SIMKELMA 5G oral süspansiyon hazırlamak için toz-11 saşe Kısa Ürün Bilgisi
{ Sodyum Zirkonyum Siklosilikat }
1. BEŞERİ TIBBİ ÜRÜNÜN ADI
SIMKELMA 5 g oral süspansiyon hazırlamak için toz
2. KALİTATİF VE KANTİTATİF BİLEŞİM
Etkin madde
Her bir saşe 5 g sodyum zirkonyum siklosilikat içerir.
Yardımcı maddeler
SIMKELMA, yardımcı madde içermemektedir.
3. FARMASÖTİK FORMU
Oral süspansiyon hazırlamak için toz. Beyaz ila gri arası toz.
4. KLİNİK ÖZELLİKLER
4.1. Terapötik endikasyonlar
SIMKELMA, erişkin hastalarda hiperkalemi tedavisinde endikedir (Bkz. Bölüm 4.4 ve Bölüm 5.1).
4.2. Pozoloji ve uygulama şekli
ErişkinlerDüzeltme fazı
SIMKELMA'nın önerilen başlangıç dozu, su içinde süspansiyon halinde oral yolla günde üç kez uygulanan 10 g'dır. Normokalemiye ulaşıldığında idame rejimine geçilmesi gerekmektedir (aşağıdaki açıklamaya bakınız).
Normokalemiye genel olarak 24 ila 48 saat içinde ulaşılır. Hastalar, 48 saatlik tedaviden sonra halen hiperkalemik ise, aynı rejime 24 saat daha devam edilebilir. Eğer 72 saatlik tedaviden sonra normokalemiye ulaşılamıyorsa başka tedavi yaklaşımlarının göz önünde bulundurulması gerekmektedir.
İdame fazı
Sürekli idame tedavisinde, hiperkalemi nüksünü önlemek için minimum etkili doz oluşturulmalıdır. Normal bir potasyum seviyesini korumak için, günde bir kez 5 g'lık doz önerilir, gerekirse günde birkez10g'akadardozarttırılabilir veya gün aşırı 5 g'a kadar doz
Tedavi sırasında serum potasyum düzeylerinin düzenli olarak izlenmesi gerekmektedir. İzlem sıklığı; diğer ilaçlar, kronik böbrek hastalığının progresyonu ve diyetle alınan potasyum miktarı gibi çeşitli faktörlere bağlı olacaktır.
Eğer şiddetli hipokalemi meydana gelirse, SIMKELMA kesilmeli ve hasta yeniden değerlendirilmelidir.
Kronik hemodiyaliz uygulanan hastalar
Diyaliz görmekte olan hastalara SIMKELMA yalnızca diyaliz uygulanmayan günlerde verilmelidir. Önerilen başlangıç dozu günde bir defa 5 g'dir. Normokalemi (4,0-5,0 mmol/L) sağlamak amacıyla doz, uzun diyaliz arasından (LIDI) sonra ölçülen diyaliz öncesi serum potasyum değerine bağlı olarak haftalık olarak yukarı veya aşağı titre edilebilir. Doz, bir haftalık aralıklarla ve 5 g'lik basamaklarla diyaliz uygulanmayan günlerde günde bir defa 15 g'ye kadar ayarlanabilir. Doz ayarlandığı sırada serum potasyum seviyelerinin her hafta kontrol edilmesi önerilir; normokalemi sağlandığında potasyum düzenli şekilde (örneğin her ay veya diyetsel potasyum veya serum potasyum düzeyini etkileyen ilaçları içeren klinik değerlendirmeye bağlı olarak daha sık) takip edilmelidir.
Unutulan doz
Hasta bir dozu almayı unutursa, kendisine bir sonraki dozu normal zamanında alması söylenmelidir.
Uygulama şekli:
SIMKELMA oral kullanım içindir. Süspansiyon aç veya tok karnına alınabilir.
Süspansiyonun hazırlanmasına ilişkin talimatlar için bkz. bölüm 6.6.
Özel popülasyonlara ilişkin ek bilgiler:
Böbrek yetmezliği:
Böbrek yetmezliği olan hastalar için normal dozlarda herhangi bir değişiklik gerekmemektedir.
Karaciğer yetmezliği:
Karaciğer yetmezliği olan hastalar için normal dozlarda herhangi bir değişiklik gerekmemektedir.
Pediyatrik popülasyon:
SIMKELMA'nın güvenliliği ve etkililiği 18 yaşın altındaki ergenlerde ve çocuklarda belirlenmemiştir. Veri bulunmamaktadır.
Geriyatrik popülasyon:
Geriyatrik hastalar için doz ayarlaması gerekmez.
4.3. Kontrendikasyonlar
SIMKELMA, sodyum zirkonyum siklosilikata karşı aşırı duyarlılığı olan hastalarda kontrendikedir.
4.4. Özel kullanım uyarıları ve önlemleri
Serum potasyum düzeyleri
Serum potasyum konsantrasyonunu etkileyen tıbbi ürünlerde (örn. renin-anjiyotensin- aldosteron sistemi (RAAS) inhibitörleri veya diüretikler) değişiklik yapıldıktan sonra ve SIMKELMA dozunun titre edilmesinden sonra klinik olarak gerektiğinde serum potasyum düzeyi izlenmelidir.
Hipokalemi
Hipokalemi gözlenebilir (Bkz. Bölüm 4.8). Bu gibi durumlarda orta ila şiddetli hipokalemiyi önlemek için idame pozolojisi başlığı altında verilen doz titrasyonu gerekli olabilir. Şiddetli hipokalemisi olan hastalarda SIMKELMA kesilmeli ve hasta yeniden değerlendirilmelidir.
QT Uzaması
Hiperkalemi düzeltildiği sırada, serum potasyum konsantrasyonundaki düşüşün fizyolojik sonucu olarak QT aralığında bir uzama gözlenebilir.
