semptomlar bildirirlerse ya da psikiyatrik olaylara neden olmas� olas� di�er t�bbi �r�nlerle e�zamanl� tedavi yap�lmas� planlan�yorsa apremilast ile tedaviye ba�laman�n veya devam etmenin riskleri ve faydalar� dikkatli bir �ekilde de�erlendirilmelidir. Hastalara ve bak�c�lar�na davran�� veya ruh halinde herhangi bir de�i�ikli�i veya herhangi bir intihar d���ncesini re�ete yazan hekimlerine bildirmeleri s�ylenmelidir. Hastalar�n yeni veya k�t�le�en psikiyatrik semptomlardan muzdarip olmas� veya intihar d���ncesi ya da intihar giri�imi tespit edildi�i durumlarda apremilast ile tedavinin b�rak�lmas� �nerilir.
�iddetli b�brek yetmezli�i
Apremilast dozu, �iddetli b�brek yetmezli�i olan hastalarda g�nde bir kez 30 mg'a d���r�lmelidir (b�l�m 4.2 ve 5.2'ye bak�n�z).
Zay�f hastalar
Tedavinin ba�lang�c�nda olmas� gerekenden zay�f hastalar�n v�cut a��rl�klar� d�zenli olarak izlenmelidir. A��klanmayan ve klinik anlaml� kilo kayb� durumunda bu hastalar bir hekim taraf�ndan de�erlendirilmeli ve tedavinin kesilmesi d���n�lmelidir.
Nadir kal�t�msal galaktoz intolerans�, Lapp laktaz yetmezli�i ya da glukoz-galaktoz malabsorpsiyon problemi olan hastalar�n bu ilac� kullanmamalar� gerekir.
4.5. Di�er t�bbi �r�nler ile etkile�imler ve di�er etkile�im �ekilleri
G��l� sitokrom P450 3A4 (CYP3A4) enzim ind�kleyicisi rifampisinin e�zamanl� uygulanmas� apremilast�n sistemik maruziyetinde bir azalma ile sonu�lanm��t�r, bu da apremilast etkinli�inin kayb�yla sonu�lanabilir. Bu nedenle g��l� CYP3A4 enzim ind�kleyicilerinin (�rn., rifampisin, fenobarbital, karbamazepin, fenitoin ve sar� kantaron) apremilast ile kullan�m� �nerilmemektedir. Apremilast�n birden fazla rifampisin dozu ile e�zamanl� uygulanmas� apremilast�n konsantrasyon zaman e�risi alt�ndaki alan (EAA) ve maksimum serum konsantrasyonu (Cmaks) de�erlerinde s�ras�yla yakla��k %72 ve %43 azalma ile sonu�lanm��t�r. Apremilast maruziyeti g��l� CYP3A4 ind�kleyicileri (�rn., rifampisin) ile e�zamanl� uyguland���nda azal�r ve azalm�� klinik yan�tla sonu�lanabilir.
Klinik �al��malarda, apremilast topikal tedavi (kortikosteroidler, katranl� �ampuan ve salisilik asit kafa derisi preparatlar� dahil) ve UVB fototerapisi ile e�zamanl� olarak uygulanm��t�r. Ketokonazol ve apremilast aras�nda klinik olarak anlaml� bir ila�-ila� etkile�imi olmam��t�r. Apremilast ketokonazol gibi potent bir CYP3A4 inhibit�r� ile birlikte uygulanabilir.
Ps�riatik artrit hastalar�nda apremilast ve metotreksat aras�nda farmakokinetik ila�-ila� etkile�imi yoktur. Apremilast metotreksat ile birlikte uygulanabilir.
Apremilast ve etinil �stradiol ve norgestimat i�eren oral kontraseptifler aras�nda farmakokinetik ila�-ila� etkile�imi olmam��t�r. Apremilast oral kontraseptiflerle birlikte uygulanabilir.
�zel pop�lasyonlara ili�kin ek bilgiler Pediyatrik pop�lasyon:
�ocuklarda herhangi bir etkile�im �al��mas� yap�lmam��t�r.
4.6. Gebelik ve laktasyon
Gebelik kategorisi: C
�ocuk do�urma potansiyeli bulunan kad�nlar/Do�um kontrol� (Kontrasepsiyon) Tedaviye ba�lamadan �nce hastan�n gebe olup olmad��� mutlaka kontrol edilmelidir. �ocuk do�urma potansiyeli olan kad�nlar tedavi s�ras�nda gebeli�i �nlemek i�in etkili bir do�um kontrol y�ntemi kullanmal�d�r.
Gebelik d�nemi
Gebe kad�nlarda apremilast kullan�m� ile ilgili veriler s�n�rl�d�r.
Apremilast gebelik s�ras�nda kontrendikedir. Apremilast�n gebelik �zerindeki etkileri halihaz�rda �nerilen en y�ksek insan dozundan daha y�ksek dozlarda fareler ve maymunlarda embriyofetal kayb� ve farelerde azalm�� fet�s a��rl��� ve gecikmi� kemikle�meyi i�ermi�tir. Klinik maruziyetin 1.3 kat� maruziyetle hayvanlarda bu tip etkiler g�zlenmemi�tir (b�l�m 5.3'e bak�n�z).
Laktasyon d�nemi
Apremilast laktasyondaki farelerin s�t�nde tespit edilmi�tir (b�l�m 5.3'e bak�n�z). Apremilast�n ya da metabolitlerinin insanlarda anne s�t�ne ge�ip ge�medi�i bilinmemektedir. Emzirilen bebekler i�in risk d��lanamaz, bu nedenle apremilast emziren annelerde kullan�lmamal�d�r.
�reme yetene�i/Fertilite
�nsanlarda fertilite verisi yoktur. Farelerde y�r�t�len hayvan �al��malar�nda klinik maruziyetin 3 kat� maruziyet d�zeylerinde erkeklerde ve klinik maruziyetin 1 kat� maruziyet d�zeylerinde di�ilerde hi�bir advers etki g�zlenmemi�tir. Klinik �ncesi fertilite verileri i�in b�l�m 5.3'e bak�n�z.
4.7. Ara� ve makine kullan�m� �zerindeki etkiler
Apremilast�n ara� ve makine kullanma becerisi �zerinde bir etkisi yoktur ya da ihmal edilebilir d�zeydedir.
4.8. �stenmeyen etkiler
G�venlilik profilinin �zetiPsA ve PSOR'da apremilast�n en yayg�n bildirilen yan etkileri, ishal (%15.7) ve mide bulant�s� (%13.9) dahil olmak �zere gastrointestinal (GI) bozukluklard�r. En s�k bildirilen di�er yan etkiler aras�nda �st solunum yolu enfeksiyonlar� (%8.4), ba� a�r�s� (%7.9) ve gerilim tipi ba�
a�r�s� (%7.2) yer al�r ve �o�unlukla hafif ila orta �iddettedir.
Gastrointestinal yan etkiler genellikle tedavinin ilk 2 haftas�nda meydana gelir ve genellikle 4 hafta i�inde d�zelir.
A��r� duyarl�l�k reaksiyonlar� apremilast klinik �al��malar�nda nadiren g�zlenmi�tir (b�l�m 4.3'e bak�n�z).
Apremilast ile tedavi edilen hastalarda ortaya ��kan istenmeyen etkiler Tablo 2'de MedDRA organ sistemine g�re s�ralanm��t�r.
