SEVASTRA 100 mg film kapl� tablet K�sa �r�n Bilgisi
{ Erlotinib }
1. BE�ER� TIBB� �R�N�N ADI
SEVASTRA 100 mg film kapl� tablet
2. KAL�TAT�F VE KANT�TAT�F B�LE��M
Etkin madde
Bir film kapl� tablet 100 mg erlotinibe e�de�er miktarda 109,3 mg erlotinib hidroklor�r i�erir.
Yard�mc� maddeler
Sodyum ni�asta glikolat 24 mg Sodyum lauril s�lfat 3 mg
Susuz laktoz 3 mg
Yard�mc� maddeler i�in B�l�m 6.1'e bak�n�z.
3. FARMAS�T�K FORMU
Film kapl� tablet.
Bir y�z� “100” �zeri “E” bask�l�, di�er y�z� d�z, beyaz ila beyaz�ms� renkte, yuvarlak, bikonveks film kapl� tabletler halindedir.
4. KL�N�K �ZELL�KLER
4.1. Terap�tik endikasyonlar
K���k h�creli d��� akci�er kanseri (KHDAK):
SEVASTRA, epidermal b�y�me fakt�r� resept�r� (EGFR) gen exon 19 delesyonu ve/veya exon 21(L858R) mutasyonu akredite bir laboratuvarda g�sterilen, metastatik non-skuam�z k���k h�creli d��� akci�er kanseri hastalar�n�n birinci basamak tedavisinde ve yukar�da tan�mlanan mutasyon ve delesyonu olan non-skuam�z k���k h�creli d��� akci�er kanseri hastalar�nda bir basamak kemoterapi sonu progresyonunda ikinci basamak tedavisinde progresyona kadar kullan�m� endikedir.
4.2. Pozoloji ve uygulama �ekli
SEVASTRA tedavisi, antikanser terapilerin kullan�m�nda deneyimli olan bir hekim taraf�ndan ba�lat�lmal�d�r.
Doktor taraf�ndan ba�ka �ekilde tavsiye edilmedi�i takdirde; Standart doz:
K���k H�creli D��� Akci�er Kanseri:
�leri veya metastatik evre k���k h�creli d��� akci�er kanseri (KHDAK) olan birinci basamak kemoterapi almam�� hastalardaSEVASTRAtedavisineba�lamadan �nce EGFR mutasyon
�nerilen g�nl�k SEVASTRA dozu yemeklerden en az bir saat �nce veya en az iki saat sonra al�nmak �zere 150 mg'd�r.
Uygulama �ekli:
A��zdan al�n�r.
�zel pop�lasyonlara ili�kin ek bilgiler:
Doz ayarlamas� gerekti�inde, dozu 50 mg'l�k ad�mlarla d���rmeniz tavsiye edilmektedir (Bkz. B�l�m 4.4).
CYP3A4 substratlar� ve d�zenleyicileri ile e� zamanl� kullan�m�nda doz ayarlamas� gerekebilir (Bkz. B�l�m 4.5).
Karaci�er yetmezli�i:
Erlotinib birincil olarak karaci�er arac�l���yla metabolize edilir ve safra ile itrah edilir. Hafif derece karaci�er fonksiyon bozuklu�u (Child-Pugh skoru 7-9) olan hastalar karaci�er fonksiyonu yeterli olan hastalar ile kar��la�t�r�ld���nda, erlotinib at�l�m� benzer olmas�na ra�men, karaci�er yetmezli�i olan hastalarda SEVASTRA uygulan�rken dikkatli olunmal�d�r. E�er ciddi advers olaylar geli�irse, doz azalt�m� veya SEVASTRA'ya ara verilmesi d���n�lmelidir. Ciddi karaci�er fonksiyon bozuklu�u olan hastalarda (AST/SGOT ve ALT/SGPT de�erleri > 5 x normal �st s�n�r) erlotinibin g�venlili�i ve etkilili�i �al���lmam��t�r (Bkz. B�l�m 4.4 ve 5.2). Total bilirubini normal �st limitten 3 kat y�ksek olan hastalarda SEVASTRA kullan�lmamal�d�r.
B�brek yetmezli�i:
Erlotinibin g�venlilik ve etkilili�i b�brek yetmezli�i bulunan hastalarda (serum kreatinin konsantrasyonu >1,5 x normal �st s�n�r) ara�t�r�lmam��t�r. Farmakokinetik �al��malara g�re hafif veya orta derece b�brek yetmezli�i hastalar�nda doz ayarlamas� gerekmemektedir (Bkz. B�l�m 5.2). �leri derece b�brek yetmezli�i hastalar�nda SEVASTRA kullan�m� �nerilmemektedir.
Pediyatrik pop�lasyon:
Erlotinibin g�venlilik ve etkilili�i onayl� endikasyonlarda 18 ya��n alt�ndaki hastalarda ara�t�r�lmam��t�r. Pediyatrik hastalarda kullan�m� �nerilmemektedir.
Geriyatrik pop�lasyon:
Erlotinibin g�venlilik ve etkilili�i ya�l� hastalarda ara�t�r�lmam��t�r.
Sigara i�enler:
Sigara i�menin erlotinib maruziyetini %50-60 azaltt��� g�sterilmi�tir. Halihaz�rda sigara i�en k���k h�creli d��� akci�er kanseri (KHDAK) hastalar�nda tolere edilebilen maksimum SEVASTRA dozu 300 mg'd�r.
Sigara i�meye devam eden hastalarda, ba�ar�s�z kemoterapi sonras� ikinci basamak tedavide �nerilen 150 mg doz ile kar��la�t�r�ld���nda 300 mg doz kullan�m� etkililikte art�� g�stermemi�tir. G�venlilik verileri 300 mg ve 150 mg dozlar� aras�nda kar��la�t�r�labilirdir ancak, daha y�ksek erlotinib dozu alan hastalarda d�k�nt�, interstisyel akci�er hastal��� ve diyare insidans�nda say�sal bir art�� olmu�tur. Halihaz�rda sigara i�en hastalara sigaray� b�rakmalar� �nerilmelidir (bk z.B�l�m4.4,4.5,5.1ve5.2).
4.3. Kontrendikasyonlar
SEVASTRA, erlotinib veya B�l�m 6.1'de listelenen yard�mc� maddelerden herhangi birine kar�� a��r� duyarl�l�k durumunda kontrendikedir.
4.4. �zel kullan�m uyar�lar� ve �nlemleri
EGFR mutasyon durumunun saptanmas�
SEVASTRA'n�n lokal ileri ya da metastatik KHDAK'de birinci basamak tedavide kullan�m� de�erlendirilirken, hastan�n EGFR mutasyon durumunun belirlenmesi �nemlidir.
Doku �rne�inden elde edilen t�m�r DNA's�n� veya kan (plazma) �rne�inden elde edilen dola��mdaki serbest DNA'y� (cfDNA) kullanan, �nceden belirlenmi� pozitiflik limitleri olan ve EGFR mutasyon durumunun belirlenmesi konusunda faydas� kan�tlanm��, valide, sa�lam, g�venilir ve duyarl� bir test uygulanmal�d�r.
E�er plazma bazl� cfDNA kullan�lm�� ve sonu� aktive edici mutasyonlar y�n�nden negatif bulunmu�sa, plazma bazl� testle yalanc� negatif sonu�lar ��kmas� muhtemel oldu�undan m�mk�nse doku testi de yap�lmal�d�r.
Sigara i�enler
Sigara i�meyenlere k�yasla sigara i�enlerde erlotinib plazma konsantrasyonlar�n�n d���k olmas� sebebiyle, halihaz�rda sigara i�enlere sigaray� b�rakmalar� �nerilmelidir. Konsantrasyondaki d���� derecesinin klinik olarak anlaml� olmas� beklenmektedir (bkz. B�l�m 4.2, 4.5, 5.1 ve 5.2).
�nterstisyel Akci�er Hastal���
K���k h�creli d��� akci�er kanseri (KHDAK) tedavisi i�in erlotinib almakta olan hastalarda �ok seyrek olarak, baz�lar� �l�mc�l olabilen, interstisyel akci�er hastal��� (�AH) benzeri olgular bildirilmi�tir. KHDAK'deki BR.21 isimli �al��mada ciddi interstisyel akci�er hastal��� benzeri olgular�n g�r�lme s�kl��� plasebo ve erlotinib gruplar�nda %0,8 olmu�tur.
KHDAK i�in randomize kontroll� klinik �al��malar�n (kontrol grubu olmamas� nedeniyle faz I ve tek kollu faz II �al��malar� hari�) meta analizinde, �AH benzeri olgular�n insidans� erlotinib grubunda %0,9 ve kontrol grubundaki hastalarda %0,4 olmu�tur.