X-ışınları ile etkileşim riski
Sodyum zirkonyum siklosilikat, X-ışınlarını geçirmeyebilir. Eğer hasta batın röntgeni çektiriyorsa, radyologların bu durumu göz önünde bulundurması gerekmektedir.
İntestinal perforasyon
SIMKELMA kullanımı ile intestinal perforasyon riski halihazırda bilinmemektedir. SIMKELMA ile herhangi bir intestinal perforasyon olayı bildirilmemiştir. Gastrointestinal sistemde etki gösteren polimerler ile intestinal perforasyon bildirilmiş olduğundan, intestinal perforasyonun bulgu ve belirtilerine karşı özel dikkat gösterilmesi gerekmektedir.
Sodyum içeriği
Bu tıbbi ürün her 5g'lık dozunda 400 mg sodyum ihtiva eder. Bu durum, kontrollü sodyum diyetinde olan hastalar için göz önünde bulundurulmalıdır.
Klinik verilerin kısıtlamaları
Şiddetli hiperkalemi
Serum potasyum konsantrasyonları 6,5 mmol/L'nin üzerinde olan hastalarda sınırlı deneyim mevcuttur.
Uzun süreli maruziyet
SIMKELMA ile klinik çalışmalar bir yıldan daha uzun süreli maruziyeti içermemektedir.
4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri
Diğer tıbbi ürünlerin sodyum zirkonyum siklosilikat üzerindeki etkisi
Sodyum zirkonyum siklosilikat vücut tarafından emilmediğinden veya metabolize edilmediğinden, diğer tıbbi ürünlerin sodyum zirkonyum siklosilikatın farmakolojik aktivitesi üzerinde beklenen bir etkisi bulunmamaktadır.
Sodyum zirkonyum siklosilikatın diğer tıbbi ürünler üzerindeki etkisi
Sodyum zirkonyum siklosilikat vücut tarafından emilmediğinden veya metabolize edilmediğinden ve diğer tıbbi ürünlere anlamlı düzeyde bağlanmadığından, diğer tıbbi ürünlerin üzerinde sınırlı etkileri bulunmaktadır.Sodyumzirkonyumsiklosilikat, hidrojen iyonlarını
biyoyararlanımı pH değerine bağlı tıbbi ürünlerin çözünürlüğünde ve emilim kinetiklerinde değişikliklere yol açabilmektedir. Sağlıklı gönüllülerde yürütülen bir klinik ilaç-ilaç etkileşimi çalışmasında sodyum zirkonyum siklosilikat ile amlodipin, klopidogrel, atorvastatin, furosemid, glipizid, varfarin, losartan veya levotiroksin ile birlikte uygulanması, klinik olarak önemli ilaç-ilaç etkileşimleri ile sonuçlanmamış. Diğer gastrik asit modifiye ediciler ile dabigatranın eşzamanlı uygulaması ile uyumlu şekilde, sodyum zirkonyum siklosilikat ile birlikte uygulandığında dabigatranın Cve EAA değerleri yaklaşık %40 daha düşük bulunmuştur. Bu tıbbi ürünlerin hiçbiri için herhangi bir doz ayarlaması veya doz uygulama zamanlarının birbirinden ayrılması gerekmemektedir. Fakat sodyum zirkonyum siklosilikat, mide pH'ına bağımlı biyoyararlanımı klinik açıdan anlamlı olan oral ilaçlardan en az 2 saat önce veya 2 saat sonra uygulanmalıdır.
Artmış mide pH'ı ile ilaçlar arasındaki olası etkileşimden kaçınmak adına sodyum zirkonyum siklosilikattan 2 saat önce veya 2 saat sonra uygulanması gereken tıbbi ürünlere verilebilecek örnekler arasında azol antifungaller (ketokonazol, itrakonazol ve posakonazol), anti-HIV ilaçlar (atazanavir, nelfinavir, indinavir, ritonavir, sakuinavir, raltegravir, ledipasvir ve rilpivirin) ve tirozin kinaz inhibitörleri (erlotinib, dasatinib ve nilotinib) yer almaktadır.
Sodyum zirkonyum siklosilikat, pH'a bağımlı biyoyararlanım sergilemeyen oral ilaçlarla doz uygulama zamanları arasında aralık bırakılmadan eşzamanlı şekilde uygulanabilir.
Sağlıklı gönüllülerde yapılan başka bir ilaç-ilaç etkileşim çalışmasında, 15g SIMKELMA'nın 5mg takrolimus ile birlikte uygulanması takrolimus EAA ve C'ında sırasıyla %37 ve %29 azalma ile sonuçlanmıştır. Bu nedenle takrolimus SIMKELMA'dan en az 2 saat önce veya sonra alınmalıdır. Aynı çalışmada, SIMKELMA ve siklosporinin birlikte uygulanması klinik olarak anlamlı bir etkileşim göstermemiştir.
Özel popülasyonlara ilişkin ek bilgiler Pediyatrik popülasyon:
Etkileşim çalışmaları sadece yetişkinlerde yapılmıştır.
4.6. Gebelik ve laktasyon
Gebelik Kategorisi: B
Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon)
Çocuk doğurma potansiyeli olan kadınlarda SIMKELMA'nın kullanımı hakkında veri yoktur. Kontrasepsiyona gerek duyulup duyulmadığı bilinmemektedir.
Gebelik dönemi
SIMKELMA'nın gebe kadınlarda kullanımına ilişkin veri bulunmamaktadır. Hayvan çalışmaları üreme toksisitesine bağlı olarak doğrudan veya dolaylı zararlı etkileri göstermemektedir (Bkz. Bölüm 5.3). Tedbir amaçlı olarak gebelik sırasında SIMKELMA kullanılmamalıdır.
Laktasyon dönemi
Emzirmekte olan kadının sodyum zirkonyum siklosilikata sistemik maruz kalması, ihmal edilebilir düzeyde olduğu için, emzirilen çocuk üzerinde herhangi bir etki öngörülmemektedir. SIMKELMA emzirme döneminde kullanılabilir.