Advers ila� reaksiyonlar� apremilast klinik geli�tirme program�ndan veriler temelinde belirlenmi�tir. Advers ila� reaksiyonlar�n�n s�kl�klar� ps�riatik artritte y�r�t�len d�rt Faz III �al��man�n (n=1945) veya ps�riaziste y�r�t�len iki Faz III �al��man�n (n=1184) ) apremilast kollar�nda bildirilenlerdir (Tablo 2'de her iki veri havuzuna ait en y�ksek s�kl�k g�sterilmektedir).
Advers ila� reaksiyonlar� a�a��da tan�mlanan s�kl��a g�re listelenmi�tir:
�ok yayg�n (1/10); yayg�n (1/100 ila <1/10); yayg�n olmayan (1/1.000 ila <1/100); seyrek (1/10.000 ila <1/1.000); �ok seyrek (<1/10.000); bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor) olarak s�n�fland�r�l�r.
Tablo 2. Ps�riatik artrit ve/veya ps�riazisteki advers reaksiyonlar�n �zeti
Sistem Organ S�n�f� | S�kl�k | Advers reaksiyon |
Enfeksiyonlar ve enfestasyonlar | �ok yayg�n | �st solunum yolu enfeksiyonu |
Yayg�n | Bron�it | |
Nazofarenjit* | ||
Ba����kl�k sistemi hastal�klar� | Yayg�n olmayan | A��r� duyarl�l�k |
Metabolizma ve beslenme hastal�klar� | Yayg�n | ��tah azalmas� * |
Psikiyatrik hastal�klar | Yayg�n | Uykusuzluk Depresyon |
Yayg�n olmayan | �ntihar d���ncesi ve davran���# | |
Sinir sistemi hastal�klar� | �ok yayg�n | Ba� a�r�s�* |
Yayg�n | Gerilim tipi ba� a�r�s�* | |
Migren* | ||
Solunum, g���s bozukluklar� ve mediastinal hastal�klar | Yayg�n | �ks�r�k |
Gastrointestinal hastal�klar | �ok Yayg�n | �shal* Bulant�* |
Yayg�n | Kusma* Dispepsi S�k ba��rsak hareketleri �st kar�n a�r�s�* Gastrointestinal refl� hastal��� | |
Yayg�n olmayan | Gastrointestinal hemoraji | |
Deri ve deri alt� doku hastal�klar� | Yayg�n olmayan | D�k�nt� �rtiker |
Bilinmiyor |
Kas-iskelet bozukluklar�, ba� doku ve kemik hastal�klar� | Yayg�n | S�rt a�r�s�* |
Genel bozukluklar ve uygulama b�lgesine ili�kin hastal�klar | Yayg�n | Yorgunluk |
Ara�t�rmalar | Yayg�n olmayan | Kilo kayb� |
*Bu advers reaksiyonlardan en az biri ciddi olarak bildirilmi�tir.
Se�ili advers reaksiyonlar�n tan�m� Psikolojik bozukluklar
#Klinik �al��malarda ve pazarlama sonras� deneyimde, yayg�n olmayan intihar d���ncesi ve davran��� vakalar� bildirilirken, pazarlama sonras�nda ger�ekle�tirilmi� intihar bildirilmi�tir. Hastalara ve bak�c�lar�na herhangi bir intihar d���ncesini hekimlerine bildirmeleri s�ylenmelidir (b�l�m 4.4'e bak�n�z).
Kilo kayb�
Hasta kilosu klinik �al��malarda rutin olarak �l��lm��t�r. Apremilast ile 52 haftaya kadar tedavi edilen hastalarda ortalama g�zlenen kilo kayb� 1.99 kg'd�r. Apremilast alan hastalar�n toplamda %14.3'�nde %5-10 aras� kilo kayb� g�zlenirken, %5.7'sinde %10'dan fazla kilo kayb� g�zlenmi�tir. Bu hastalar�n hi�birinde kilo kayb�na ba�l� a�ikar klinik sonu�lar geli�memi�tir. Apremilast ile tedavi edilen hastalar�n toplamda %0.1'i azalm�� kilo advers reaksiyonu nedeniyle ilac� b�rakm��t�r.
Tedavinin ba�lang�c�nda zay�f olan hastalar i�in b�l�m 4.4'teki ilave uyar�ya bak�n�z.
�zel pop�lasyonlara ili�kin ek bilgiler:
Geriyatrik pop�lasyon:
Pazarlama sonras� deneyime g�re 65 ya� ve �st�ndeki hastalar �iddetli ishal, bulant� ve kusma komplikasyonlar� a��s�ndan daha y�ksek risk ta��yabilir.
Karaci�er yetmezli�i:
Karaci�er yetmezli�i olan ps�riatik artritli veya ps�riazisli hastalarda apremilast�n g�venlili�i de�erlendirilmemi�tir.
B�brek yetmezli�i:
Ps�riatik artrit veya ps�riazis klinik �al��malar�nda, hafif b�brek yetersizli�i olan hastalarda g�zlenen g�venlilik profili normal b�brek fonksiyonuna sahip hastalardakine benzerdir.
Klinik �al��malarda orta �iddette veya �iddetli b�brek yetersizli�i olan ps�riatik artritli veya ps�riazisli hastalarda apremilast�n g�venlili�i de�erlendirilmemi�tir.
��pheli advers reaksiyonlar�n raporlanmas�
Ruhsatland�rma sonras� ��pheli ila� advers reaksiyonlar�n�n raporlanmas� b�y�k �nem ta��maktad�r. Raporlama yap�lmas�, ilac�n yarar/risk dengesinin s�rekli olarak izlenmesine olanak sa�lar. Sa�l�k mesle�i mensuplar�n�n herhangi bir ��pheli advers reaksiyonu T�rkiye Farmakovijilans Merkezi (T�FAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e- posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks: 0 312 218 35 99).
4.9. Doz a��m� ve tedavisi
Apremilast sa�l�kl� g�n�ll�lerde4.5g�nboyunca100mg'l�k (50 mg BID olarak verilen)
Bir doz a��m� durumunda hastan�n advers etki bulgu ve belirtileri a��s�ndan izlenmesi ve uygun semptomatik tedavinin ba�lat�lmas� �nerilir. Doz a��m� durumunda semptomatik ve destekleyici bak�m tavsiye edilir.
5. FARMAKOLOJ�K �ZELL�KLER
5.1. Farmakodinamik �zellikler
Farmakoterap�tik grup: Selektif imm�nos�presantlar ATC kodu: L04AA32
Etki Mekanizmas�
Bir oral k���k molek�ll� fosfodiesteraz 4 (PDE4) inhibit�r� olan apremilast bir pro- inflamatuar ve anti-inflamatuar mediyat�r a��n� d�zenlemek �zere intrasel�ler olarak i� g�r�r. PDE4, siklik adenozin monofosfata (cAMP) �zg� bir PDE'dir ve inflamatuar h�crelerdeki bask�n olan PDE'dir. PDE4 inhibisyonu intrasel�ler cAMP d�zeylerini art�r�r, bu da TNF-⍺, IL-23, IL-17 ve di�er inflamatuar sitokinlerin ortaya ��kmas�n� d�zenleyerek inflamatuar yan�t� azalt�r. Siklik AMP ayn� zamanda IL-10 gibi anti-inflamatuar sitokinlerin d�zeylerini de d�zenler. Bu pro-inflamatuar ve anti-inflamatuar mediyat�rlerin ps�riatik artrit ve ps�riaziste i�e kar��t�klar� g�sterilmi�tir.