�nterstisyel akci�er hastal��� benzeri olgu bulundu�undan ��phe edilen hastalarda bildirilen tan�lara baz� �rnekler, pn�moni, radyasyon pn�monisi, a��r� duyarl�l�k pn�monisi, interstisyel pn�moni, interstisyel akci�er hastal���, obliteratif bron�iyolit, pulmoner fibrozis, akut solunum s�k�nt�s� sendromu (ASSS), akci�er infiltrasyonu, alveolittir. Bu semptomlar, erlotinib tedavisine ba�lad�ktan birka� g�n sonra ile birka� ay aras�nda ortaya ��km��t�r. E� zamanl� veya �ncesindeki kemoterapi, �ncesinde radyoterapi, daha �nceden mevcut olan parankimal akci�er hastal���, metastatik akci�er hastal��� veya pulmoner enfeksiyonlar gibi kar��t�r�c� fakt�rler s�k g�r�lm��t�r. Japonya'da yap�lan �al��malarda daha y�ksek interstisyel akci�er hastal��� insidans� (%1,5 mortalite oran� ile yakla��k olarak %5) g�r�lm��t�r.
Dispne, �ks�r�k ve ate� gibi ani ba�lang��l� yeni ve/veya ilerleyici a��klanamayan pulmoner semptomlar geli�en hastalarda, SEVASTRA tedavisi tan�sal de�erlendirmeler tamamlanana dek kesilmelidir. �nterstisyel akci�er hastal��� tan�s� konacak olursa, SEVASTRA tedavisi kesilmeli ve gereken uyguntedaviyeba�lanmal�d�r(Bkz.B�l�m 4.8).
Diyare, dehidratasyon, elektrolit dengesizli�i, b�brek yetmezli�i
Erlotinib kullanmakta olan hastalar�n yakla��k %50'sinde diyare (baz� nadir vakalarda �l�mle sonu�lanabilen) g�zlenmi� olup, orta ve �iddetli diyarenin loperamid ile tedavi edilmesi gerekir. Baz� olgularda dozun d���r�lmesi gerekli olabilir. Klinik �al��malarda 50 mg'l�k ad�mlar �eklinde doz d���r�lmesi yap�lm��t�r. 25 mg'l�k ad�mlar �eklinde doz azaltma �zerine �al���lmam��t�r. �iddetli veya inat�� diyare, bulant�, i�tahs�zl�k ve dehidratasyon ile birlikte kusma g�r�lmesi halinde SEVASTRA tedavisi kesilmeli ve dehidratasyonu tedavi etmek i�in gerekli �nlemler al�nmal�d�r (Bkz. B�l�m 4.8). Hipokalemi ve akut renal yetmezli�i vakalar� (baz�lar� �l�mc�l olabilen) seyrek olarak bildirilmi�tir. Baz� renal yetmezlikler e� zamanl� kemoterapi uygulamas� ile i� i�e girerken, baz�lar� da diyareye, kusma ve/veya i�tahs�zl��a ba�l� dehidratasyona sekonder olmu�tur. Daha �iddetli veya inat�� diyare vakalar�nda veya dehidratasyona yol a�an vakalarda, �zellikle k�t�le�tiren risk fakt�r� (�zellikle beraber kullan�lan kemoterapi ve di�er ila�lar, semptomlar veya hastal�klar veya ileri ya� dahil di�er yatk�nl�k durumlar�) bulunan hasta gruplar�nda, SEVASTRA tedavisi kesilmelidir ve hastay� intraven�z olarak yo�un bir �ekilde rehidrate etmek i�in gerekli �nlemler al�nmal�d�r. Ek olarak, dehidratasyon riski bulunan hastalarda, b�brek fonksiyonlar� ve potasyum dahil serum elektrolitleri periyodik olarak izlenmelidir.
Hepatit, hepatik yetmezlik
Erlotinib tedavisi s�ras�nda, �l�mc�l de olabilen seyrek hepatik bozukluk vakalar� bildirilmi�tir. Kar��t�r�c� fakt�rler �nceden var olan karaci�er hastal��� veya e�lik eden hepatotoksik medikasyonlar� i�ermektedir. Bu y�zden, bu hastalarda periyodik karaci�er fonksiyon testleri d���n�lmelidir. Karaci�er fonksiyonlar�ndaki de�i�iklikler ciddi oldu�unda SEVASTRA dozlamas�na ara verilmelidir (Bkz. B�l�m 4.8). SEVASTRA'n�n ciddi hepatik disfonksiyonu olan hastalarda kullan�lmas� �nerilmemektedir.
Gastrointestinal perforasyon
Erlotinib kullanan hastalarda yayg�n olmayan (baz� vakalarda �l�mle sonu�lanabilen) �ekilde g�r�len gastrointestinal perforasyonun geli�me riski y�ksektir. Antianjiyogenik ila�lar, kortikosteroid, NSA�� ve/veya taksan bazl� kemoterapi ile e� zamanl� tedavi alan veya daha �nceden peptik �lserasyon veya divertik�ler hastal�k ge�mi�i olan hastalarda risk y�ksektir. Gastrointestinal perforasyon geli�en hastalarda, SEVASTRA tedavisi kal�c� olarak kesilmelidir (Bkz. B�l�m 4.8).
B�ll�z veya eksfoliyatif deri hastal�klar�
Steven Johnson sendromu/toksik epidermal nekrolizi i�aret eden �ok seyrek vakalar� i�eren, baz�lar� �l�mc�l olabilen b�ll�z, kabart�l� ve eksfolyatif deri rahats�zl�klar� bildirilmi�tir (Bkz. B�l�m 4.8). Hastalar ciddi b�ll�z, kabart�l� ve eksfoliyatif deri rahats�zl�klar� geli�tirirse, SEVASTRA tedavisine ara verilmelidir veya kesilmelidir. B�ll�z ve eksfolyatif deri rahats�zl�klar� bulunan hastalar deri enfeksiyonuna kar�� test edilmeli ve lokal tedavi k�lavuzlar�na g�re tedavi edilmelidir.
Ok�ler hastal�klar
Hastalarda g�z enflamasyonu, lakrimasyon, ����a hassasiyet, bulan�k g�rme, g�zde a�r� ve/veya k�zar�kl�k gibi akut ya da k�t�le�en keratit olu�umuna i�aret eden semptomlar�n g�r�lmesi halinde derhal g�z hekimine ba�vurulmal�d�r. E�er �lseratif keratit te�his edilmi�se SEVASTRA tedavisine ara verilmelidir veya kesilmelidir. E�er keratit te�his edilmi�se tedaviye devam edilmesinin risk-yarar de�erlendirmesi dikkatlice yap�lmal�d�r. SEVASTRA keratit, �lseratif keratit ve �iddetli g�z kurulu�u �yk�s� olanlarda dikkatle kullan�lmal�d�r. Kontakt lens, keratit ve �lserasyoni�inriskfakt�r�d�r.SEVASTRA kullan�m� s�ras�nda �ok
�lserasyon i�in risk fakt�rleri; kirpik de�i�iklikleri, keratokonjunktivit siccay� i�eren di�er
ok�ler hastal�klar ve kontakt lens kullan�m�d�r (bkz. B�l�m 4.8).
Di�er t�bbi �r�nlerle etkile�im
CYP3A4'�n potent ind�kleyicileri SEVASTRA'n�n etkilili�ini azaltabilirken CYP3A4'�n potent inhibit�rleri toksisitede art��a yol a�abilir. Bu tip tedavi ajanlar�n�n birlikte kullan�m�ndan ka��n�lmal�d�r (bkz. B�l�m 4.5).
Di�er etkile�im �ekilleri
Erlotinib 5'ten y�ksek pH seviyesinde ��z�n�rl�kte azalma ile karakterizedir. Proton pompas� inhibit�rleri, Hantagonistleri ve antasidler gibi �st gastrointestinal sistemin pH's�n� de�i�tiren t�bbi �r�nler, erlotinibin ��z�n�rl���n� dolay�s�yla biyoyararlan�m�n� de�i�tirebilir. Bu �r�nlerle birlikte kullan�ld���nda SEVASTRA dozunun art�r�lmas�, maruziyet d�����n� telafi etmeyecektir. Erlotinibin proton pompas� inhibit�rleriyle birlikte kullan�m�ndan ka��n�lmal�d�r. Erlotinibin, Hantagonistleri ve antasidlerle birlikte kullan�m�ndaki etki bilinmemektedir, ancak d���k biyoyararlan�m beklenmektedir. Dolay�s�yla, bu kombinasyonlar�n birlikte kullan�lmas�ndan ka��n�lmal�d�r (bkz. B�l�m 4.5). SEVASTRA tedavisi s�ras�nda antasid kullan�m� gerekliyse, g�nl�k SEVASTRA dozundan en az 4 saat �nce veya 2 saat sonra al�nmal�d�r.
Di�er
SEVASTRA, daha �nce herhangi bir EGFR yola�� inhibit�r� kullanm�� hastalarda kullan�lmaz.
Yard�mc� maddeler
Her bir SEVASTRA tablet, susuz laktoz i�erir. Nadir kal�t�msal galaktoz intolerans�, Lapp- laktoz yetmezli�i veya glukoz-galaktoz malabsorpsiyon problemi olan hastalar�n bu ilac� kullanmamalar� gerekir.
Her bir SEVASTRA 100 mg film kapl� tablet, her “doz”unda 1 mmol (23 mg)'dan daha az sodyum ihtiva eder; yani asl�nda “sodyum i�ermez”.