Sıçanlardaki bir postnatal çalışmada, sodyum zirkonyum siklosilikata maternal maruziyetin postnatal gelişim üzerinde bir etkisi olmamıştır. Fizikokimyasal özelliklerine bağlı olarak sodyum zirkonyum siklosilikatın sistemik emilimi bulunmadığından anne sütüne atılması beklenmez.
Üreme yeteneği/Fertilite
İlacın uygulandığı sıçanlarda ya da tavşanlarda embriyo-fetal gelişim üzerinde advers etki olmamıştır.
4.7. Araç ve makine kullanımı üzerindeki etkiler
SIMKELMA'nın araç ve makine kullanımı üzerinde etkisi bulunmamakta ya da etkisi ihmal edilebilecek kadar azdır.
4.8. İstenmeyen etkiler
Güvenlilik profilinin özeti
En yaygın sıklıkla bildirilen advers reaksiyonlar hipokalemi (%4,1) ve ödem ile ilişkili olaylar (%5,7)'dır.
Advers reaksiyonların tablolaştırılmış özeti
SIMKELMA'nın güvenlilik profili, 507'si bir yıl boyunca maruz kalan 1760 hastayı içeren klinik çalışmalarda değerlendirilmiştir.
Kontrollü çalışmalarda tanımlanan advers reaksiyonlar Tablo 1'de gösterilmektedir. Advers reaksiyonların sıklığı için aşağıdaki standart kullanılmıştır: Çok yaygın (≥1/10); yaygın (≥1/100 ila <1/10); yaygın olmayan (≥1/1.000 ila <1/100); seyrek (≥1/10.000 ila <1/1.000); çok seyrek (<1/10.000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor).
Tablo 1. Klinik çalışmalardaki advers reaksiyonların listesi
Sistem Organ sınıfı |
|
Metabolizma ve beslenme hastalıkları | Yaygın: Hipokalemi |
Genel bozukluklar ve uygulama bölgesine ilişkin hastalıklar | Yaygın: Ödem ile ilişkili olaylar |
Seçili advers reaksiyonların tanımı
Hipokalemi
Klinik çalışmalarda SIMKELMA hastalarının %4,1'i 3,5 mmol/L'nin altında serum potasyum değerleriyle seyreden ve SIMKELMA dozunda ayarlama veya SIMKELMA'nın kesilmesiyle düzelen hipokalemi geliştirmiştir.
Ödem ile ilişkili olaylar
Aşırı sıvı yükü, sıvı tutulumu, jeneralize ödem, hipervolemi, lokalize ödem, ödem, periferik ödem ve periferik şişmeyi içeren ödem ile ilişkili olaylar, SIMKELMA kullanan hastaların
%5,7'si tarafından bildirilmiştir. Bu olaylar sadece idame fazında gözlenmiş ve 15 g ile tedavi edilen hastalarda daha yaygın olarak görülmüştür. Olayların %53 kadarı diüretik tedavisine başlanarak veya diüretik dozunda ayarlama yapılarak kontrol edilmiş, kalanı ise tedavi gerektirmemiştir.
Uzun süreli maruziyet
874 gönüllüde 1 yıla varan süreyle SIMKELMA'ya açık etiketli maruziyetin söz konusu olduğu 2 klinik çalışmada araştırmacılar tarafından şu olaylar ilgili olarak bildirilmiştir: gastrointestinal olaylar [konstipasyon (%2,9), diyare (%0,9), abdominal ağrı/distansiyon (%0,5), bulantı (%1,6) ve kusma (%0,5)]; aşırı duyarlılık reaksiyonları [döküntü (%0,3) ve prürit (%0,1)]. Bu olaylar hafif ila orta şiddetli bir yapı sergilemiştir, hiçbiri ciddi şeklinde bildirilmemiştir ve genel olarak hasta tedaviye devam ederken çözümlenmiştir. Açık etiketli çalışma tasarımı nedeniyle bu olaylar ile SIMKELMA arasında bir nedensel ilişki kesin olarak ortaya konamamaktadır.
Şüpheli advers reaksiyonların raporlanması
Ruhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar / risk dengesinin sürekli olarak izlenmesine olanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu Türkiye Farmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e-posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks: 0 312 218 35 99).
4.9. Doz aşımı ve tedavisi
Sodyum zirkonyum siklosilikat ile doz aşımı hipokalemiye neden olabilir. Hastaların serum potasyum düzeyleri kontrol edilmeli ve gerekirse potasyum takviyesi yapılmalıdır.
5. FARMAKOLOJİK ÖZELLİKLER
5.1. Farmakodinamik özellikler
Farmakoterapötik grup: Hiperkalemi ve hiperfosfatemi tedavisinde kullanılan ilaçlar ATC kodu: V03AE10
Etki mekanizması:
Sodyum zirkonyum siklosilikat, hidrojen ve sodyum katyonlarının değiş-tokuş değişiminde potasyumu tercihli bir şekilde yakalayan tekdüze mikrogözenekli yapıya sahip absorbe olmayan, polimer olmayan bir inorganik tozdur. Sodyum zirkonyum siklosilikat, in vitro ortamda kalsiyum ve magnezyum gibi diğer katyonların varlığında dahi potasyum iyonları için yüksek düzeyde seçicidir. Sodyum zirkonyum siklosilikat, gastrointestinal (Gİ) sistemin tamamında potasyumu yakalar ve Gİ lümende serbest potasyum konsantrasyonunu düşürür, böylece hiperkalemiyi düzeltmek için serum potasyum düzeylerini azaltır ve feçes ile potasyum atılımını artırır.