Farmakodinamik Etkiler
Ps�riatik artritli hastalarda y�r�t�len klinik �al��malarda, apremilast IL-1⍺, IL-6, IL-8, MCP- 1, MIP-1ß, MMP-3 ve TNF-⍺'n�n plazma protein d�zeylerini tamamen inhibe etmemekle birlikte �nemli �l��de d�zenlemi�tir. Apremilast ile 40 haftal�k tedaviden sonra IL-17 ve IL- 23'�n plazma protein d�zeylerinde bir azalma ve IL-10'da bir art�� g�r�lm��t�r. Ps�riazisli hastalarda y�r�t�len klinik �al��malarda apremilast lezyonlu deri epidermal kal�nl���, inflamatuar h�cre infiltrasyonu ve ind�klenebilir nitrik oksit sentaz (iNOS), IL-12/IL-23p40, IL-17A, IL-22 ve IL-8 i�in olanlar dahil pro-inflamatuar genlerin ekspresyonunu azaltm��t�r.
50 mg BID'e varan dozlarda uygulanan apremilast sa�l�kl� g�n�ll�lerde QT aral���n� uzatmam��t�r.
Klinik �al��malar Ps�riatik Artrit
Apremilast�n g�venlili�i ve etkilili�i, k���k molek�ll� veya biyolojik DMARD'larla �nceki tedaviye ra�men aktif ps�riatik artritli (≥3 �i� eklem ve ≥3 hassas eklem) yeti�kin hastalarda benzer tasar�ma sahip 3 �ok merkezli, randomize, �ift k�r, plasebo kontroll� �al��mada (PALACE 1, PALACE 2 ve PALACE 3 �al��malar�) de�erlendirilmi�tir. Toplamda 1493 hasta randomize edilmi� ve plasebo, apremilast 20 mg ya da apremilast 30 mg ile g�nde iki kez oral yolla tedavi edilmi�tir.
Bu �al��malardaki hastalar en az 6 ayl�k ps�riatik artrit tan�s�na sahipti. PALACE 3'te ayr�ca niteleyici bir ps�riazis deri lezyonu da (en az 2 cm �ap�nda) gerekliydi. Apremilast k���k molek�ll� DMARD'lar�n stabil dozlar� ile kombinasyon halinde (%65.2) veya monoterapi (%34.8) olarak kullan�lm��t�r. Hastalar �unlar�n biri veya daha fazlas� ile kombinasyon halinde apremilast alm��t�r: metotreksat (MTX, ≤ 25 mg/hafta, %54.5), s�lfasalazin (SSZ, ≤ 2 g/g�n, %9.0) ve leflunomid (LEF; ≤ 20 mg/g�n, %7.4). TNF blokerleri dahil olmak �zere biyolojik DMARD'larla e�zamanl� tedaviye izin verilmemi�tir. Simetrik poliartrit (%62.0), asimetrik oligoartrit (%26.9), distal interfalangeal (DIP) eklem artriti (%6.2), artritis mutilans (%2.7) ve bask�n olarak spondilit (%2.1) dahil her bir ps�riatik artrit alt tipindeki hastalar bu 3 �al��maya kaydedilmi�tir. �nceden mevcut entezopati (%63) veya daktilit (%42) g�r�len hastalar kaydedilmi�tir. Hastalar�ntoplamda%7Saklamaya y�nelik �zel tedbirler tedbirler'�daha�nce sadece k���k molek�ll�
DMARD'larla tedavi edilirken, hastalar�n %22.4'� daha �nce biyolojik DMARD'larla tedavi edilmi�ti (bunlar�n %7.8'inde �nceki biyolojik DMARD ile terap�tik ba�ar�s�zl�k vard�). Medyan ps�riatik artrit hastal�k s�resi 5 y�ld�.
�al��ma tasar�m�na dayal� olarak, hassas ve �i� eklem say�mlar� 16. haftada en az %20 iyile�me g�stermemi� hastalar yan�t vermeyenler olarak kabul edilmi�tir. Yan�t vermeyenler olarak kabul edilen plasebo hastalar� 1:1 oran�nda k�rlenmi� bir �ekilde ya g�nde iki kez apremilast 20 mg ya da g�nde iki kez 30 mg'a yeniden randomize edilmi�tir. 24. Haftada, t�m geri kalan plasebo ile tedavi edilen hastalar ya apremilast 20 ya da 30 mg BID'ye ge�irilmi�tir. 52 haftal�k tedaviden sonra hastalar PALACE 1, PALACE 2 ve PALACE 3 �al��malar�n�n uzun vadeli uzat�lmas� kapsam�nda a��k etiketli apremilast 20 mg veya 30 mg'a 5 y�la varan toplam tedavi s�resince (260 hafta) devam edebilmi�tir.
Birincil sonlan�m noktas� 16. haftada Amerikan Romatoloji Derne�i (ACR) 20 yan�t�n� elde eden hastalar�n y�zdesiydi.
Apremilast ile tedavi 16. haftada plaseboya k�yasla ACR 20 yan�t kriterleri ile de�erlendirilen ps�riatik artrit bulgu ve semptomlar�nda anlaml� iyile�melerle sonu�lanm��t�r. 16. Haftada g�nde iki kez 30 mg apremilast i�in ACR 20/50/70 elde eden hastalar�n oran� (PALACE 1, PALACE 2 ve PALACE 3 �al��malar�ndaki yan�tlar ve PALACE 1, PALACE 2 ve PALACE 3 �al��malar� i�in birle�tirilmi� veriler) Tablo 3'te g�sterilmektedir. ACR 20/50/70 yan�tlar�
24. haftada devam etmi�tir.
Ba�lang��ta g�nde iki kez 30 mg apremilast tedavisine randomize edilmi� hastalar aras�nda ACR 20/50/70 yan�t oranlar� birle�tirilmi� PALACE 1, PALACE 2 ve PALACE 3 �al��malar�nda 52. haftaya kadar korunmu�tur (�ekil 1).
Tablo 3. 16. Haftada PALACE 1, PALACE 2 ve PALACE 3 �al��malar� ve birle�tirilmi� �al��malarda ACR yan�tlar�na sahip hastalar�n oran�
| PALACE 1 | PALACE 2 | PALACE 3 | B�RLE�T�R�LM�� | ||||
N | Plasebo | Apremilast | Plasebo | Apremilast | Plasebo | Apremilast | Plasebo | Apremilast |
| ± | 30 mg BID | ± | 30 mg BID | ± | 30 mg BID | ± | 30 mg BID |
| DMARD | ± | DMARD | ± | DMARD | ± | DMARD | ± |
| N=168 | DMARD | N=159 | DMARD | N=169 | DMARD | N=496 | DMARD |
|
| N=168 |
| N=162 |
| N=167 |
| N=497 |
ACR 20 |
|
|
|
|
|
|
|
|
16. Hafta | %19.0 | %38.1** | %18.9 | %32.1* | %18.3 | %40.7** | %18.8 | %37.0** |
ACR 50 |
|
|
|
|
|
|
|
|
16. Hafta | %6.0 | %16.1* | %5.0 | %10.5 | %8.3 | %15.0 | %6.5 | %13.9** |
ACR 70 |
|
|
|
|
|
|
|
|
16. Hafta | %1.2 | %4.2 | %0.6 | %1.2 | %2.4 | %3.6 | %1.4 | %3.0 |
* Plaseboya kar�� apremilast i�in p≤0.01
** Plaseboya kar�� apremilast i�in p≤0.001
�ekil 1 PALACE 1, PALACE 2 ve PALACE 3 �al��malar�n�n birle�tirilmi� analizinde
52. Haftaya kadar ACR 20/50/70 yan�t� verenlerin oran� (NRI*)
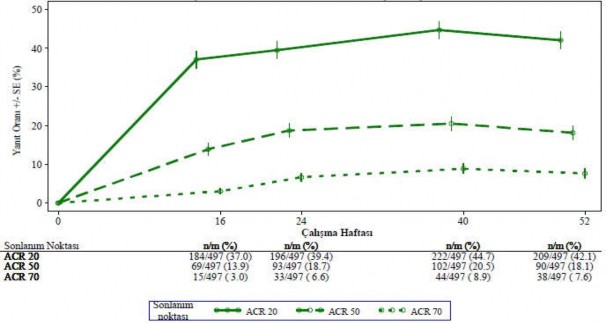
*NRI: Yan�t vermeyen. Zaman noktas�ndan �nce ayr�lan ve zaman noktas�nda yan�t durumunun kesin bir �ekilde belirlenmesi i�in yeterli veriye sahip olmayan denekler yan�t vermeyenler olarak say�lmaktad�r.