4.5. Di�er t�bbi �r�nler ile etkile�imler ve di�er etkile�im �ekilleri
Etkile�im �al��malar� yaln�zca yeti�kinler �zerinde ger�ekle�tirilmi�tir. Erlotinib ve di�er CYP substratlar�Erlotinib CYP1A1'in potent bir inhibit�r�, CYP3A4 ve CYP2C8'in orta derecede inhibit�r� ve in vitro UGT1A1 ile glukoronidasyonun g��l� bir inhibit�r�d�r. CYP1A1'in g��l� inhibisyonunun fizyolojik ba�lam� CYP1A1'in insan dokular�ndaki �ok s�n�rl� ekspresyonu sebebiyle bilinmemektedir.
Erlotinib, CYP1A2'nin orta d�zey inhibit�r� olan siprofloksasin ile birlikte uyguland���nda, erlotinibe maruziyet [EAA] %39 oranda anlaml� d�zeyde artm��, �te yandan maksimum konsantrasyon (C) seviyelerinde istatistiksel olarak anlaml� herhangi bir de�i�im bulunmam��t�r. Benzer �ekilde, aktif metabolite maruziyet s�ras�yla EAA ve Cseviyeleri i�in yakla��k %60 ve %48 oran�nda artm��t�r. S�z konusu art��lar�n klinik anlaml�l��� saptanmam��t�r. Siprofloksasin veya g��l� CYP1A2 inhibit�rleri (�rn. fluvoksamin) erlotinib ile kombine edildi�inde dikkatli olunmal�d�r. Erlotinib kaynakl� advers reaksiyonlar�n g�zlenmesi halinde, erlotinibin dozu azalt�labilir.
Erlotinibin �n tedavisi veya e� zamanl� uygulamas�, prototip CYP3A4 substratlar� midazolam
ve eritromisinin klerensini de�i�tirmemi�tir, ancak midazolam�n oral biyoyararlan�m�n�
%24'e kadar azaltt��� g�r�lm��t�r. Ba�ka bir klinik �al��mada, erlotinibin e� zamanl� uygulanan CYP3A4/2C8 substrat� paklitakselin farmakokineti�ini etkilemedi�i g�sterilmi�tir. Bu nedenle di�er CYP3A4 substratlar�n�n klerensi ile anlaml� etkile�imlerin olmas� pek m�mk�n de�ildir.
Glukoronidasyonun inhibisyonu, UGT1A1'in substratlar� olan ve yaln�zca bu yolla at�lan t�bbi �r�nlerle etkile�imlere neden olabilir. D���k UGT1A1 ekspresyon seviyeleri bulunan veya genetik glukoronidasyon bozukluklar�na (�rn. Gilbert hastal���) sahip olan hastalar, artm�� bilirubin serum konsantrasyonu ortaya koyabilir ve bu hastalar dikkatle tedavi edilmelidir.
Erlotinib insanlarda karaci�erde hepatik sitokromlar, birincil olarak CYP3A4 ve daha az �l��de CYP1A2 ile metabolize edilmektedir. Ba��rsakta CYP3A4 ile, akci�erde CYP1A1 ile ve t�m�r dokusunda CYP1B1 taraf�ndan ger�ekle�tirilen ekstrahepatik metabolizma da erlotinibin metabolik klerensine ayr�ca katk�da bulunmaktad�r. Bu enzimler taraf�ndan metabolize edilen veya bu enzimlerin inhibit�r� veya ind�kleyicisi olan ila�larla potansiyel etkile�imler ortaya ��kabilir.
CYP3A4 aktivitesinin potent inhibit�rleri, erlotinib metabolizmas�n� azalt�r ve erlotinib plazma konsantrasyonlar�n� artt�r�rlar. CYP3A4 metabolizmas�n�n ketokonazol ile inhibisyonu (5 g�n s�reyle, a��zdan g�nde iki kez 200 mg) artm�� bir erlotinibe maruz kalma (medyan erlotinib maruziyetinde %86 art�� [EAA-e�ri alt� alan]) ve yaln�zca erlotinibe k�yasla Cde�erinde %69'luk bir art��a yol a�m��t�r. Bu nedenle, erlotinib azol antifungalleri (ba�ka deyi�le ketakonazol, itrakonazol, vorikonazol) proteaz inhibit�rleri, eritromisin veya klaritromisin gibi g��l� CYP3A4 inhibit�rleri ile kombine edildi�inde dikkatli olunmal�d�r. Gerekli ise, �zellikle toksisite g�r�l�yorsa, erlotinib dozu azalt�lmal�d�r.
CYP3A4 aktivitesinin g��l� ind�kleyicileri, erlotinib metabolizmas�n� art�r�r ve erlotinib plazma konsantrasyonlar�n� anlaml� d�zeyde d���r�rler. Bir klinik �al��mada erlotinib ve g��l� bir CYP3A4 ind�kleyicisi olan rifampisinin (7 g�n s�reyle, a��zdan g�nde 1 kez 600 mg) e� zamanl� kullan�m� medyan erlotinib EAA seviyelerinde %69'luk d���� ile sonu�lanm��t�r. Rifampisinin 450 mg tek erlotinib dozu ile e� zamanl� uygulamas�, erlotinibe ortalama maruziyeti (EAA) rifampisin tedavisi olmaks�z�n uygulanan tek doz 150 mg erlotinib dozu ile g�zlenen seviyenin %57,5'i ile sonu�lanm��t�r. Bu nedenle, SEVASTRA'n�n CYP3A4 ind�kleyicileriyle e� zamanl� uygulamas�ndan ka��n�lmal�d�r. SEVASTRA'n�n rifampisin gibi g��l� bir CYP3A4 ind�kleyicisi ile e� zamanl� uygulanmas� gerekti�i hastalarda g�venlilik (b�brek ve karaci�er fonksiyonlar� ve serum elektrolitleri dahil) yak�ndan izlenerek dozda 300 mg'a kadar art�� yap�lmas� de�erlendirilmelidir ve 2 haftadan uzun s�reyle iyi tolere edilmesi halinde, yak�n g�venlilik izlemesi ile 450 mg'a kadar bir art�� daha yap�lmas� dikkate al�nabilir. �rne�in fenitoin, karbamazepin, barbit�ratlar veya St. John's Wort (hypericum perforatum) gibi di�er ind�kleyicilerle de maruziyet d�zeyinde azalma olu�abilir. S�z konusu etkin maddeler erlotinib ile kombine edildi�inde dikkat edilmelidir. M�mk�n oldu�u zamanlarda g��l� CYP3A4 ind�kleyici aktivitesi bulunmayan alternatif tedavi se�enekleri de�erlendirilmelidir.
Erlotinib ve kumarin t�revi antikoag�lanlar
Erlotinib alan hastalarda y�kselmi� Uluslararas� Normalle�tirilmi� Oran (INR-International Normalized Ratio) ve baz� durumlarda �ld�r�c� bulunmu� kanama olgular�na sebep olan varfarin dahil kumarin t�revi antikoag�lanlar ile etkile�imler bildirilmi�tir. Kumarin t�revi antikoag�lan ila�lar� kullanmakta olan hastalar protrombin zaman� veya INR de�i�iklikleri a��s�ndan d�zenli olarak izlenmelidir.
Erlotinib ve statinler
SEVASTRA ile bir statin kombinasyonu, seyrek g�r�len rabdomiyoliz dahil statin kaynakl� miyopati potansiyelini art�rabilir.
Erlotinib ve sigara i�enler
Bir farmakokinetik etkile�im �al��mas�n�n sonu�lar� sigara kullananlarda erlotinib uygulamas� sonras�nda EAA, Cve plazma konsantrasyonunda sigara kullanmayanlara k�yasla 24 saatte s�ras�yla anlaml� 2,8-, 1,5- ve 9 kat� azalma oldu�unu g�stermi�tir. Bu nedenle halihaz�rda sigara i�mekte olan hastalar SEVASTRA ile tedavi ba�lat�lmadan �nce m�mk�n olan en k�sa s�rede sigaray� b�rakmalar� i�in te�vik edilmelidir, ��nk� aksi takdirde plazma erlotinib konsantrasyonlar� azalmaktad�r.
CURRENTS �al��mas�ndan elde edilen veriler, sigara i�en hastalarda �nerilen 150 mg doz ile kar��la�t�r�ld���nda, 300 mg'l�k y�ksek erlotinib dozunun fayda g�sterdi�ine dair herhangi bir kan�t g�stermemi�tir. G�venlilik verileri 300 mg ve 150 mg dozlar� aras�nda kar��la�t�r�labilirdir ancak, daha y�ksek erlotinib dozu alan hastalarda d�k�nt�, interstisyel akci�er hastal��� ve diyare insidans�nda say�sal bir art�� olmu�tur (bkz. B�l�m 4.2, 4.4, 5.1 ve 5.2).
Erlotinib ve p-glikoprotein inhibit�rleri
Erlotinib, P-glikoprotein etkin maddesi ta��y�c�s� i�in bir substratt�r. P-gp inhibit�rleri (�r: siklosporin ve verapamil) ile e� zamanl� uygulama erlotinib da��l�m�n� ve /veya eliminasyonunu de�i�tirebilir. Bu etkile�imin sonucunda, �r. MSS toksisitesi a��s�ndan neler oldu�u saptanmam��t�r. Bu t�r durumlarda dikkatli olunmal�d�r.