Farmakodinamik etkiler
Sodyum zirkonyum siklosilikat, alımından sonraki 1 saat kadar kısa bir süre içinde serum potasyum konsantrasyonlarını düşürmeye başlar ve normokalemiye genel olarak 24 ila 48 saat
konsantrasyonlarını ya da idrarla sodyum atılımını etkilemez. Başlangıç serum potasyum düzeyleri ile etkinin büyüklüğü arasında yakın bir korelasyon vardır; başlangıç serum potasyum düzeyleri daha yüksek hastalarda serum potasyum düzeylerindeki azalmalar daha fazla olmaktadır. Serum potasyum konsantrasyonunda azalmanın bir sonucu olarak idrarla potasyum atılımında bir düşüş meydana gelir. Dört gün süreyle günde bir kez SIMKELMA 5 g veya 10 g verilen sağlıklı gönüllülerle gerçekleştirilen bir çalışmada, serum potasyum konsantrasyonu ve idrarla toplam potasyum atılımındaki doza bağımlı azalmaya, feçes ile potasyum atılımında ortalama artışlar eşlik etmiştir. İdrarla sodyum atılımında istatistiksel olarak anlamlı değişiklikler gözlenmemiştir.
Sodyum zirkonyum siklosilikatın aç veya tok karnına uygulandığı durumlarda farmakokinetiğini inceleme amaçlı çalışmalar gerçekleştirilmemiştir.
Sodyum zirkonyum siklosilikatın ayrıca in vitro ve in vivo koşullarda amonyumu bağladığı, böylelikle amonyumu uzaklaştırıp serum bikarbonat düzeylerini artırdığı gösterilmiştir. SIMKELMA ile tedavi edilen hastalar bikarbonat düzeyinde günde bir kez 5 g dozunda 1,1 mmol/L, günde bir kez 10 g dozunda 2,3 mmol/L ve günde bir kez 15 g dozunda 2,6 mmol/L artış yaşamış, plasebo uygulanan hastalarda ise ortalama artış 0,6 mmol/L olmuştur. Renin ve aldosteronu etkileyen diğer faktörlerin kontrol edilmediği bir ortamda SIMKELMA, plasebo grubu (+%14) ile karşılaştırıldığında, ortalama serum aldosteron düzeylerinde dozdan bağımsız bir değişim sağlamıştır (aralık: -%30 ila -%31). Ancak bununla ilişkili olarak sistolik ve diyastolik kan basıncında herhangi bir etki gözlenmemiştir.
Ek olarak, plasebo (0,8 mg/dL) ve düşük doz sodyum zirkonyum siklosilikat (0,3 mg/dL) gruplarındaki küçük ortalama artışlar ile karşılaştırıldığında günde üç kez 5 g (1,1 mg/dL) ve
10 g (2,0 mg/dL) gruplarında serum üre nitrojen (BÜN) değerlerinde ortalama düşüşler gözlenmiştir.
Klinik etkililik ve güvenlilik:
SIMKELMA'nın potasyum düşürücü etkisi, hiperkalemisi olan hastalarda yürütülen randomize, çift kör, plasebo kontrollü üç çalışmada gösterilmiştir. Üç çalışma 48 saatlik bir periyot boyunca SIMKELMA'nın hiperkalemiyi düzeltmedeki ilk etkisini test etmiş ve iki çalışma ayrıca elde edilen normokalemi etkisinin idamesini test etmiştir. İdame çalışmalarına, kronik böbrek hastalığı (% 58), kalp yetmezliği (% 10), diabetes mellitus (% 62) ve RAAS inhibitör tedavisi (% 68) olan hastalar dahil edilmiştir. Ek olarak iki açık etiketli idame çalışmasında SIMKELMA'nın uzun süreli güvenliliği test edilmiştir. Bu beş çalışmada SIMKELMA dozları verilen 1760 hasta yer almıştır; bu hastaların 507'si ilaca en az 360 gün maruz kalmıştır. Ek olarak, SIMKELMA'nın etkililiği ve güvenliliği, 8 hafta boyunca SIMKELMA dozları alan hiperkalemili 196 kronik hemodiyaliz hastası ile yürütülen çift kör, plasebo kontrollü bir çalışmada incelenmiştir. Bu çalışmalarda SIMKELMA, altta yatan hiperkalemi sebebi, yaş, cinsiyet, ırk, komorbid hastalık ve eşzamanlı RAAS inhibitörleri kullanımından bağımsız olarak serum potasyumu düzeyini düşürmüş ve normal serum potasyum düzeylerini korumuştur. Herhangi bir diyet kısıtlaması getirilmemiş olup, hastalara özel bir değişiklik olmaksızın her zamanki diyetlerine devam etmeleri söylenmiştir.
![]()
İki fazlı, plasebo kontrollü düzeltme ve idame kullanımı çalışması
Hiperkalemisi (5 ila ≤6,5 mmol/L, başlangıçtaki potasyum ortalaması 5,3 mmol/L) olan ve kronik böbrek hastalığı, kalp yetmezliği ve diyabeti olan ve RAAS inhibitör tedavisinde olan hastaların dahil olduğu 753 hastada (ortalama yaş 66 yıl, aralık 22 ila 93) yürütülen 2 kısımlı, çift kör, randomize, plasebo kontrollü bir çalışmadır.
Düzeltme fazı sırasında hastalar, ilk 48 saat süreyle günde üç kez uygulanan SIMKELMA (1,25 g, 2,5 g, 5 g veya 10 g) ya da plasebo almak üzere randomize edilmiştir (Tablo 2).