Ba�lang��ta g�nde iki kez 30 mg apremilasta randomize edilmi� 497 hasta i�erisinden 375 hasta (%75) 52. haftada halen bu tedaviye devam etmekteydi. Bu hastalarda 52. haftadaki ACR 20/50/70 yan�tlar� s�ras�yla %57, %25 ve %11'di. Ba�lang��ta g�nde iki kez 30 mg apremilasta randomize edilen 497 hasta aras�nda 375 hasta (%75) uzun vadeli uzatma �al��malar�na girmi� olup, bunlardan 221 hasta (%59) 260. haftada halen bu tedaviyi almaktayd�. ACR yan�tlar� uzun vadeli a��k etiketli uzatma �al��malar�nda 5 y�la kadar korunmu�tur.
Apremilast ile tedavi edilen grupta g�zlenen yan�tlar MTX dahil e�zamanl� DMARD kullanan ve kullanmayan hastalarda benzerdi. Daha �nce DMARD'lar veya biyolojiklerle tedavi edilmi� apremilast alan hastalar 16. haftada plasebo alan hastalardan daha y�ksek ACR 20 yan�t� elde etmi�tir.
DIP dahil farkl� ps�riatik artrit alt tiplerinin g�r�ld��� hastalarda benzer ACR yan�tlar� g�zlenmi�tir. Artritis mutilans ve bask�n spondilit alt tipleri g�r�len hastalar�n say�s� anlaml� de�erlendirilebilmek i�in �ok k���kt�.
PALACE 1, PALACE 2 ve PALACE 3'te Hastal�k Aktivitesi �l�e�i (DAS) 28 C-reaktif protein (CRP) ve modifiye ps�riatik artrit yan�t kriterlerine (PsARC) elde eden hastalar�n oran�ndaki iyile�meler 16. haftada plaseboya k�yasla apremilast grubunda daha y�ksekti (nominal p-de�eri s�ras�yla p≤0.0017, p≤0.0004). Bu iyile�meler 24. haftada devam etmi�tir. �al��man�n ba�lang�c�nda randomize edildikleri apremilast tedavisini s�rd�ren hastalar aras�nda DAS28(CRP) skoru ve PsARC yan�t� 52. haftaya kadar korunmu�tur.
16 ve 24. haftalarda apremilast ile tedavi edilen hastalarda ps�riazis artritin periferik aktivite �zelli�ine ili�kin parametrelerde (�rn., �i� eklem say�s�, a�r�l�/hassas eklem say�s�, daktilit ve entezit) ve ps�riazisin deri bulgular�nda iyile�meler g�r�lm��t�r. �al��man�n ba�lang�c�nda randomize edildikleri apremilast tedavisini s�rd�ren hastalarda bu iyile�meler 52. haftaya kadar korunmu�tur.
A��k etiketli uzatma �al��malar�nda 5 y�la varan tedavi boyunca ps�riazisin deri bulgular�nda ve ayn� periferik aktivite parametrelerindeki klinik yan�tlar korunmu�tur.
Fiziksel fonksiyon ve sa�l�klaili�kiliya�amkalitesi
birle�tirilmi� �al��malarda 16. haftada plaseboya k�yasla sa�l�k de�erlendirme anketinin engellilik indeksinde (HAQ-DI) ba�lang�ca g�re de�i�iklik de�erlendirildi�inde fiziksel fonksiyonda istatistiksel olarak anlaml� bir iyile�me g�stermi�tir. HAQ-DI skorlar�ndaki iyile�me 24. haftada devam etmi�tir.
Ba�lang��ta g�nde iki kez 30 mg apremilast tedavisine randomize edilmi� hastalar aras�nda, PALACE 1, PALACE 2 ve PALACE 3 �al��malar�n�n a��k etiketli faz�n�n birle�tirilmi� analizinde 52. haftadaki HAQ-DI skorunda ba�lang�ca g�re de�i�iklik g�nde iki kez 30 mg apremilast grubunda -0.333 idi.
PALACE 1, PALACE 2 ve PALACE 3 �al��malar�nda, 16 ve 24. haftalarda plaseboya k�yasla apremilast ile tedavi edilen hastalarda K�sa Form Sa�l�k Anketi versiyon 2'nin (SF-36v2) fiziksel i�levsellik (PF) domeninde ve Fonksiyonel Kronik Hastal�k Tedavisi De�erlendirmesi – Yorgunluk (FACIT-yorgunluk) skorlar�nda ba�lang�ca g�re de�i�iklikte sa�l�kla ili�kili ya�am kalitesinde anlaml� iyile�meler g�sterilmi�tir. �al��man�n ba�lang�c�nda randomize edildikleri apremilast tedavisini s�rd�ren hastalar aras�nda fiziksel fonksiyon ve FACIT- yorgunluktaki iyile�me 52. haftaya kadar korunmu�tur.
HAQ-DI ve SF36v2PF domeni ve FACIT-yorgunluk skorlar� ile de�erlendirildi�inde iyile�mi� fiziksel fonksiyon a��k etiketli uzatma �al��malar�nda 5 y�ll�k tedavi boyunca korunmu�tur.
Ps�riazis
Apremilast�n g�venlili�i ve etkilili�i, orta �iddette ila �iddetli plak ps�riazisli, (≥ %10 v�cut y�zey alan� (BSA) tutulumu g�r�len, Ps�riazis Alan ve Ciddiyet �ndeks (PASI) skoru ≥ 12 olan, statik Hekim Genel De�erlendirmesi (sPGA) ≥ 3 (orta �iddette veya �iddetli) ve fototerapi ya da sistemik tedaviye aday, toplamda 1257 hastan�n kaydedildi�i iki �ok merkezli, randomize, �ift k�r, plasebo kontroll� �al��mada (ESTEEM 1 ve ESTEEM 2 �al��malar�) de�erlendirilmi�tir.
Bu �al��malar 32. haftaya kadar benzer tasar�ma sahiptir. Her iki �al��mada da hastalar 2:1 oran�nda 16 hafta boyunca apremilast 30 mg BID veya plaseboya (plasebo kontroll� faz) randomize edilmi� ve 16-32. haftalar aras� t�m hastalar apremilast 30 mg BID (idame faz�) alm��t�r. Randomize Tedavinin Geri �ekilme Faz� s�ras�nda (32-52. haftalar) orijinal olarak apremilasta randomize edilmi�, PASI skorlar�nda %75 azalma (PASI-75) (ESTEEM 1) veya
PASI skorlar�nda %50 azalma (PASI-50) (ESTEEM 2) elde etmi� hastalar 32. haftada yeniden plasebo veya apremilast 30 mg BID'ye randomize edilmi�tir. Plaseboya yeniden randomize edilmi� ve 32. haftada PASI-75 yan�t�n� (ESTEEM 1) kaybetmi� veya PASI iyile�mesinin
%50'sini kaybetmi� hastalar (ESTEEM 2) apremilast 30 mg BID ile yeniden tedavi edilmi�tir.