Erlotinib ve pH de�i�tiren t�bbi �r�nler
Erlotinib 5'ten y�ksek pH seviyesinde ��z�n�rl�kte azalma ile karakterizedir. �st sindirim kanal�n�n pH's�n� de�i�tiren ila�lar, erlotinib ��z�n�rl���n� ve buna ba�l� olarak biyoyararlan�m�n� de�i�tirebilir. Erlotinibin bir proton pompas� inhibit�r� olan omeprazol ile birlikte uygulanmas� erlotinib maruziyetini [EAA] ve maksimum konsantrasyonunu [C] s�ras�yla %46 ve %61 azaltm��t�r. Tve yar� �mr�nde herhangi bir de�i�iklik olmam��t�r. Erlotinib ve bir H2-resept�r antagonisti olan ranitidinin 300 mg'� ile beraber kullan�lmas�, erlotinibe maruziyeti [EAA] ve C'� s�ras�yla %33 ve %54 oran�nda azaltm��t�r. Bu tip ajanlarla e� zamanl� uyguland���nda erlotinibin dozunun art�r�lmas�n�n maruziyetteki bu kayb� telafi etmesi pek m�mk�n de�ildir. Bununla birlikte, erlotinib 150 mg g�nde iki kere, ranitidinden 2 saat �nce veya 10 saat sonras�nda ayr� saatlere b�l�nerek kullan�l�rsa, erlotinib maruziyeti [EAA] ve Cs�ras�yla sadece %15 ve %17 oran�nda azalm��t�r. Antasitlerin erlotinib emilimi �zerindeki etkisi ara�t�r�lmam��t�r, ancak emilim bozularak plazma seviyelerinde d����e yol a�abilir. �zet olarak, erlotinibin proton pompas� inhibit�rleri ile kombinasyonundan ka��n�lmal�d�r. SEVASTRA ile tedavi s�ras�nda antasitlerin kullan�m� d���n�l�yorsa, bu ila�lar�n erlotinibin g�nl�k dozundan en az 4 saat �nce veya 2 saat sonra al�nmas� gerekir. Ranitidin kullan�m� d���n�l�yorsa, ayr� saatlere b�l�nerek kullan�lmal�, ba�ka deyi�le erlotinib, ranitidin dozu al�nmadan en az 2 saat �nce veya al�nd�ktan 10 saat sonra al�nmal�d�r.
Erlotinib ve Gemsitabin
Bir Faz Ib �al��mada, gemsitabinin erlotinib farmakokineti�i �zerinde anlaml� herhangi bir etkisine veya erlotinibin gemsitabin farmakokineti�i �zerinde anlaml� herhangi bir etkisine rastlanmam��t�r.
Erlotinib ve Karboplatin/Paklitaksel
Erlotinib, platin konsantrasyonlar�n� art�r�r. Bir klinik �al��mada karboplatin ve paklitaksel ile birlikte erlotinib kullan�m�, toplam platin EAA de�erinde %10,6'l�k bir art��a yol a�m��t�r. �statistiksel olarak anlaml� olmas�na ra�men, bu fark�n klinik a��dan �nemli oldu�u d���n�lmemektedir. Klinik uygulamada karboplatin maruziyetini art�ran, b�brek yetmezli�i gibi di�er kofakt�rler olabilir. Erlotinib farmakokineti�i �zerine karboplatin veya paklitakselin anlaml� etkisi bulunmamaktad�r.
Erlotinib ve Kapesitabin
Kapesitabin, erlotinib konsantrasyonlar�n� art�rabilir. Tek ajan olarak erlotinib uygulanan �al��madan elde edilen de�erlerle kar��la�t�r�ld���nda, kapesitabin ile birlikte uygulanan erlotinib �al��mas�nda, erlotinib EAA's� istatistiksel olarak anlaml� derecede artmaktad�r ve Cd�zeyi s�n�rda bir art�� g�stermektedir. Kapesitabin farmakokineti�i �zerinde erlotinibin anlaml� etkisi bulunmamaktad�r.
Erlotinib ve proteazom inhibit�rleri
�al��ma mekanizmas�na ba�l� olarak, bortezomib dahil proteazom inhibit�rlerinin erlotinib gibi EGFR inhibit�rlerinin etkisini de�i�tirmesi beklenebilir. Bu de�i�im, proteazomdan EGFR degradasyonunu g�steren s�n�rl� klinik veriler ve preklinik �al��malarla desteklenmektedir.
�zel pop�lasyonlara ili�kin ek bilgiler:
�zel pop�lasyonlara ili�kin etkile�im �al��mas� yap�lmam��t�r.
Pediyatrik pop�lasyon:
Erlotinibin 18 ya��n alt�ndaki hastalarda etkile�im �al��mas� yap�lmam��t�r.
4.6. Gebelik ve laktasyon
Gebelik kategorisi: D
�ocuk do�urma potansiyeli bulunan kad�nlar/ Do�um kontrol� (Kontrasepsiyon) Do�urganl�k potansiyeline sahip kad�nlar�n SEVASTRA kullan�rken gebe kalmaktan ka��nmalar� konusunda uyar�lmalar� gereklidir. Tedavi s�ras�nda ve tedavinin tamamlanmas�ndan sonraki en az iki hafta boyunca, yeterli do�um kontrol y�ntemleri kullan�lmal�d�r.
Gebelik d�nemi
Gebe kad�nlarda erlotinib kullan�m� ile ilgili yeterince veri bulunmamaktad�r. Hayvanlar �zerinde yap�lan �al��malar teratojenisite veya abnormal parturisyon ile ilgili bir sonu� g�stermemi�tir. Buna kar��n, gebelik �zerinde olas� bir advers olay, tav�an ve s��anlarda artan embriyo/fetal letalite g�r�ld���nden (bkz. B�l�m 5.3), g�z ard� edilememektedir. �nsanlar i�in potansiyel risk bilinmemektedir. SEVASTRA kesinlikle gerekli olmad�k�a gebelik d�neminde kullan�lmamal�d�r. Gebe kad�nlarda tedavi, ancak anne i�in beklenen faydalar�n, fet�s i�in do�abilecek risklerden daha �st�n olmas� halinde s�rd�r�lmelidir.
Laktasyon d�nemi
Erlotinibin anne s�t�ne ge�ip ge�medi�i bilinmemektedir. Erlotinibin s�t �retimindeki
etkisini veya anne s�t�nde bulunmas�n� de�erlendiren herhangi bir �al��ma yap�lmam��t�r. Bebek i�in potansiyel zarar bilinmedi�inden, anneler SEVASTRA kullan�rken ve son dozu ald�ktan en az 2 hafta sonraya kadar emzirmemeleri konusunda uyar�lmal�d�r.
�reme yetene�i/Fertilite
Hayvanlarda yap�lan �al��malarda fertilite bozuklu�u g�r�lmemi�tir. Ancak, hayvanlardaki �al��malar, �reme parametreleri �zerinde etkileri oldu�u g�sterildi�inden fertilite �zerine advers etkiler g�z ard� edilemez (bkz. B�l�m 5.3). �nsanlar i�in potansiyel risk bilinmemektedir.
4.7. Ara� ve makine kullan�m� �zerindeki etkiler
4.7. Ara� ve makine kullan�m� �zerindeki etkiler
4.8. �stenmeyen etkiler
Erlotinibin g�venlilik de�erlendirmesi, en az bir doz 150 mg erlotinib monoterapi alan 1500 hastadan ve gemsitabinle kombinasyon halinde 100 mg veya 150 mg erlotinib alan 300 hastan�n verilerine dayanmaktad�r.
Erlotinib ile monoterapi veya kombinasyon halinde kemoterapi alan hastalarda ortaya ��kan advers reaksiyonlar�n insidans� Ulusal Kanser Enstit�s� - Ortak Toksisite Kriterleri (NCI- CTC) Derecesine g�re Tablo 1'de �zetlenmektedir. Listelenen istenmeyen etkiler erlotinib ile tedavi edilen hastalarda plasebo grubuna g�re daha s�k (≥%3) ve erlotinib grubunda hastalar�n en az %10'unda ortaya ��kan advers reaksiyonlard�r. Di�er klinik �al��malarda ortaya ��kan istenmeyen etkiler Tablo 2'de �zetlenmektedir.
Klinik ara�t�rmalarda ortaya ��kan istenmeyen etkiler Tablo 1'de MedDRA organ sistemine g�re s�ralanm��t�r. �stenmeyen etkileri s�kl�klar�na g�re s�ralamak i�in �u terimler kullan�lm��t�r: �ok yayg�n (1/10); yayg�n (1/100, <1/10); yayg�n olmayan (1/1,000,
<1/100); seyrek (1/10,000, <1/1000); �ok seyrek (<1/10,000); bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor).