Tablo 2. Düzeltme fazı (Çalışma 1): 48 saat SIMKELMA sonrasında normokalemik olguların yüzdesi
SIMKELMA dozu (günde üç kez) | |||||
| Plasebo | 1,25 g | 2,5 g | 5 g | 10 g |
N | 158 | 154 | 141 | 157 | 143 |
Başlangıç serum potasyum, mmol/L | 5,3 | 5,4 | 5,4 | 5,3 | 5,3 |
48 saatte normokalemik, % | 48 | 51 | 68 | 78 | 86 |
Plaseboya karşı p değeri |
| AD | <0,001 | <0,001 | <0,001 |
AD: anlamlı değil
Günde üç kez uygulanan SIMKELMA 10 g, 48 saatte serum potasyum düzeyini 0,7 mmol/L düşürmüştür (plaseboya karşı p<0,001); ilk dozdan 1 saat sonra istatistiksel olarak anlamlı %14 potasyum düşüşü gözlenmiştir. Başlangıçta potasyum düzeyleri daha yüksek olan hastaların SIMKELMA'ya yanıtı daha fazla olmuştur. Tedavi öncesi potasyum düzeyleri 5,5 mmol/L'nin üzerinde olan hastalar (ortalama başlangıç değeri 5,8 mmol/L) 48 saatte 1,1 mmol/L'lik ortalama bir düşüş elde ederken başlangıçtaki potasyum düzeyleri 5,3 mmol/L veya bu değerin altında olanlarda ortalama düşüş, en yüksek dozda 0,6 mmol/L olmuştur.
Düzeltme fazı sırasında SIMKELMA aldıktan sonra normokalemik duruma geçen hastalar, günde bir kez plasebo veya düzeltme fazı sırasında günde üç kez aldıkları ile aynı doz düzeyinde günde bir kez SIMKELMA almak üzere tekrar randomize edilmiştir (Tablo 3).
Tablo 3. İdame fazı (12 gün, Çalışma 1): Ortalama normokalemik gün sayısı
İdame fazı tedavisi (günde bir kez) | |||||
| Plasebo |
| SIMKELMA | Plaseboya p değeri | |
Düzeltme fazı SIMKELMA dozu | n | Gün | n | Gün |
|
1,25 g günde üç kez | 41 | 7,6 | 49 | 7,2 | AD |
2,5 g günde üç kez | 46 | 6,2 | 54 | 8,6 | 0,008 |
5 g günde üç kez | 68 | 6,0 | 64 | 9,0 | 0,001 |
10 g günde üç kez | 61 | 8,2 | 63 | 10,2 | 0,005 |
AD: anlamlı değil
SIMKELMA'nın artık uygulanmadığı idame periyodunun sonunda, ortalama potasyum düzeyleri, başlangıç düzeylerineyakınbirseviyeyeyükselmiştir.
![]()
Ek bir açık etiket fazlı, çok merkezli, plasebo kontrollü idame çalışması
Çalışmanın düzeltme fazında, hiperkalemisi olan 258 hasta (başlangıç ortalaması 5,6, aralık 4,1
- 7,2 mmol/L), 48 saat süreyle günde üç kez 10 g SIMKELMA almıştır. SIMKELMA'nın
10 g'lık ilk dozundan 1 saat sonra potasyum düzeylerinde düşüşler gözlenmiştir. Normokalemiye kadar geçen medyan süre 2,2 saat olmuş, hastaların %66'sı 24 saatte ve %88'i 48 saatte normokalemiye ulaşmıştır. Hiperkalemisi daha şiddetli olan hastalarda yanıtlar daha büyük olmuştur; başlangıç serum potasyum düzeyleri <5,5, 5,5-5,9 ve ≥6 mmol/L olan hastalarda serum potasyum düzeyi sırasıyla 0,8, 1,2 ve 1,5 mmol/L düşmüştür.
Normokalemiye ulaşan hastalar (potasyum düzeyleri 3,5 ile 5 mmol/L arasında) çift kör olarak 28 gün süreyle (çift kör randomize doz azaltma fazı) günde bir kez uygulanan (çift kör randomize doz azaltma fazı) SIMKELMA'nın üç dozundan birine [5 g (n=45), 10 g (n=51) veya 15 g (n=56)] ya da plaseboya (n=85) randomize edilmiştir.
Çalışmada Gün 8'den Gün 29'a (üç haftalık periyot) ortalama serum potasyum düzeyi
<5,1 mmol/L olan olguların oranı, plasebo (%46) ile karşılaştırıldığında, SIMKELMA'nın günde bir kez 5 g, 10 g ve 15 g dozlarında (sırasıyla %80, %90 ve %94) daha yüksek bulunmuştur. Serum potasyum değerinde sırasıyla 0,44 mmol/L, 0,77 mmol/L, 1,10 mmol/L ve 1,19 mmol/L ortalama düşüş olmuştur ve normokalemik kalan olguların oranı, günde bir doz SIMKELMA 5 g, 10 g, 15 g ve plasebo gruplarında sırasıyla %71, %76, %85 ve %48 olmuştur.
SIMKELMA titrasyonu (açık etiketli) ile idame fazı sonuçları: 123 hasta 11 aylık açık etiketli faza girmiştir. Serum potasyum değerini etkileyebilen diğer faktörlere bakılmaksızın ortalama serum potasyum düzeyi <5,1 mmol/L olan olguların oranı %88 ve ortalama serum potasyum düzeyi 4,66 mmol/L idi. Serum potasyum düzeyi 3,5 mmol/L'nin altında olan olguların oranı
%1'in altında; serum potasyum düzeyi 3,5 ile 5,1 mmol/L arasında olan olguların oranı %77 veya serum potasyum seviyesi 3,5 ile 5,5 mmol/L arası olan olguların oranı %93 olmuştur. Çalışma tamamlandığında (Gün 365) tedavi kesilmiştir.
İdame fazında relapsa kadar geçen zamana ilişkin Kaplan-Meier tahminleri, relapsa kadar geçen zamanda doza bağımlılık olduğunu göstermiş, 5 g dozu için medyan süre başlangıçtaki serum potasyum düzeylerine bağlı olarak 4 ila 21 gün arasında değişmiştir. Serum potasyum düzeyinin periyodik olarak izlenmesi ve SIMKELMA dozunun Bölüm Pozoloji ve uygulama şekli başlığı altında belirtildiği şekilde titre edilmesi gerekmektedir.