32. Haftaya kadar belirlenen PASI yan�t�na ula�mam�� veya ba�lang��ta plaseboya randomize edilmi� hastalar, 52. haftaya kadar apremilast tedavisinde kalm��t�r. �al��malarda y�z, koltukalt� ve kas�kta d���k potensli topikal kortikosteroid, katranl� �ampuan ve/veya salisilik asit sa� derisi preparatlar�n�n kullan�lmas�na izin verilmi�tir. Buna ilaveten, 32. haftada, ESTEEM 1'de PASI-75 yan�t� veya ESTEEM 2'de PASI-50 yan�t� elde etmemi� deneklerin apremilast 30 mg BID tedavisine ek olarak topikal ps�riazis tedavileri ve/veya fototerapi kullanmalar�na izin verilmi�tir.
52 haftal�k tedaviden sonra, hastalar ESTEEM 1 ve ESTEEM 2 �al��malar�n�n uzun vadeli uzatma �al��malar�yla 5 y�la kadar toplam tedavi s�resi boyunca (260 hafta) a��k etiketli apremilast 30mg kullanmaya devam edebilmi�tir.
Her iki �al��mada da birincil sonlan�m noktas� 16. haftada PASI-75 elde eden hastalar�n oran�d�r. Maj�r ikincil sonlan�m noktas� 16. haftada temiz (0) ya da neredeyse temiz (1) sPGA skoru elde etmi� hastalar�n oran�d�r.
Ortalama ba�lang�� PASI skoru 19.07 (medyan 16.80) olup, ba�lang��ta sPGA skoru 3 (orta �iddette) ve 4 (�iddetli) olan hastalar�n oran� %25.19'luk ortalama BYA tutulumu (medyan
%21.0) ile s�ras�yla %70.0 ve%29.8idi.T�mhastalar�nyakla��k %30'u ps�riazis tedavisi i�in
daha �nce fototerapi ve %54'� daha �nce konvansiyonel sistemik ve/veya biyolojik tedavi g�rm�� olup (tedavi ba�ar�s�zl�klar� dahil), %37'si daha �nce konvansiyonel sistemik tedavi ve
%30'u daha �nce biyolojik tedavi g�rm��t�. Hastalar�n yakla��k ��te biri daha �nce fototerapi, konvansiyonel sistemik veya biyolojik tedavi g�rmemi�ti. Hastalar�n toplamda %18'i ps�riatik artrit �yk�s�ne sahipti.
PASI-50, -75 ve -90 yan�tlar� elde eden ve sPGA skoru temiz (0) veya neredeyse temiz (1) olan hastalar�n oran� a�a��daki Tablo 4'te sunulmaktad�r. Apremilast ile tedavi plaseboya k�yasla 16. haftada PASI-75 yan�t�na sahip hastalar�n oran� ile g�sterildi�i �zere orta �iddette ila �iddetli plak tipi ps�riaziste anlaml� iyile�me ile sonu�lanm��t�r. 16. Haftada sPGA, PASI- 50 ve PASI- 90 yan�tlar� ile �l��len klinik iyile�me g�sterilmi�tir. Buna ilaveten apremilast ka��nt�, t�rnak hastal���, sa� derisi tutulumu ve ya�am kalitesi �l��mleri dahil olmak �zere �e�itli ps�riazis bulgular�nda tedavi faydas� g�stermi�tir.
Tablo 4. ESTEEM 1 ve ESTEEM 2 �al��malar�nda 16. haftadaki klinik yan�t (FAS a, LOCFb)
| ESTEEM 1 | ESTEEM 2 | ||
| Plasebo | 30 mg BID APR* | Plasebo | 30 mg BID APR* |
N | 282 | 562 | 137 | 274 |
PASI, n (%) | 15 (5.3) | 186 (33.1) | 8 (5.8) | 79 (28.8) |
sPGA Temiz veya Neredeyse Temiz, n (%) | 11 (3.9) | 122 (21.7) | 6 (4.4) | 56 (20.4) |
PASI 50, n (%) | 48 (17.0) | 330 (58.7) | 27 (19.7) | 152 (55.5) |
PASI 90, n (%) | 1 (0.4) | 55 (9.8) | 2 (1.5) | 24 (8.8) |
Y�zde De�i�iklik BYA (%) ortalama ± SD | -6.9 ±38.95 | -47.8 ±38.48 | -6.1 ±47.57 | -48.4 ±40.78 |
Ka��nt� VAS'�nda de�i�iklik (mm), ortalama±SD | -7.3 ±27.08 | -31.5 ±32.43 | -12.2 ±30.94 | -33.5 ±35.46 |
DLQI'da de�i�iklik, ortalama±SD | -2.1 ±5.69 | -6.6 ±6.66 | -2.8 ±7.22 | -6.7 ±6.95 |
SF-36 MCS'de de�i�iklik, ortalama±SD | -1.02 ±9.161 | 2.39 ±9.504 | 0.00 ±10.498 | 2.58 ±10.129 |
Apremilast�n klinik faydas� ba�lang�� demografikleri ve ba�lang�� klinik hastal�k �zellikleri ile tan�mlanan (ps�riazis hastal��� s�resi ve ps�riatik artrit �yk�s� olan hastalar dahil) �ok say�da alt grup aras�nda g�sterilmi�tir. Apremilast�n klinik faydas� ayn� zamanda �nceki ps�riazis ilac� kullan�m� ve �nceki ps�riazis tedavilerine verilen yan�ta bak�lmaks�z�n g�sterilmi�tir. T�m kilo aral�klar�nda benzer yan�t oranlar� g�zlenmi�tir.
Apremilasta yan�t, 2. hafta itibariyle plaseboya k�yasla PASI, deride rahats�zl�k/a�r� ve ka��nt�
Genelde PASI yan�tlar� 16. hafta itibariyle elde edilmi� ve 32. haftaya kadar devam etmi�tir. Her iki �al��mada da PASI'da ba�lang�ca g�re ortalama y�zde iyile�me 32. Haftada apremilasta yeniden randomize edilmi� hastalar i�in Randomize Tedaviden �ekilme Faz� s�ras�nda stabil kalm��t�r (Tablo 5).
Tablo 5. 0. Haftada APR 30 BID'ye randomize edilmi� ve 32. hafta ila 52. haftada APR 30 BID'ye yeniden randomize edilmi� deneklerdeki etkinin kal�c�l���
| Zaman Noktas� | ESTEEM 1 | ESTEEM 2 |
32. Haftada PASI-75 elde eden hastalar | 32. Haftada PASI-50 elde eden hastalar | ||
PASI'da ba�lang�ca g�re Y�zde De�i�iklik (%) ± SD | 16. Hafta | -77.7±20.30 | -69.7±24.23 |
32. Hafta | -88±8.30 | -76.7±13.42 | |
52. Hafta | -80.5±12.60 | -7.8±Saklamaya y�nelik �zel tedbirler tedbirler1 | |
DLQI'da ba�lang�ca g�re de�i�iklik, ortalama±SD | 16. Hafta | -8.3±6.26 | -74.4±18.91 |
32. Hafta | -8.9±6.68 | -7.8±Saklamaya y�nelik �zel tedbirler tedbirler1 | |
52. Hafta | -7.8±5.75 | -7.7±5.92 | |
Sa� Derisi Ps�riazisi PGA's� (ScPGA) 0 veya 1 olan g�n�ll�lerin oran�, n/N (%) | 16. Hafta | 40/48 (83.3) | 21/37 (56.8) |
32. Hafta | 39/48 (81.3) | 27/37 (73.0) | |
52. Hafta | 35/48 (72.9) | 20/37 (54.1) |
ESTEEM 1 �al��mas�nda, 32. haftada apremilasta yeniden randomize edilmi� hastalar�n yakla��k %61'i 52. haftada PASI-75 yan�t�na sahipti. Randomize Tedaviden Geri �ekilme Faz� s�ras�nda 32. haftada plaseboya yeniden randomize edilmi� en az�ndan PASI-75 yan�t�na sahip hastalardan %11.7'si 52. haftada PASI-75 yan�t� veren hastalard�. Plaseboya yeniden randomize edilmi� hastalar aras�nda PASI-75 yan�t� kayb�na kadar ge�en medyan s�re 5.1 haftayd�.