Her bir s�kl�k gruplamas�nda, advers reaksiyonlar azalan ciddiyet s�ras�na g�re sunulmaktad�r. K���k H�creli D��� Akci�er Kanseri (KHDAK)
(SEVASTRA, monoterapi olarak kullan�l�r.)
EGFR Mutasyonlu Hastalar�n Birinci Basamak Tedavisi
154 hastada ger�ekle�tirilen a��k etiketli, randomize, Faz III ML20650 �al��mas�nda EGFR aktive edici mutasyonlar� olan KHDAK hastalar�n�n birinci basamak tedavisinde erlotinibin g�venlili�i 75 hastada de�erlendirilmi�tir. Bu hastalarda yeni g�venlilik sinyalleri g�zlenmemi�tir.
ML20650 �al��mas�nda erlotinib ile tedavi edilen hastalarda en s�k g�r�len yan etkiler d�k�nt� ve diyare olmu�tur (s�ras�yla %80 ve %57), �o�u Evre 1/2'dir ve giri�im olmadan y�netilebilmi�tir. Evre 3 d�k�nt� ve diyare, hastalar�n s�ras�yla %9'u ve %4'�nde
g�r�lm��t�r. Evre 4 d�k�nt� veya diyare g�r�lmemi�tir. Hem d�k�nt� hem de diyare,
hastalar�n %1'inde erlotinibin b�rak�lmas�na neden olmu�tur.
D�k�nt� ve diyare i�in doz modifikasyonlar� (kesilmeler veya azaltmalar), s�ras�yla hastalar�n
%11'i ve %7'sinde gerekli olmu�tur.
�kinci ve �leri Basamak Tedavi
Randomize, �ift-k�r bir �al��mada (BR.21; erlotinib ikinci se�enek tedavi olarak uygulanm��t�r), d�k�nt� (%75) ve diyare (%54) en yayg�n rapor edilen advers reaksiyonlar olmu�tur. �o�u �iddet a��s�ndan evre 1 veya evre 2 d�zeyinde olmu� ve m�dahaleye gerek kalmaks�z�n d�zelmi�lerdir. Evre 3/4 d�k�nt� ve diyare erlotinib ile tedavi edilen hastalar�n s�ras�yla %9'unda ve %6's�nda ortaya ��km��t�r ve her biri hastalar�n %1'inin �al��madan ayr�lmas� ile sonu�lanm��t�r. D�k�nt� ve diyare i�in hastalar�n s�ras�yla %6 ve %1'inde doz d�����ne ihtiya� olmu�tur. BR.21 �al��mas�nda, d�k�nt�n�n ba�lamas�na kadar ge�en medyan s�re 8 g�n ve diyarenin ba�lamas�na kadar ge�en medyan s�re 12 g�n olarak bulunmu�tur.
Genel olarak, d�k�nt� g�n ����� alan b�lgelerde ortaya ��kabilen veya k�t�le�ebilen hafif veya orta �iddette eritemat�z ve papulopust�ler d�k�nt� olarak kendini belli etmektedir. G�n �����na maruz kalan hastalarda, koruyucu giysiler ve/veya g�ne� koruyucu preparat (�rn. mineral i�eren) kullan�m� �nerilebilir.
Tablo 1: BR.21 (Erlotinib ile tedavi) ve PA.3 (Erlotinib+gemsitabin ile tedavi) �al��malar�nda hastalar�n %10'unda g�r�len ve plasebo grubuna g�re daha s�k (≥ 3%) g�r�len yan etkiler
|
Erlotinib (BR.21) N = 485 |
Erlotinib (PA.3) N = 259 | S�kl�k kategorisi ve en y�ksek insidans | ||||
NCI-CTC Evresi | T�m Evreler | 3 | 4 | T�m Evreler | 3 | 4 | |
MedDRA Tercih Edilen Terimi |
% |
% |
% |
% |
% |
% | |
Enfeksiyonlar ve enfestasyonlar
Enfeksiyon* |
24 |
4 |
0 |
31 |
3 |
<1 |
�ok yayg�n |
Metabolizma ve beslenme hastal�klar� Anoreksiya Kilo azalmas� |
52 |
8 |
1 |
- |
- |
- |
�ok yayg�n �ok yayg�n |
- | - | - | 39 | 2 | 0 | ||
| Erlotinib (BR.21) N = 485 | Erlotinib (PA.3) N = 259 | S�kl�k kategorisi ve en y�ksek insidans | ||||
NCI-CTC Evresi | T�m Evreler | 3 | 4 | T�m Evreler | 3 | 4 | |
MedDRA Tercih Edilen Terimi |
% |
% |
% |
% |
% |
% | |
G�z hastal�klar� |
|
|
|
|
|
|
|
Kuru g�z sendromu | 12 | 0 | 0 | - | - | - | �ok yayg�n |
Konjuktivit | 12 | <1 | 0 | - | - | - | �ok yayg�n |
Psikiyatrik hastal�klar
Depresyon |
- |
- |
- |
19 |
2 |
0 |
�ok yayg�n |
Sinir sistemi hastal�klar� |
|
|
|
|
|
|
|
N�ropati | - | - | - | 13 | 1 | <1 | �ok yayg�n |
Ba� a�r�s� | - | - | - | 15 | <1 | 0 | �ok yayg�n |
Solunum, g���s bozukluklar� ve mediastinal hastal�klar |
|
|
|
|
|
|
|
Dispne | 41 | 17 | 11 | - | - | - | �ok yayg�n |
�ks�r�k | 33 | 4 | 0 | 16 | 0 | 0 | �ok yayg�n |
Gastrointestinal hastal�klar |
|
|
|
|
|
|
|
Diyare** | 54 | 6 | <1 | 48 | 5 | <1 | �ok yayg�n |
Bulant� | 33 | 3 | 0 | - | - | - | �ok yayg�n |
Kusma | 23 | 2 | <1 | - | - | - | �ok yayg�n |
Stomatit | 17 | <1 | 0 | 22 | <1 | 0 | �ok yayg�n |
Kar�n a�r�s� | 11 | 2 | <1 | - | - | - | �ok yayg�n |
Dispepsi | - | - | - | 17 | <1 | 0 | �ok yayg�n |
Flatulans | - | - | - | 13 | 0 | 0 | �ok yayg�n |
Deri ve deri alt� doku hastal�klar� |
|
|
|
|
|
|
|
D�k�nt�*** | 75 | 8 | <1 | 69 | 5 | 0 | �ok yayg�n |
Ka��nt� | 13 | <1 | 0 | - | - | - | �ok yayg�n |
Kuru cilt | 12 | 0 | 0 | - | - | - | �ok yayg�n |
Alopesi | - | - | - | 14 | 0 | 0 | �ok yayg�n |
Genel bozukluklar ve uygulama b�lgesine ili�kin hastal�klar |
|
|
|
|
|
|
|
Yorgunluk | 52 | 14 | 4 | 73 | 14 | 2 | �ok yayg�n |
Pireksi | - | - | - | 36 | 3 | 0 | �ok yayg�n |
Titreme | - | - | - | 12 | 0 | 0 | �ok yayg�n |
* N�tropeni ile birlikte olan veya olmayan ciddi enfeksiyonlar pn�moni, sepsis ve deri alt� dokusu iltihab�n� i�ermektedir.
** Dehidrasyon, hipokalemi ve renal yetmezli�e yol a�abilir.
*** Dermatit akneiform dahil d�k�nt�
Tablo 2: S�kl��a G�re Advers Etkilerin �zeti
Sistem Organ S�n�f� | �ok yayg�n (≥1/10) | Yayg�n (≥1/100 ila<1/10) | Yayg�n olmayan (≥1/1,000 ila <1/100) | Seyrek (≥1/10,000 ila <1/1,000) | �ok seyrek (<1/10,000) |
G�z hastal�klar� |
| |
| perforasyonlar �lserasyon | |
Solunum, g���s bozukluklar� ve mediastinal hastal�klar� |
| | |
|
|
Gastrointestinal hastal�klar� | Diyare | | |
|
|
Hepato-biliyer hastal�klar� | |
|
| |
|
Deri ve deri alt� doku hastal�klar� | | deri reaksiyonlar� | | | |
B�brek ve idrar yolu hastal�klar� |
| |
|
|
Keratit
4.9. Doz a��m� ve tedavisi
Sa�l�kl� ki�ilerde 1000 mg'a varan ve kanser hastalar�nda 1600 mg'a varan tek oral dozlar
tolere edilmi�tir. Sa�l�kl� ki�ilerde tekrarlanan g�nde iki kez 200 mg dozu, doz uygulamas�n�n hen�z birka� g�n sonras�ndan itibaren k�t� tolere edilmi�tir. Bu �al��malar�n verilerine dayanarak, �nerilen dozun �zerinde diyare, d�k�nt� ve olas� karaci�er transaminazlar� y�kselmesi gibi �iddetli advers olaylar ortaya ��kabilir.
Y�netim
Doz a��m�ndan ��phelenilmesi durumunda, SEVASTRA kesilmeli ve semptomatik tedavi ba�lat�lmal�d�r.