Şekil 1 çalışmanın düzeltme ve idame fazlarındaki ortalama serum potasyumu göstermektedir.
Şekil 1. Düzeltme ve idame fazları (Çalışma 2): zaman içinde gözlenen ortalama serum potasyum düzeyleri ve %95 CI değerleri
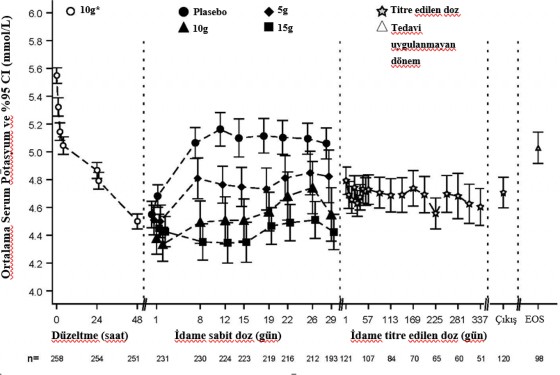
Çıkış=Son Dozun uygulanmasını takip eden 1 gün içindeki Son Ziyaret, EOS=Çalışma Sonu (Son Dozdan sonra 7 gün +/- 1 gün)
*Günde üç defa
Çalışma 3
Hiperkalemili kronik böbrek hastalığı olan hastalarda bir çalışma
Bu çalışma, başlangıç eGFR'si 30 ila 60 ml/dk/1,73 m ve hiperkalemisi (başlangıç serum potasyum değeri 5,2 mmol/L, aralık: 4,6 - 6 mmol/L) olan 90 hastada (60 SIMKELMA hastası; 30 kontrol) yürütülen çift kör, plasebo kontrollü bir doz yükseltme çalışmasıdır. Hastalar, iki ila dört gün süreyle günde üç kez yemek ile birlikte uygulanan SIMKELMA'nın yükselen dozlarına (0,3 g, 3 g ve 10 g) veya plasebo kollarına randomize edilmiştir. Birincil sonlanım noktası tedavinin ilk 2 günü boyunca serum potasyum düzeyinde başlangıca göre değişim oranı olmuştur. Çalışma, plasebo ile karşılaştırıldığında SIMKELMA'nın 3 g ve 10 g dozlarında birincil etkililik sonlanım noktasına ulaşmıştır. SIMKELMA 10 g dozunda ve 3 g dozunda sırasıyla 0,92 mmol/L ve 0,43 mmol/L'lik ortalama maksimum düşüşler ile sonuçlanmıştır. Yirmi dört saatlik idrar örnekleri SIMKELMA'nın idrarla potasyum atılımını başlangıca kıyasla düşürdüğünü göstermiştir; plasebo 8,9 mmol/24 saat ile karşılaştırıldığında 15,8 mmol/24 saat, (p <0,001). Sodyum atılımı plaseboya göre değişmemiştir (plasebo 36,9 mmol/24 saat (AD) ile karşılaştırıldığında 10 g için 25,4 mmol/24 saat).
Çalışma 4
İki fazlı, çok merkezli, çok dozlu, açık etiketli bir güvenlik ve etkinlik çalışması SIMKELMA'nın uzun süreli (12 aya kadar) etkileri bu çalışmada hiperkalemili 751 gönüllü üzerinde araştırılmıştır (başlangıç ortalaması 5,59 mmol/L; aralık 4,3 – 7,6 mmol/L). Komorbid durumlar kronik böbrek hastalığı (%65), diabetes mellitus (%64), kalp yetmezliği (%15) ve hipertansiyonu içermiştir (%83). Gönüllülerin sırasıyla %51 ve 70'inde diüretik ve RAAS inhibitörü kullanımı bildirilmiştir. Düzeltme fazı sırasında en az 24 saat süresince ve 72 saate varan süreyle günde üç defa 10 g SIMKELMA uygulanmıştır. Normokaleminin (3,5 – 5,0 mmol/L, sınır değerleri dahil) 72 saat içinde sağlandığı gönüllüler çalışmanın idame fazına girmiştir. İdame fazındaki tüm gönüllüler günde bir defa 5 g şeklinde bir başlangıç dozuyla SIMKELMA almıştır ve bu doz günde bir defa 5 g'lik artışlarla yükseltilebilmiştir (günde bir defa maksimum 15 g'ye kadar) veya titrasyon rejimine göre azaltılabilmiştir (iki günde bir uygulanan minimum 5 g'ye kadar).
Düzeltme fazı dozunun uygulamasından 24, 48 ve 72 saat sonra gönüllülerin 494/748 (%66), 563/748 (%75) ve 583/748'inde (%78) normokalemi sağlanmıştır ve serum potasyumdaki ortalama azalma 24 (n=748), 48 (n=104) ve 72 (n=28) saatte sırasıyla 0,81 mmol/L, 1,02 mmol/L ve 1,10 mmol/L olmuştur. Normokaleminin başlangıçtaki potasyum konsantrasyonuna bağlı olduğu görülmüştür ve başlangıçta en yüksek serum potasyum konsantrasyonlarına sahip olan gönüllülerin çalışma ilacına başladıktan sonra en belirgin azalmayı yaşadığı fakat normokalemi sağlanan gönüllülerin oranının bu grupta en düşük olduğu gözlenmiştir. Yüz yirmi altı hastanın başlangıç serum potasyum değerinin ≥ 6,0 mmol/L olduğu belirlenmiştir (başlangıçtaki ortalama potasyum 6,28 mmol/L). Bu gönüllülerde ortalama azalmanın düzeltme fazının sonunda 1,37 mmol/L olduğu tespit edilmiştir.