ESTEEM 2 �al��mas�nda, 32. haftada apremilasta yeniden randomize edilmi� hastalar�n yakla��k %80.3'� 52. haftada PASI-50 yan�t�na sahipti. 32. Haftada plaseboya yeniden randomize edilmi� en az PASI-50 yan�t�na sahip hastalardan %24.2'si 52. haftada PASI-50 yan�t� veren hastalard�. 32. Hafta PASI iyile�melerinde %50 kayba kadar ge�en medyan s�re
12.4 haftayd�.
32. Haftada randomize tedaviden geri �ekilme sonras�nda, ESTEEM 1 �al��mas�ndaki hastalar�n yakla��k %70'i ve ESTEEM 2 �al��mas�ndaki hastalar�n %65.6's� apremilast tedavisinin yeniden ba�lat�lmas�ndan sonra PASI-75 (ESTEM 1) veya PASI-50 (ESTEEM 2) yan�tlar�n� yeniden kazanm��t�r. �al��ma tasar�m�ndan dolay� yeniden tedavinin s�resi de�i�ken olup, 2.6 ile 22.1 hafta aras�nda de�i�mi�tir.
ESTEEM 1 �al��mas�nda, �al��man�n ba�lang�c�nda apremilasta randomize edilmi� 32. haftada PASI-75 yan�t� elde etmemi� hastalar�n, 32 ve 52. haftalar aras�nda e�zamanl� topikal tedaviler ve/veya UVB fototerapisi kullanmalar�na izin verilmi�tir. Bu hastalardan %12'si 52. haftada apremilast art� topikal tedavi ve/veya fototerapi ile PASI-75 yan�t� elde etmi�tir.
ESTEEM 1 ve ESTEEM 2 �al��malar�nda, 16. haftada plasebo ile tedavi edilen hastalara k�yasla apremilast alan hastalarda T�rnak Ps�riazis �iddet �ndeksinde (NAPSI) ba�lang�ca g�re ortalama de�i�iklik y�zdesi ile �l��ld����zeret�rnakps�riazisindeanlaml� iyile�meler (azalmalar)
hastalarda 32. haftada t�rnak ps�riazisinde ilave iyile�meler g�zlenmi�tir.
ESTEEM 1 ve ESTEEM 2 �al��malar�nda plasebo ile tedavi edilen hastalara k�yasla apremilast alan hastalarda 16. haftada Sa� Derisi Ps�riazisi Hekimin Genel De�erlendirmesi (ScPGA) temiz (0) ya da minimum (1) skoru elde etmi� hastalar�n oran� ile �l��ld��� �zere en az orta �iddette (≥3) sa� derisi ps�riazisinde anlaml� iyile�meler g�zlenmi�tir. �yile�meler Apremilasta yeniden randomize edilmi� deneklerde 32. haftadan 52. haftaya genellikle korunmu�tur (Tablo 5).
ESTEEM 1 ve ESTEEM 2 �al��malar�nda, plasebo ile tedavi edilen hastalara k�yasla apremilast alan hastalarda Dermatoloji Ya�am Kalitesi �ndeksi (DLQI) ve SF-36v2MCS ile �l��len ya�am kalitesinde anlaml� iyile�meler g�r�lm��t�r (Tablo 4). DLQI'daki iyile�meler32. haftada apremilasta yeniden randomize edilmi� deneklerde 52. haftaya kadar korunmu�tur (Tablo 5). Buna ilaveten, ESTEEM 1 �al��mas�nda, plaseboya k�yasla apremilast alan hastalarda �� K�s�tlamalar� Anketinde (WLQ-25) anlaml� iyile�me elde edilmi�tir.
Ba�lang��ta g�nde iki kez 30 mg apremilasta randomize edilmi� 832 hasta aras�nda 443 hasta (%53) ESTEEM 1 ve ESTEEM 2'nin a��k etiketli uzatma �al��malar�na girmi� olup, bunlardan
115 hasta (%26) 260. haftada halen tedavi g�rmekteydi. ESTEEM 1 ve ESTEEM 2 �al��malar�n�n a��k etiketli uzatmas�nda apremilasta devam eden hastalar i�in PASI skoru, etkilenmi� BSA, ka��nt�, t�rnak ve ya�am kalitesi �l��mlerinde iyile�meler genellikle 5 y�la kadar korunmu�tur.
Ps�riatik artrit ve ps�riazis g�r�len hastalarda g�nde iki kez 30 mg apremilast�n uzun d�nem g�venlili�i 5 y�la varan toplam tedavi s�resince de�erlendirilmi�tir. Apremilast ile a��k etiketli uzatma �al��malar�nda uzun d�nemli deneyim genellikle 52 haftal�k �al��malardakine benzer olmu�tur.
5.2. Farmakokinetik �zellikler
Genel �zelliklerEmilim
Apremilast yakla��k 2.5 saatlik medyan s�rede (tmaks) meydana gelen pik plazma konsantrasyonlar� (Cmaks) e�li�inde yakla��k %73 mutlak oral biyoyararlan�m ile iyi d�zeyde emilmektedir. Apremilast farmakokineti�i do�rusal olup, g�nl�k 10 ile 100 mg doz aral���nda sistemik maruziyette dozla orant�l� bir art�� s�z konusudur. Birikim apremilast g�nde bir kez uyguland���nda minimum ve g�nde iki kez uyguland���nda sa�l�kl� g�n�ll�lerde yakla��k
%53 ve ps�riazisli g�n�ll�lerde %68'dir. G�dalarla birlikte uygulama biyoyararlan�m� de�i�tirmedi�inden apremilast a� veya tok karn�na uygulanabilir.
Da��l�m
Apremilast�n insan plazma proteinine ba�lanmas� yakla��k %68'dir. Ortalama g�r�n�r da��l�m hacmi (Vd) 87L olup, ekstravask�ler da��l�ma i�aret eder.
Biyotransformasyon
Apremilast oksidasyon, hidroliz ve konjugasyon dahil yayg�n olarak hem CYP arac�l� hem de CYP d��� yolaklarla metabolize edilir, bu da tek bir klirens yola��n�n inhibisyonunun belirgin ila�-ila� etkile�imine neden olmas�n�n muhtemel olmad���n� d���nd�r�r. Apremilast�n oksidatif metabolizmas�na ba�l�ca CYP3A4 arac�l�k ederken CYP1A2 ve CYP2A6'dan min�r katk�lar s�z konusudur. Apremilast oral uygulamay� takiben dola��mdaki maj�r bile�endir. Apremilast, uygulanan ana bile�i�in s�ras�yla sadece % 3 ve % 7'sinin idrar ve d��k�da geri kazan�lmas�yla yo�un bir metabolizmaya maruz kal�r. Dola��mdaki maj�r inaktif metabolit O- demetile apremilast�n glukuronid konjugat�d�r(M12). Apremilast�nbir CYP3A4 substrat� olmas� ile
e�zamanl� uyguland���nda azal�r.