5. FARMAKOLOJ�K �ZELL�KLER
5.1. Farmakodinamik �zellikler
Farmakoterap�tik grup: Antineoplastik ila�, protein kinaz inhibit�r� ATC kodu: L01EB02
Etki mekanizmas�:
Erlotinib epidermal b�y�me fakt�r resept�r�/insan epidermal b�y�me fakt�r tip 1 resept�r�n�n (EGFR, ayn� zamanda HER1 olarak bilinen) inhibit�r�d�r. EGFR'nin intrasell�ler fosforilizasyonunu etkili bir �ekilde inhibe eder. EGFR/HER1 resept�r� normal h�cre ve kanser h�crelerinin h�cre y�zeylerinde eksprese edilir. Klinik d��� modellerde, EGFR fosfotirozininin inhibisyonu h�cre staz� ve/veya �l�m� ile sonlanmaktad�r.
EGFR mutasyonlar� antiapoptotik ve proliferatif sinyal yollar�nda konstit�tif aktivasyona yol a�abilir. Erlotinibin bu EGFR mutasyon pozitif t�m�rlerde EGFR arac�l� sinyalleri engelleyen g��l� etkisi, EGFR'nin mutant kinaz b�lgesindeki ATP ba�layan b�lgesine erlotinibin s�k�ca ba�lanmas�na atfedilmi�tir. A�a�� ak�m sinyalin engellenmesi nedeniyle, h�cre �o�almas� durmakta ve intrinsik apoptotik yolla h�cre �l�m� ba�lamaktad�r. EGFR'yi aktive eden bu mutasyonlar�n ekspresyonunun tetiklendi�i fare modellerinde t�m�r regresyonu g�zlenmi�tir.
Klinik etkililik
- EGFR aktive eden mutasyonlar� olan hastalarda k���k h�creli d��� akci�er kanserinin (KHDAK) birinci basamak tedavisi (monoterapi olarak erlotinib uygulamas�):
EGFR aktive eden mutasyonlar� olan KHDAK hastalar�n�n birinci basamak tedavisinde erlotinib ilac�n�n etkinli�i faz III, randomize, a��k etiketli �al��mada g�sterilmi�tir (ML20650, EURTAC). Bu �al��ma metastatik veya lokal olarak ileri evre (evre IIIB ve IV) KHDAK olan, daha �nce ilerlemi� hastal��� i�in kemoterapi veya sistemik antit�m�r tedavisi almam��, EGFR tirozin kinaz b�lgesinde mutasyonlar� (ekson 19 delesyonu veya ekson 21 mutasyonu) bulunan beyaz �rktan hastalarda ger�ekle�tirilmi�tir. Hastalar g�nl�k erlotinib 150 mg veya d�rt d�ng� platin bazl� ikili kemoterapi almak �zere 1:1 oran�nda randomize edilmi�lerdir.
Birincil sonlan�m noktas�, ara�t�rmac�n�n de�erlendirdi�i progresyonsuz sa�kal�m (PFS) olan �al��maya ait etkililik sonu�lar� Tablo 3'te verilmektedir.
|
| Erlotinib | Kemoterapi | Risk Oran� (95% GA) | p-de�eri |
�nceden |
| n=77 | n=76 |
|
|
planlanm�� ara analiz (%35 OS mat�rite) (n=153) | |||||
Birincil sonlan�m noktas�: Progresyonsuz Sa�kal�m (PFS, ay olarak medyan) * |
9,4 |
5,2 |
0,42 [0,27-0,64] |
p<0,0001 | |
| Ara�t�rmac� taraf�ndan | 10,4 | 5,4 | 0,47 | p=0,003 |
| de�erlendirilen ** |
|
| [0,27-0,78] |
|
Kesim tarihi: | Ba��ms�z inceleme ** |
|
|
|
|
A�u 2010 | En �yi Genel Yan�t | %54,5 | %10,5 |
| p<0,0001 |
| Oran� (CR/PR) | ||||
| Genel Sa�kal�m (OS) (ay) | 22,9 | 18,8 | 0,80 [0,47-1,37] | p=0,4170 |
Ara�t�rma ama�l� analiz (%40 OS mat�rite) (n=173) |
| n=86 | n=87 |
|
|
PFS (ay olarak medyan), Ara�t�rmac� taraf�ndan de�erlendirilen |
9,7 |
5,2 | 0,37 [0,27-0,54] |
p<0,0001 | |
Kesim tarihi: Oca 2011 | En �yi Genel Yan�t Oran� (CR/PR) | %58,1 | %14,9 |
| p<0,0001 |
19,5 | [0,65-1,68] | p=0,8702 | |||
| OS (ay) | 19,3 |
�ekil 1: ML20650 (EURTAC) �al��mas�nda ara�t�rmac� taraf�ndan de�erlendirilen PFS i�in Kaplan-Meier e�risi (Nisan 2012 kesimli)
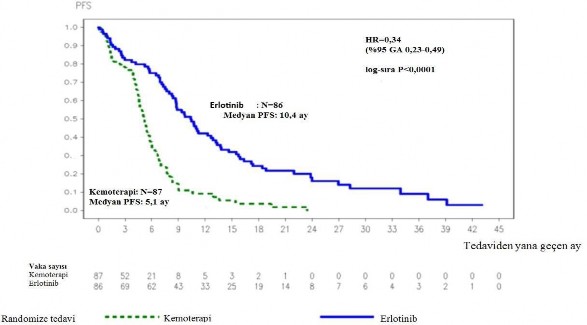
HR: Risk oran�, GA: G�ven aral���, PFS: Progresyonsuz sa�kal�m
Tablo 3: ML20650 (EURTAC) �al��mas�nda Erlotinibin kemoterapi ile kar��la�t�rmal� etkililik sonu�lar�
G�ncellenmi� analiz (%62 OS mat�rite) (n=173)
Kesim tarihi: Nisan 2012 |
| n=86 | n=87 |
|
|
PFS (ay olarak medyan) | 10,4 | 5,1 | 0,34 [0,23-0,49] | p<0,0001 | |
OS*** (ay) |
22,9 |
20,8 | 0,93 [0,64-1,36] |
p=0,7149 |
-En az bir ba�ar�s�z kemoterapi sonras� KHDAK tedavisi (monoterapi olarak erlotinib uygulamas�):
�kinci-���nc� basamak tedavi olarak erlotinib ilac�n�n etkililik ve g�venlili�i randomize, �ift k�r, plasebo kontroll� �al��mada (BR.21) g�sterilmi�tir. Bu �al��mada yer alan 731 hastada en az bir kemoterapi rejiminden sonra lokal olarak ilerlemi� veya metastatik KHDAK vard�r. Hastalar g�nde bir kere oral olarak erlotinib 150 mg veya plasebo almak �zere 2:1 oran�nda randomize edilmi�lerdir. �al��ma son noktalar�: genel sa�kal�m, progresyonsuz sa�kal�m (PFS), yan�t oran�, yan�t s�resi, akci�er kanserine ba�l� belirtilerin (�ks�r�k, dispne ve a�r�) k�t�le�mesine dek ge�en s�re ve g�venliliktir. Birincil sonlan�m noktas� sa�kal�md�r.
Demografik �zellikler iki tedavi grubu aras�nda dengelidir. Hastalar�n ��te ikisi erkektir ve yakla��k ��te birinde ba�lang�� Do�u Kooperatif Onkoloji Grubu (ECOG) performans durumu (PS) 2 olup, %9'unda ba�lang�� ECOG PS 3't�r. Erlotinib ve plasebo grubundaki hastalar�n s�ras�yla %93 ve 92 kadar� daha �nce platin i�erenrejim ve s�ras�yla %36 ve %37 kadar� taksan tedavisi alm��t�r.
Erlotinib grubunda plaseboya g�re �l�m i�in ayarlanan tehlike oran� (HR) 0,73 (%95 GA, 0,60 – 0,87) (p =0.001) olarak saptanm��t�r. 12. ayda ya�ayan hastalar�n oran� erlotinib ve plasebo gruplar� i�in s�ras�yla %31,2 ve %21,5 olarak saptanm��t�r. Medyan genel sa�kal�m erlotinib grubunda 6,7 ay iken (%95 GA, 5,5– 7,8 ay) plasebo grubunda 4,7 ayd�r (%95 GA,
4,1 – 6,3 ay).