Tablo 4. Düzeltme fazı (Çalışma 4): düzeltme fazındaki çalışma gününe göre serum potasyum konsantrasyonları 3,5 ile 5,0 mmol/L arasında (sınır değerleri dahil) veya 3,5 ve 5,5 mmol/L (sınır değerleri dahil) arasında olan gönüllülerin oranı – ITT popülasyon
Düzeltme Fazı (DF) | Günde üç defa 10 g SIMKELMA (N=749) | |||||
Serum potasyum 3,5 ila 5,0 mmol/L (sınır değerleri dahil) | Serum potasyum 3,5 ila 5,5 mmol/L (sınır değerleri dahil) | |||||
n/N | Oran | %95 CI | n/N | Oran | %95 CI | |
DF 24 saat | 494/748 | 0,660 | 0,625, 0,694 | 692/748 | 0,925 | 0,904, 0,943 |
DF 48 saat | 563/748 | 0,753 | 0,720, 0,783 | 732/748 | 0,979 | 0,965, 0,988 |
DF 72 saat / DF Son | 583/748 | 0,779 | 0,748, 0,809 | 738/748 | 0,987 | 0,976, 0,994 |
Not: Bir gönüllünün doz sonrası değeri son dozu takip eden 1 günden daha sonrasına aitti. Dolayısıyla bu gönüllü Düzeltme Fazının ITT Popülasyonu için uygundu; fakat ilgili zaman noktası analize dahil edilmedi.
Hastalar ilacı almaya devam ederken normokalemi korunmuştur ve ortalama serum potasyum değeri ilacın bırakılmasını takiben artmıştır. Başlangıçta RAAS inhibitörleri kullanan hastaların
%89'u RAAS inhibitörü tedavisini bırakmamıştır, %74'ü idame fazı sırasında aynı dozda kalabilmiştir, başlangıçta RAAS inhibitörleri kullanmayanların ise %14'ü bu tedaviye başlayabilmiştir. İdame fazı sırasında gönüllülerin %75,6'sı RAAS inhibitörü kullanımına rağmen normokalemiyi korumuştur.
Şekil 2'de çalışmanın düzeltme ve idame fazlarındaki ortalama serum potasyum gösterilmektedir.

%95 CI ile serum potasyum (mmol/L)
Şekil 2: 12 aylık açık etiketli çalışmanın (Çalışma 4) düzeltme ve idame fazları – %95 CI ile zaman içindeki ortalama serum potasyum
CPBL=Düzeltme Fazı Başlangıcı, MPBL=İdame Fazı Başlangıcı
Çıkış=Son Dozu Takip Eden 1 Gün İçindeki Son Ziyaret, EOS=Çalışma Sonu (Son Dozun Ardından 7 gün +/- 1 gün)
Çalışma 5
Kronik hemodiyaliz uygulanan hastalar üzerinde gerçekleştirilen randomize, çift kör, plasebo kontrollü çalışma
Bu çalışmada son dönem böbrek hastalığı görülen, en az 3 aydır stabil diyaliz gören ve persistan diyaliz öncesi hiperkalemi sergileyen 196 hasta (ortalama yaş 58, aralık 20 ila 86 yaş), diyaliz uygulanmayan günlerde günde bir defa 5 g SIMKELMA veya plasebo almak üzere randomize edilmiştir. Randomizasyon sırasındaki ortalama serum potasyum seviyelerinin SIMKELMA grubunda 5,8 mmol/L (aralık 4,2-7,3 mmol/L), plasebo grubunda ise 5,9 mmol/L (aralık 4,2– 7,3 mmol/L) olduğu belirlenmiştir. Doz ayarlama periyodu sırasında (ilk 4 hafta) 4,0-5,0 mmol/L arası bir diyaliz öncesi serum potasyum seviyesine ulaşmak amacıyla doz, LIDI sonrası serum potasyum ölçümüne göre, haftalık olarak 5 g'lık artışlarla ayarlanabilmiştir. Doz ayarlama periyodunun sonunda ulaşılan doz takip eden 4 haftalık değerlendirme periyodu süresince devam ettirilmiştir. Doz ayarlama periyodunun sonunda hastaların %37, %43 ve
%19'unun 5 g, 10 g ve 15 g SIMKELMA almakta olduğu belirlenmiştir. Değerlendirme periyodu sırasında kurtarma tedavisi görmeyenler ve LIDI sonrası 4 diyaliz tedavisinin en az 3'ünde diyaliz öncesi serum potasyum düzeyinin 4,0 ile 5,0 mmol/L arasında korunduğu gönüllüler şeklinde tanımlanan yanıt verenlerin oranı SIMKELMA grubunda %41, plasebo grubunda ise %1 olmuştur (p<0,001)(Bkz.Şekil 3).
LIDI sonrası post-hoc analizlerde değerlendirme periyodu sırasında hastaların serum potasyum seviyesinin 4,0 ile 5,0 mmol/L arasında olduğu durumların sayısı SIMKELMA grubunda daha yüksek olmuştur. SIMKELMA grubunda 4 ziyaretin tümünde değerleri bu aralıkta olan hastaların oranının %24 olduğu belirlenmiştir, plasebo grubunda ise bu oran %0 olmuştur. Post- hoc analiz, değerlendirme periyodu sırasında LIDI sonrası 4 diyaliz tedavisinin en az 3'ünde serum potasyum düzeyinin 3,5 ile 5,5 mmol/L arasında korunduğu hastaların oranının SIMKELMA grubunda %70, plasebo grubunda ise %21 olduğunu göstermiştir.
Tedavinin sonunda, ortalama diyaliz sonrası serum potasyum seviyesi SIMKELMA grubunda 3,6 mmol/L (aralık 2,6-5,7 mmol/L), plasebo grubunda ise 3,9 mmol/L (aralık 2,2-7,3 mmol/L) olmuştur. Diyaliz arası kilo artışı (IDWG) açısından SIMKELMA ve plasebo grupları arasında fark gözlenmemiştir. IDWG, diyaliz öncesi kilo eksi önceki diyaliz seansındaki diyaliz sonrası kilo şeklinde tanımlanmıştır ve LIDI sonrasında ölçülmüştür.