�n vitro apremilast sitokrom P450 enzimlerinin inhibit�r� ya da ind�kleyicisi de�ildir. Bu nedenle CYP enzimlerinin substratlar� ile birlikte uygulanan apremilast�n CYP enzimleri ile metabolize edilen etkin maddelerin klirensini ve maruziyetini etkilemesi olas� de�ildir.
�n vitro apremilast bir P-glikoprotein substrat� ve zay�f inhibit�r�d�r (IC50>50µM), bununla birlikte P-gp arac�l���yla klinik a��dan ilgili ila� etkile�imlerinin meydana gelmesi beklenmemektedir.
�n vitro apremilast Organik Anyon Ta��y�c�s� (OAT)1 ve OAT3, Organik Katyon Ta��y�c�s� (OKT)2, Organik Anyon Ta��y�c�s� Polipeptid (OATP)1B1 ve OATP1B3 veya meme kanseri diren� proteini (BCRP) �zerinde inhibe edici etkiye sahip de�ildir veya �ok az etki s�z konusudur ve bu ta��y�c�lar i�in bir substrat de�ildir. Bu nedenle apremilast bu ta��y�c�lar�n substratlar� veya inhibit�rleri ile birlikte uyguland���nda klinik a��dan ilgili ila�-ila� etkile�imleri olas� de�ildir.
Eliminasyon
Apremilast�n plazma klirensi yakla��k 9 saatlik bir terminal eliminasyon yar�lanma �mr� ile sa�l�kl� ki�ilerde ortalama yakla��k 10 L/saattir. Radyo-etiketli apremilast�n oral yolla uygulanmas�n� takiben radyoaktivitenin yakla��k %58 ve %39'u s�ras�yla idrar ve fe�este geri kazan�l�rken, radyoaktif dozun s�ras�yla %3'� ve %7'si idrar ve fe�este apremilast olarak geri kazan�lm��t�r.
�zel pop�lasyonlar:
Geriyatrik pop�lasyon:
Apremilast gen� ve ya�l� sa�l�kl� g�n�ll�lerde ara�t�r�lm��t�r. Ya�l� g�n�ll�lerde (65 ila 85 ya�) maruziyet gen� g�n�ll�lere (18 ila 55 ya�) k�yasla apremilast i�in EAA'da yakla��k %13 daha y�ksek ve C'ta yakla��k %6 daha y�ksektir. Klinik �al��malarda 75 ya� �zerindeki g�n�ll�lerde farmakokinetik veriler s�n�rl�d�r. Ya�l� hastalar i�in herhangi bir doz ayarlamas� gerekli de�ildir.
B�brek yetmezli�i:
Hafif veya orta �iddette b�brek yetmezli�i olan g�n�ll�ler ve e�le�tirilmi� sa�l�kl� g�n�ll�ler aras�nda apremilast�n FK's�nda anlaml� bir fark yoktur (her biri N=8). Bulgular hafif ve orta �iddette b�brek yetmezli�i olan hastalarda doz ayarlamas� gerekmedi�ini desteklemektedir. �iddetli b�brek yetmezli�i (eGFR <30 mL/dakika/1.73 m2 veya CLcr < 30 mL/dakika) olan hastalarda apremilast dozu g�nde bir kez 30 mg'a azalt�l�r. 30 mg'l�k tek doz apremilast uygulanan �iddetli b�brek yetmezli�i olan 8 g�n�ll�de apremilast�n EAA ve Cmaks de�erleri s�ras�yla yakla��k %89 ve %42 artm��t�r.
Karaci�er yetmezli�i:
Apremilast ve maj�r metaboliti M12'nin farmakokineti�i orta �iddette veya �iddetli karaci�er yetmezli�inden etkilenmez. Karaci�er yetmezli�i olan hastalar i�in herhangi bir doz ayarlamas� gerekli de�ildir.
5.3. Klinik �ncesi g�venlilik verileri
Klinik d��� veriler konvansiyonel g�venlilik farmakolojisi ve tekrarlanan doz toksisitesi �al��malar� temelinde insanlar i�in �zel bir tehlikeyi ortaya koymam��t�r. �mm�notoksik potansiyel, dermal irritasyon veya fototoksik potansiyel i�in kan�t yoktur.
![]()
Fertilite ve erken embriyonikgeli�im
fertilitesi �zerinde bir etkiye yol a�mam��t�r; erkek fertilitesi i�in advers etki g�zlenmeyen d�zey (NOAEL) 50 mg/kg/g�nden fazlad�r (klinik maruziyetin 3 kat�).
10, 20 ,40 ve 80 mg/kg/g�nl�k oral dozajlarla birle�tirilmi� bir di�i fare fertilitesi ve embriyo- fetal geli�imsel toksisite �al��mas�nda 20 mg/kg/g�n ve �zerinde �strus sikluslarda uzama ve �iftle�meye kadar ge�en s�rede art�� g�zlenmi�tir; buna ra�men farelerin t�m� �iftle�mi� ve gebelik oranlar� etkilenmemi�tir. Di�i fertilitesi i�in advers etki g�zlenmeyen d�zey (NOEL) 10 mg/kg/g�nd�r (klinik maruziyetin 1.0 kat�).
Embriyo-fetal geli�im
10, 20, 40 ve 80 mg/kg/g�nl�k oral dozajlarla birle�tirilmi� bir di�i fare fertilitesi ve embriyo- fetal geli�imsel toksisite �al��mas�nda maternal hayvanlar�n mutlak ve/veya nispi kalp a��rl�klar� 20, 40 ve 80 mg/kg/g�nde artm��t�r. Artm�� erken rezorpsiyon say�s� ve azalm�� kemikle�mi� tarsal say�s� 20, 40 ve 80 mg/kg/g�nde g�zlenmi�tir. 40 ve 80 mg/kg/g�nde supraoksipital kafatas� kemi�inin osifikasyonunda gecikme ve azalm�� fetal a��rl�klar g�zlenmi�tir. Farede maternal ve geli�imsel NOEL 10 mg/kg/g�nd�r (klinik maruziyetin 1.3 kat�).
Bir maymun embriyo-fetal geli�imsel toksisite �al��mas�nda 20, 50, 200 ve 1000 mg/kg/g�nl�k oral dozajlar 50 mg/kg/g�n ve �zerindeki dozajlarda prenatal kay�pta (d���k) dozla ili�kili bir art��la sonu�lanm��t�r; 20 mg/kg/g�nde (klinik maruziyetin 1.4 kat�) prenatal kay�pta test maddesi ile ili�kili bir etki g�zlenmemi�tir.