Genel sa�kal�m �zerine etki farkl� hasta alt k�melerinde incelenmi�tir. Genel sa�kal�m �zerine erlotinib etkisi �u hastalarda benzerdir: ba�lang�� performans durumu (ECOG) 2-3 (HR = 0,77, %95 GA, 0,6-1,0) veya 0-1 (HR = 0,73, %95 GA, 0,6-0,9), erkek (HR = 0,76, %95 GA,
0,6-0,9) veya kad�n (HR = 0,80, %95 GA, 0,6-1,1), < 65 ya� (HR = 0,75, %95 GA, 0,6-0,9)
veya daha ya�l� hastalar (HR = 0,79, %95 GA, 0,6- 1,0), daha �nce bir rejim alan hastalar (HR
= 0,76, %95 GA, 0,6-1,0) veya daha �nce birden fazla rejim alan (HR = 0,75, %95 GA, 0,6-
1,0), beyazlar (HR = 0,79, %95 GA, 0,6-1,0) veya Asyal� hastalar (HR = 0,61, %95 GA, 0,4-
1,0), adenokarsinomu olanlar (HR = 0,71, %95 GA, 0,6-0,9) veya skuam�z h�creli karsinomu olanlar (HR = 0,67, %95 GA, 0,5-0,9). �u hastalarda ise benzer de�ildir: di�er histolojileri olanlar (HR 1,04, %95 GA, 0,7-1,5), tan�da hastal��� evre IV olan hastalar (HR = 0,92, %95 GA, 0,7-1,2) veya tan�da hastal��� evre < IV olanlar (HR = 0,65, %95 GA, 0,5-0,8). Daha �nce hi� sigara i�memi� olan hastalar, �u anda veya eskiden sigara i�enlere nazaran (HR
= 0,87, %95 GA, 0,71-1,05) erlotinibden daha fazla fayda sa�lam��t�r (sa�kal�m HR = 0,42,
%95 GA, 0,28-0,64).
EGFR ekspresyon durumu bilinen hastalar�n %45 kadar�nda, EGFR pozitif t�m�rleri olanlar�n
tehlike oran� 0,68 (% 95 GA, 0,49-0,94) ve EGFR-negatif t�m�r� olanlar�n tehlike oran� 0,93
(% 95 GA, 0,63-1,36) olarak saptanm��t�r (EGFR pharmDx kit kullan�lan IHC ile
tan�mlanm��t�r ve y�zde ondan az boyal� t�m�r h�cresi EGFR negatif olarak tan�mlanm��t�r).
Kalan %55 hastan�n EGFR ekspresyon durumu bilinmemektedir ve HR 0,77 (%95 GA, 0,61-
0,98) olarak saptanm��t�r.
Erlotinib grubunda ortalama PFS 9,7 haftad�r (%95 GA, 8,4 – 12,4 hafta) ve plasebo grubunda 8,0 haftad�r (%95 GA, 7,9 – 8,1 hafta).
Objektif yan�t oran�, Solid T�m�rlerde Yan�t De�erlendirme Kriterleri'ne (RECIST) g�re erlotinib grubunda %8,9 olarak saptanm��t�r (%95 GA, 6,4 – 12,0). �lk 330 hasta merkezi olarak de�erlendirilmi�tir (yan�t oran� %6,2); 401 hasta ara�t�rmac� taraf�ndan de�erlendirilmi�tir (yan�t oran� %11,2).
Medyan yan�t s�resi 34,3 hafta olup, 9,7 ila 57,6+ hafta aras�ndad�r. Tam yan�t, k�smi yan�t veya stabil hastal�k ya�ayan hasta oran� s�ras�yla erlotinib ve plasebo gruplar�nda %44,0 ve
%27,5 olarak saptanm��t�r (p = 0,004).
Erlotinib i�in sa�kal�m faydas�, (RECIST'e g�re) nesnel t�m�r yan�t� elde etmeyen hastalarda da g�zlenmi�tir. Bunun kan�t�, en iyi yan�t� stabil hastal�k veya progresif hastal�k olan hastalar aras�nda �l�m i�in HR'nin 0,82 (%95 GA, 0,68 – 0,99) olmas�yla ortaya konmu�tur.
Erlotinib, plaseboya nazaran semptomlar �zerinde de fayda g�stermi� olup �ks�r�k, dispne ve a�r�n�n k�t�le�mesi i�in ge�en s�re anlaml� derecede uzam��t�r.
Lokal ileri veya metastatik, sigara i�en (y�lda ortalama 38 paket) KHDAK hastalar�nda kemoterapi sonras� ikinci basamak tedavide iki erlotinib dozunun kar��la�t�r�ld��� (300 mg'a kar��l�k 150 mg) �ift-k�r, randomize Faz III �al��mada (MO22162, CURRENTS), 300 mg doz progresyonsuz sa�kal�m faydas� g�stermemi�tir (s�ras�yla 7 hafta ve 6,86 hafta).
Sekonder sonlan�m noktalar�n�n t�m� primer sonlan�m noktalar�yla tutarl�d�r ve g�nde 300 mg ve 150 mg erlotinib ile tedavi edilen hastalar aras�nda sa�kal�m fark� g�r�lmemi�tir (HR 1,03, %95 GA 0,80 – 1,32). G�venlik verileri, 300 mg ve 150 mg dozlar� aras�nda kar��la�t�r�labilirdir ancak, daha y�ksek erlotinib dozu alan hastalarda d�k�nt�, interstisyel akci�er hastal��� ve diyare insidans�nda say�sal bir art�� olmu�tur. CURRENTS �al��mas�ndan elde edilen veriler, sigara i�en hastalarda �nerilen 150 mg doz ile kar��la�t�r�ld���nda 300 mg dozla herhangi bir yarar g�r�lmedi�ini g�stermi�tir.
CURRENT �al��mas�ndaki hastalar EGFR mutasyon durumuna g�re se�ilmemi�tir (bkz. B�l�m 4.2, 4.4, 4.5 ve 5.2).
5.2. Farmakokinetik �zellikler
Genel �zelliklerEmilim:
Oral uygulama sonras�nda erlotinib ortalama doruk plazma d�zeylerine oral dozdan yakla��k
4 saat sonra ula��r. Normal sa�l�kl� g�n�ll�lerdeki bir �al��mada yakla��k %59'luk bir biyoyararlan�m elde edilmi�tir. Bir oral doz sonras�ndaki biyoyararlan�m, yiyeceklerle birlikte artt�r�labilir.
Da��l�m:
Erlotinib, 232 L'lik ortalama sanal da��l�m hacmine sahiptir ve insan t�m�r dokular�na da��l�r. G�nl�k 150 mg oral erlotinibdozualmaktaolan4hastada(3'� k���k h�creli d��� akci�er
cerrahi eksizyonlardan al�nan t�m�r �rnekleri, t�m�rdeki erlotinib konsantrasyonlar�n�n
ortalama 1,185 ng/g doku oldu�unu ortaya koymu�tur. Bu da kararl� durumda g�zlenen doruk plazma konsantrasyonlar�n�n genel ortalamas�n�n %63'�ne kar��l�k gelmektedir. (%5-161 aral���). Primer aktif metabolitler ortalama 160 ng/g doku konsantrasyonlar�nda tespit edilmi� olup bu de�er de kararl� durumda g�zlenen doruk plazma konsantrasyonlar�n�n %113'l�k genel ortalamas�na denk gelmektedir (%88-130). Plazma protein ba�lanmas� yakla��k olarak
%95'tir. Erlotinib serum albumine ve alfa-1 asit glikoproteine (AAG) ba�lanmaktad�r.
Biyotransformasyon:
Erlotinib insanlarda karaci�erde hepatik sitokrom enzimleri taraf�ndan, birincil olarak CYP3A4 ve daha az �l��de CYP1A2 taraf�ndan metabolize edilmektedir. CYP4A4'�n ba��rsaktaki, CYP1A1'in akci�erdeki, CYP1B1'in t�m�r dokusundaki ekstrahepatik metabolizmas� erlotinibin metabolik klerensine potansiyel olarak yard�m eder.
Tespit edilen 3 ana metabolik yol mevcuttur: 1) yan zincirlerden biri veya her ikisinin O- demetilasyonu ve bunu takiben karboksilik aside oksidasyon; 2) asetilen k�sm�n�n oksidasyonu ve takiben aril karboksilik aside hidrolizi; ve 3) fenil-asetilen k�sm�n�n aromatik hidroksilasyonu. Yan zincirlerden birinin O-demetilasyonu ile olu�an OSI 420 ve OSI 413 primer metabolitleri preklinik in vitro deneyler ve in vivo t�m�r modellerindeki erlotinib ile kar��la�t�r�labilir etkiye sahiptir. Bunlar plazmada erlotinibin <%10'u oran�nda mevcut bulunup, erlotinib ile benzer farmakokinetik g�sterirler.
Eliminasyon:
Erlotinib b�y�k oranda metabolitleri halinde birincil olarak fe�es ile at�l�rken (>%90), renal eliminasyon bir oral dozun yaln�zca k���k bir miktar�na (yakla��k %9) kar��l�k gelir. Tek ajan olarak erlotinib verilen 591 hastadaki bir pop�lasyon farmakokinetik analizi, 36,2 saatlik ortalama yar�-�m�rle, 4,47 L/saatlik ortalama g�r�nen klerens ortaya koymu�tur. Bu nedenle, kararl�l�k durumu plazma konsantrasyonlar�na ula��lmas�n�n yakla��k 7-8 g�n i�inde ger�ekle�mesi beklenmektedir.
Do�rusall�k/do�rusal olmayan durum:
Yeterli veri yoktur.