Şekil 3: Kronik diyaliz uygulanmakta olan hastalarda ortalama diyaliz öncesi serum potasyum seviyelerinin zaman içindeki seyri
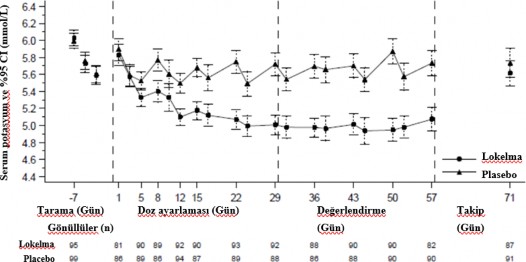
Gösterilen hata çubukları %95 güven aralıklarına karşılık gelmektedir.
n = Belirli bir ziyarette potasyum ölçümleri eksik olmayan hastaların sayısı.
5.2. Farmakokinetik özellikler
Emilim:
Klinik çalışmalar, sistemik emilimi olmadığını göstermiştir. Sıçanlardaki bir in vivo kütle dengesi çalışması, sodyum zirkonyum siklosilikatın herhangi bir sistematik emilim kanıtı olmadan feçeste tespit edildiğini göstermiştir. Bu faktörler ve çözünür olmaması nedeniyle, sitokrom P450 (CYP450) enzimleri veya taşıyıcı aktivitesi üzerindeki etkisini değerlendirmek için in vivo veya in vitro çalışma gerçekleştirilmemiştir.
Dağılım:
Sistemik dağılımı bulunmamaktadır.
Biyotransformasyon:
Sodyum zirkonyum siklosilikat, enzimatik olarak metabolize olmayan inorganik, çözünmeyen bir bileşiktir.
Eliminasyon:
Sodyum zirkonyum siklosilikat feçes yoluyla elimine edilir.
Doğrusallık/ doğrusal olmayan durum:
Uygulanabilir değildir.
5.3. Klinik öncesi güvenlilik verileri
Klinik dışı veriler; güvenlilik farmakolojisi, tekrarlı doz toksisitesi, genotoksisite, karsinojenik potansiyel, üreme ve gelişim toksisitesi standart çalışmalarına dayalı olarak insanlar için herhangi bir özel tehlike ortaya koymamaktadır.
6. FARMASÖTİK ÖZELLİKLER
6.1. Yardımcı maddelerin listesi
Bulunmamaktadır.
6.2. Geçimsizlikler
Geçerli değildir.
6.3. Raf ömrü
36 ay
6.4. Saklamaya yönelik özel tedbirler
30°C altındaki oda sıcaklığında saklayınız.
6.5. Ambalajın niteliği ve içeriği
3-katmanlı alüminyum folyo laminattan (PET/alüminyum/LLDPE) yapılmış saşeler içinde 5 g toz.
Ambalaj büyüklüklüğü: 11 veya 28 saşe
6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler
Tek dozluk saşenin tüm içeriği, yaklaşık 45 mL su içeren bir su bardağına boşaltılmalı ve iyice karıştırılmalıdır. Toz çözülmeyecektir. Tadı olmayan sıvı, henüz bulanıkken içilmelidir. Eğer toz çökerse su yeniden karıştırılmalıdır. Tüm içeriğin içildiğinden emin olunmalıdır.
Kullanılmamış olan ürünler ya da atık materyaller “Tıbbi Atıkların Kontrolü Yönetmeliği†ve “Ambalaj Atıklarının Kontrolü Yönetmelikâ€lerine uygun olarak imha edilmelidir.
 HIV ve Aids
HIV, Human Immunodeficiency Virus’dür (İnsanlarda Bağışıklık Sistemini Bozan
Virüsdür). Bu virüs AIDS hastalığına sebep olur.
HIV ve Aids
HIV, Human Immunodeficiency Virus’dür (İnsanlarda Bağışıklık Sistemini Bozan
Virüsdür). Bu virüs AIDS hastalığına sebep olur. |
 Diyabet Hastalığı
Diyabet, insülin hormonu ile ilgili problemlerden kaynaklanan bir hastalıktır.
Diyabet Hastalığı
Diyabet, insülin hormonu ile ilgili problemlerden kaynaklanan bir hastalıktır. |
 |
Pankreas Kanseri Pankreas karnın alt kısmında yatay şekilde bulunan bir organdır. Sindirime yardımcı olan enzimleri ve kan şekerini yönetmeye yardımcı olan hormonları vücuda dağıtmakla görevlidir. |
 |
Grip, Soğuk Algınlığı ve Öksürük Grip ve soğuk algınlığı (nezle) semptomları arasındaki farkı bilmek önemlidir. Soğuk algınlığı gripten daha hafif belirtiler gösteren bir solunum yolu hastalığıdır. |
 |
Lösemi Kan Kanseri Lösemi, kan kanseridir ve vücudunun kan oluşturan dokularının hastalanması anlamına gelir. Birçok lösemi türü vardır; bazı lösemi türleri çocuklarda bazıları da yetişkinlerde sık görülür. |
İLAÇ GENEL BİLGİLERİ
AstraZeneca Türkiye İlaç Sanayi ve Ticaret Ltd.Şti.
| Satış Fiyatı | 5868.47 TL [ 18 Apr 2025 ] |
| Önceki Satış Fiyatı | 5868.47 TL [ 14 Apr 2025 ] |
| Original / Jenerik | Original İlaç |
| Reçete Durumu | Normal Reçeteli bir ilaçdır. |
| Barkodu | 8699786250031 |
| Etkin Madde | Sodyum Zirkonyum Siklosilikat |
| ATC Kodu | V03AE10 |
| Birim Miktar | 5 |
| Birim Cinsi | G |
| Ambalaj Miktarı | 11 |
| Çeşitli İlaçlar > Diğer Tüm İlaçlar |
| İthal ( ref. ülke : Abd ) ve Beşeri bir ilaçdır. |