Pre-natal ve post-natal geli�im
Bir pre-natal ve post-natal �al��mada apremilast gebe di�i farelere 10, 80 ve 300 mg/kg/g�n dozajlar�nda 6. gestasyon g�n�nden (GG) laktasyonun 20. g�n�ne kadar oral yolla uygulanm��t�r. 300 mg/kg/g�nde maternal v�cut a��rl��� ve kilo art���nda azalmalar ve yavrular�n do�urulmas�nda g��l�kle ili�kili bir �l�m g�zlenmi�tir. Yavrular�n do�umu ile ili�kili maternal toksisitenin fiziksel belirtileri ayn� zamanda 80 ve 300 mg/kg/g�n�n her birinde bir farede g�zlenmi�tir. ≥80 mg/kg/g�nde (klinik maruziyetin ≥4.0 kat�) artm�� pre- natal ve postnatal yavru �l�mleri ve laktasyonun ilk haftas� s�ras�nda azalm�� yavru v�cut a��rl�klar� g�zlenmi�tir. Gebelik s�resi, gestasyon periyodunun sonunda gebe farelerin say�s�, yavru do�uran farelerin say�s� �zerinde apremilast ile ili�kili etkiler veya do�um sonras� 7. g�n �tesinde yavrularda herhangi bir geli�imsel etkiler s�z konusu de�ildir. Postnatal periyodun ilk haftas� s�ras�nda g�zlenen yavru geli�imsel etkileri muhtemelen apremilast ile ili�kili yavru toksisitesi (azalm�� yavru kilosu ve canl�l���) ve/veya maternal bak�m eksikli�i (yavrular�n midesinde s�t olmamas� a��s�ndan daha y�ksek insidans) ile ili�kilidir. T�m geli�imsel etkiler postnatal periyodun ilk haftas� s�ras�nda g�zlenmi�tir; s�tten kesme �ncesi ve sonras� periyotlar�n geri kalan�nda cinsel olgunla�ma, davran��, �iftle�me, fertilite ve uterin parametreleri dahil olmak �zere apremilastla ili�kili hi�bir etki g�r�lmemi�tir. Maternal toksisite ve F1 jenerasyonu i�in faredeki NOEL 10 mg/kg/g�nd�r (klinik EAA'n�n 1.3 kat�).
Karsinojenisite �al��malar�
Fareler ve s��anlarda karsinojenisite �al��malar� apremilast tedavisi ile ili�kili karsinojenisite kan�t� g�stermemi�tir.
Genotoksisite �al��malar�
Apremilast genotoksik de�ildir. Apremilast Ames analizinde mutasyonlar� veya metabolik aktivasyon varl���nda veya yoklu�unda k�lt�rlenmi� insan periferik kan lenfositlerinde kromozom anomalilerini ind�klememi�tir. Apremilast 2000 mg/kg/g�ne varan dozlarda in vivo fare mikronukleus analizinde klastojenik bulunmam��t�r.
Di�er �al��malar
�mm�notoksik, dermal iritasyonveyafototoksikpotansiyeledair kan�ta rastlanmam��t�r.
6. FARMAS�T�K �ZELL�KLER
6.1. Yard�mc� maddelerin listesi
APREM�LAST 10 mg film kapl� tablet Tablet �ekirde�i
Mikrokristalin sel�loz
Laktoz monohidrat (s���r kaynakl�) Kroskarmelloz sodyum
Kolloidal silikon dioksit Magnezyum stearat
Film kaplama maddesi Hipromelloz
Titanyum dioksit (E171) Makrogol 3350
Demir oksit k�rm�z� (E172)
APREM�LAST 20 mg film kapl� tablet Tablet �ekirde�i
Mikrokristalin sel�loz
Laktoz monohidrat (s���r kaynakl�) Kroskarmelloz sodyum
Kolloidal silikon dioksit Magnezyum stearat
Film kaplama maddesi Hipromelloz
Titanyum dioksit (E171) Makrogol 3350
Demir oksit k�rm�z� (E172) Demir oksit sar� (E172)
APREM�LAST 30 mg film kapl� tablet Tablet �ekirde�i
Mikrokristalin sel�loz
Laktoz monohidrat (s���r kaynakl�) Kroskarmelloz sodyum
Kolloidal silikon dioksit Magnezyum stearat
Film kaplama maddesi Hipromelloz
Titanyum dioksit (E171) Makrogol 3350
Demir oksit k�rm�z� (E172) Demir oksit sar� (E172) Demir oksit siyah (E172)
6.2. Ge�imsizlikler
Ge�erli de�il.
6.3. Raf �mr�
24 ay
6.4. Saklamaya y�nelik �zel tedbirler
25°C alt�ndaki oda s�cakl���nda saklay�n�z.
6.5. Ambalaj�n niteli�i ve i�eri�i
Tedaviye ba�lama paketi 27 film kapl� tablet i�erir (4 adet 10 mg, 4 adet 20 mg, 19 adet 30 mg). Film kapl� tabletler �effaf PVC/PE/PVdC - al�minyum folyo blister ambalajlarda kullanma talimat� ile birlikte sunulmaktad�r.
6.6. Be�eri t�bbi �r�nden arta kalan maddelerin imhas� ve di�er �zel �nlemler
Kullan�lmam�� olan �r�nler ya da at�k materyaller “T�bbi At�klar�n Kontrol� Y�netmeli�i” ve “Ambalaj At�klar�n�n Kontrol� Y�netmeli�i”ne uygun olarak imha edilmelidir.
 A��z Kanseri
A��z kanserinin en yayg�n t�rleri, dudak, dil, di�etidir. Nadiren
yanak i�i veya damak b�lgelerini de i�ine al�r.
A��z Kanseri
A��z kanserinin en yayg�n t�rleri, dudak, dil, di�etidir. Nadiren
yanak i�i veya damak b�lgelerini de i�ine al�r. |
 Deri Kanseri
Deri kanseri �ok rastlanan bir hastal�kt�r. �� ana t�r� bulunur ;genelde kemirici �lser olarak bilinen bazal h�creli karsinom, yass� h�creli karsinom ve k�t� huylu t�m�r.
Deri Kanseri
Deri kanseri �ok rastlanan bir hastal�kt�r. �� ana t�r� bulunur ;genelde kemirici �lser olarak bilinen bazal h�creli karsinom, yass� h�creli karsinom ve k�t� huylu t�m�r. |
�LA� E�DE�ERLER�
| E�de�er �la� Ad� | Barkodu | �la� Fiyat� |
|---|---|---|
| LEDEZLA | 8699511099041 | 3,743.91TL |
| OPRELAST | 8699586096068 | 3,923.41TL |
| SERONDIR | 8680972609139 | 3,027.86TL |
| Di�er E�de�er �la�lar |
 Kanseri") |
Rahim Boyu ( Serviks ) Kanseri Rahim boynu (serviks) kanseri 35 ya� alt� kad�nlarda g�r�len vakalarda meme kanserinden sonra ikinci s�ray� al�r.Serviks kanserinin geli�mesi y�llarca s�rebilir. |
 |
S�rt A�r�s� S�rt a�r�s� birden bire ortaya ��k�p �iddetli (akut) olabilir veya zamanla geli�ip daha uzun s�reli sorunlara (kronik) neden olabilir. |
 |
Tiroid Kanseri En s�k g�r�len tiroid kanseri t�r� olan papiller tiroid kanseri, t�m tiroid kanserlerinin yakla��k %70'ini olu�turur. |
�LA� GENEL B�LG�LER�
Pharmer Dan��manl�k Sa�l�k Ve Kozmetik �r�nleri Ltd.�ti
| Sat�� Fiyat� | 3027.86 TL [ 28 Jun 2024 ] |
| �nceki Sat�� Fiyat� | 3027.86 TL [ 14 Jun 2024 ] |
| Original / Jenerik | Original �la� |
| Re�ete Durumu | K�s�tlanm�� Beyaz Re�eteli bir ila�d�r. |
| Barkodu | 8680972609139 |
| Etkin Madde | Apremilast |
| ATC Kodu | L04AA32 |
| Birim Miktar | 10+20+30 |
| Birim Cinsi | MG |
| Ambalaj Miktar� | 4+4+19 |
| Antineoplastik ve �mm�nomod�lat�r Ajanlar > �mm�nsupresif Ajanlar |
| Yerli ve Be�eri bir ila�d�r. |