Hastalardaki karakteristik �zellikler
Beklenen g�r�n�r klerens ile hasta ya��, v�cut a��rl���, cinsiyet ve etnik �zellikler aras�nda anlaml� bir ili�ki g�zlenmemi�tir. Erlotinib farmakokineti�ini de�i�tiren hastaya ait fakt�rler, serum total bilirubin, alb�min ve alfa-1 asit glikoprotein konsantrasyonlar� ve sigara kullan�m�n�n devam etmesidir. Artm�� total bilirubin serum konsantrasyonlar� ve alb�min ve alfa-1 asit glikoprotein konsantrasyonlar� daha yava� h�zda erlotinib klerensi ile birliktelik g�stermi�tir. Bu farkl�l�klar�n klinik relevans� belli de�ildir. Bununla birlikte, sigara i�enlerde daha h�zl� bir erlotinib klerensi g�zlenmi�tir. Bu durum, tek bir oral doz olarak 150 mg erlotinib alan, sigara i�meyen ve halihaz�rda sigara i�en sa�l�kl� bireylerde yap�lan farmakokinetik �al��mada do�rulanm��t�r. C'�n geometrik ortalamas� sigara i�meyenlerde 1056 ng/mL iken sigara i�enlerde 689 ng/mL olmu�tur ve sigara i�enler i�in sigara i�meyenlere g�re ortalama oran %65,2'dir (95% GA: 44,3 ila 95,9, p = 0,031). EAA i�in geometrik ortalama sigara i�meyenlerde 18726 ngxh/mL ve sigara i�enlerde 6718 ngxh/mL olmu�tur ve ortalama oran %35,9'dur (%95 GA: 23,7 ila 54,3, p < 0,0001). Ci�in geometrik ortalama sigara i�meyenlerde 288 ng/mL ve sigara i�enlerde 34,8 ng/mL olmu�tur ve ortalama oran %12,1'dir (%95 GA: 4,82ila30,2,p=0,0001).
Pivotal faz III KHDAK �al��mas�nda, halihaz�rda sigara i�enlerde erlotinib i�in kararl� durum plazma konsantrasyonu 0,65 mcg/mL olmu�tur ve bu de�er sigaray� b�rakanlarda veya hi�
sigara i�memi� bireylerde g�r�len konsantrasyonun iki kat�ndan daha azd�r (1,28 mcg/mL, n=108). Bu etki ile birlikte g�r�len erlotinib plazma klerensinde %24'l�k bir art�� g�zlenmi�tir. Daha �nce sigara i�mi� olan KHDAK hastalar� �zerinde yap�lan faz I doz eskalasyon �al��mas�nda, kararl� durumdaki farmakokinetik analizleri erlotinib dozu 150 mg'den maksimum tolere edilebilir doz olan 300 mg'ye art�r�ld���nda erlotinib maruziyetinde doz orant�l� art�� g�stermi�tir. Bu �al��madaki halihaz�rda sigara i�enlerde uygulanan 300 mg dozda kararl� durum plazma konsantrasyonu 1,22 mcg/mL olmu�tur (n=17) (bkz. B�l�m 4.2, 4.4, 4.5 ve 5.1).
Populasyon farmakokinetik analizinden elde edilen sonu�lara g�re bir opioid varl���n�n maruziyeti %11 oran�nda art�rd��� g�r�lm��t�r.
Pediyatrik populasyon:
Pediyatrik hastalara �zg�n �al��malar bulunmamaktad�r.
Geriyatrik populasyon:
Ya�l� hastalara �zg�n �al��malar bulunmamaktad�r.
Karaci�er bozuklu�u:
Erlotinib birincil olarak karaci�er arac�l���yla metabolize edilir. Solid t�m�rleri olan ve orta derecede hepatik fonksiyon bozuklu�u bulunan hastalarda (Child-Pugh skoru 7-9) erlotinib EAA ve C geometrik ortalamas� s�ras� ile 27000 ngxh/mL ve 805 ng/mL olmu�tur ve bu de�erler primer karaci�er kanseri veya hepatik metastazlar� olanlar da dahil olmak �zere hepatik fonksiyonlar� yeterli olan hastalarda s�ras� ile 29300 ngxh/mL ve 1090 ng/mL �eklindedir. C de�erinin orta derecede hepatik fonksiyon bozuklu�u olan hastalarda istatiksel olarak anlaml� derecede daha d���k olmas�na kar��n bu fark�n klinik olarak anlaml� olmad��� d���n�lmektedir. �iddetli hepatik disfonksiyonun erlotinib farmakokineti�i �zerindeki etkisi ile ilgili veri bulunmamaktad�r. Pop�lasyon farmakokinetik analizinde, total bilirubinin artm�� serum konsantrasyonlar�n�n daha d���k erlotinib klerensi h�z� ile ili�kili oldu�u g�r�lm��t�r.
B�brek bozuklu�u:
Erlotinib ve metabolitlerinin b�brekler taraf�ndan at�l�m� �nemli �l��de de�ildir. Tek bir dozun %9'dan az� idrar ile at�lmaktad�r. Pop�lasyon farmakokinetik analizinde, erlotinib klerensi ve kreatinin klerensi aras�nda klinik olarak anlaml� bir ili�ki g�r�lmemi�tir ancak kreatinin klerensi 15 ml/dk'den az olan hastalar ile ilgili bir veri bulunmamaktad�r.
5.3. Klinik �ncesi g�venlilik verileri
6. FARMAS�T�K �ZELL�KLER
6.1. Yard�mc� maddelerin listesi
Susuz laktoz (inek s�t�nden elde edilmektedir) Mikrokristalin sel�loz (E460)
Sodyum ni�asta glikolat Sodyum lauril s�lfat Magnezyum stearat (E470 b) Kollodial susuz silika Opadry beyaz� (20A580000) Saf Su
Opadry beyaz� (20A580000) bile�imi:
Hipromelloz (E464) Hidroksipropil sel�loz (E463) Titanyum dioksit (E171) Sodyum lauril s�lfat
6.2. Ge�imsizlikler
Yeterli veri yoktur.
6.3. Raf �mr�
24 ay
6.4. Saklamaya y�nelik �zel tedbirler
25°C alt�ndaki oda s�cakl���nda saklay�n�z.
6.5. Ambalaj�n niteli�i ve i�eri�i
30 tabletlik OPA/AL/PVC-Alu-Alu blisterde sunulmaktad�r.
6.6. Be�eri t�bbi �r�nden arta kalan maddelerin imhas� ve di�er �zel �nlemler
Kullan�lmam��/Son kullanma tarihi ge�mi� �r�nlerin imhas�
kullan�n�z.
Kullan�lmam�� olan �r�nler ya da at�k materyallar “T�bbi At�klar�n Kontrol� y�netmeli�i” ve “Ambalaj ve Ambalaj At�klar�n�n Kontrol� y�netmelikleri”ne uygun olarak imha edilmelidir.
 Mesane Kanseri
Mesane kanseri her zaman mukozada ba�lar. Erken safhalarda bu tabakada s�n�rl� kal�r ve
h�cre i�indeki karsinom olarak nitelendirilir.
Mesane Kanseri
Mesane kanseri her zaman mukozada ba�lar. Erken safhalarda bu tabakada s�n�rl� kal�r ve
h�cre i�indeki karsinom olarak nitelendirilir. |
 Y�ksek Tansiyon
Hipertansiyon s�rekli anormal derecede y�ksek olan kan bas�nc�d�r. Tansiyon
atardamarlar�n�zdaki kan�n bas�nc�d�r.
Y�ksek Tansiyon
Hipertansiyon s�rekli anormal derecede y�ksek olan kan bas�nc�d�r. Tansiyon
atardamarlar�n�zdaki kan�n bas�nc�d�r. |
�LA� E�DE�ERLER�
| E�de�er �la� Ad� | Barkodu | �la� Fiyat� |
|---|---|---|
| BINITOL | 8680352000273 | 18,499.10TL |
| DIYAGLIP | 8699717090736 | 652.40TL |
| ERTINOB | 8699262090878 | |
| ERTIVEC | 8699828091554 | 18,499.17TL |
| ETINIB | 8699717090705 | 21,894.43TL |
| Di�er E�de�er �la�lar |
 |
Kalp Krizi Kalbe giden kan ak��� durdu�unda kalp krizi meydana gelir. |
 |
�izofrenlik �izofrenli�in psikiatrik te�hisi hakk�nda �ok fazla anla�mazl�k vard�r. Bu sayfadaki bilgiler, �izofrenli�in te�hisi, nedenleri ve tedavisi hakk�ndaki fakl� teoriler hakk�nda bilgi verecektir. |
 |
L�semi Kan Kanseri L�semi, kan kanseridir ve v�cudunun kan olu�turan dokular�n�n hastalanmas� anlam�na gelir. Bir�ok l�semi t�r� vard�r; baz� l�semi t�rleri �ocuklarda baz�lar� da yeti�kinlerde s�k g�r�l�r. |
�LA� GENEL B�LG�LER�
Turgut �la� A.�.
| Sat�� Fiyat� | 17618.88 TL [ 1 Dec 2025 ] |
| �nceki Sat�� Fiyat� | 17618.88 TL [ 24 Nov 2025 ] |
| Original / Jenerik | Original �la� |
| Re�ete Durumu | K�rm�z� Re�eteli bir ila�d�r. |
| Barkodu | 8699519090118 |
| Etkin Madde | Erlotinib |
| �thal ( ref. �lke : Yunanistan ) ve Be�eri bir ila�d�r. |