SOFEXAN 200 mg film kaplı tablet Kısa Ürün Bilgisi
{ Sorafenib }
1. BEŞERİ TIBBİ ÜRÜNÜN ADI
SOFEXAN 200 mg film kaplı tablet
2. KALİTATİF VE KANTİTATİF BİLEŞİM
Etkin madde
Sorafenib 200 mg (274,09 mg sorafenib tosilat olarak)
Yardımcı maddeler
Yardımcı maddeler için 6.1'e bakınız.
3. FARMASÖTİK FORMU
Film kaplı tablet
Koyu pembe renkli, yuvarlak, bombeli film kaplı tabletler
4. KLİNİK ÖZELLİKLER
4.1. Terapötik endikasyonlar
SOFEXAN;
Metastatik renal hücreli karsinoma (mRHK) tedavisinde interferon alfa ve/veya interlökin-2 ile yanıt alınamadığı veya bu tip tedavilerin yan etki nedeniyle uygun olmadığı durumlarda kullanılması için endikedir.
4.2. Pozoloji ve uygulama şekli
ErişkinlerPozoloji:
Önerilen SOFEXAN dozu günde toplam 800 mg'dır.
Uygulama sıklığı ve süresi:
Önerilen toplam günlük SOFEXAN dozu, günde iki kez 400 mg (2 x 2 tablet) tablet şeklinde alınır.
Tedavi, hasta artık tedaviden daha fazla klinik yarar görmeyinceye veya kabul edilemez ölçüde yan etki ortaya çıkıncaya kadar sürdürülmelidir.
Uygulama şekli:
Antikanser tedavide deneyimli bir hekim kontrolünde kullanılmalıdır.
SOFEXAN oral uygulama içindir ve bir bardak su ile yutulmalıdır. Tabletler aç karına ya da düşük veya orta yağlı bir öğün ile birlikte alınabilir. Tabletler yüksek yağlı bir öğünle alınacaksa, yemekten en az 1 saat önce veya yemekten 2 saat sonra alınmalıdır.
Doz titrasyonu, doz ayarlaması, özel izleme önerileri:
Hepatoselüler Karsinoma (HSK) ve İlerlemiş Renal Hücreli Karsinomada (RHK) Doz Azaltımı
Kuşkulu advers ilaç reaksiyonlarının tedavisinde, sorafenib tedavisinin geçici olarak durdurulması ve/veya dozun azaltılması gerekli olabilir.
HSK ve ilerlemiş RHK tedavisinde doz azaltımı gerekli olduğunda, sorafenib dozu günde bir kez 400 mg'a (1 x 2 tablet) azaltılmalıdır (bkz. Bölüm 4.4).
Deri toksisitesi durumunda önerilen doz modifikasyonları Tablo 1'de özetlenmiştir.
Tablo 1: HSK ve RHK ile Deri Toksisitesi Durumunda Önerilen Doz Modifikasyonları
Derece | Ortaya çıkış | SOFEXAN doz modifikasyonu |
Derece 1: El veya ayaklarda hastanın normal aktivitelerini etkilemeyen uyuşma, disestezi, parestezi, karıncalanma, ağrısız rahatsızlık | Herhangi bir zaman | Derhal destekleyici önlemler alınır ve SOFEXAN tedavisine devam edilir. |
Derece 2: El veya ayaklarda hastanın normal aktivitelerini etkileyen ağrılı eritem ve şişme ve/veya rahatsızlık | İlk olay | Derhal destekleyici önlemler alınır ve SOFEXAN dozu 28 gün süre ile günde 400 mg'a düşürülür. Doz azaltımından sonra toksisite derece 0-1'e gerilerse, 28 gün sonra sorafenib dozu artırılarak tam doza geçilir. Doz azaltımına karşın toksisite derece 0-1'e gerilemezse, en az 7 gün süreyle, toksisite derece 0- 1'e dönünceye kadar SOFEXAN tedavisine ara verilir. Aradan sonra tedaviye yeniden başlarken, SOFEXAN 28 gün süreyle günde bir kez 400 mg'lık azaltılmış doz şeklinde uygulanır. Doz azaltımıyla toksisite derece 0-1'de tutulabilirse, 28 gün sonra SOFEXAN dozu artırılarak tam doza geçilir. |
| 7 gün içinde iyileşme yoksa veya 2. veya 3. oluşum | Toksisite derece 0-1'e gerileyene dek SOFEXAN tedavisine ara veriniz. Tedaviye yeniden başlarken, SOFEXAN dozunu bir doz düzeyi azaltınız (günde 400 mg veya iki günde bir 400 mg). |
Dördüncü kez | SOFEXAN tedavisini sonlandırınız. | |
Derece 3: El veya ayaklarda hastanın çalışmasını ve günlük aktivitelerini gerçekleştirmesini engelleyen nemli deskuamasyon, ülserasyon, kabarma veya şiddetli ağrı veya şiddetli rahatsızlık | İlk olay | Derhal destekleyici önlemler alınır ve en az 7 gün süreyle, toksisite derece 0-1'e gerileyinceye kadar SOFEXAN tedavisine ara verilir. Aradan sonra tedaviye yeniden başlarken, SOFEXAN 28 gün süreyle günde bir kez 400 mg'lık azaltılmış doz şeklinde uygulanır. Doz azaltımıyla toksisite derece 0-1'de tutulabilirse, 28 gün sonra SOFEXAN dozu artırılarak tam doza geçilir. |
İkinci kez | İlk ortaya çıkıştaki gibi davranılır, ancak SOFEXAN tedavisi yeniden başlatıldıktan sonra, sürekli olarak azaltılmış günlük 400 mg dozu uygulanır. | |
Üçüncü kez | SOFEXAN tedavisini sonlandırınız. |
Diferansiye Tiroid Karsinomada (DTK) Doz Azaltımı
Kuşkulu advers ilaç reaksiyonlarının tedavisinde, sorafenib tedavisinin geçici olarak durdurulması ve/veya dozun azaltılması gerekli olabilir.
DTK tedavisinde doz azaltımı gerekli olduğunda, sorafenib dozu bölünmüş dozlar şeklinde günlük 600 mg'a (12 saat arayla bir seferde 200 mg'lık iki tablet ve diğer seferde 200 mg'lık bir tablet olacak şekilde günde toplam 3 tablet) azaltılmalıdır.
Ek doz azaltımı gerekli olursa, sorafenib dozu günde iki kez 200 mg'lık bir tablete, ardından günde bir kez 200 mg'lık bir tablete indirilebilir. Hematolojik olmayan advers reaksiyonların iyileşmesinden sonra, sorafenib dozu artırılabilir.
Tablo 2: Diferansiye Tiroid Kanserli Hastalarda Önerilen Doz Azaltımı Düzeyleri
Doz düzeyi | SOFEXAN dozu |
|
0 | 800 mg günlük doz | (Günde iki kez 400 mg, günde iki kez 2 tablet) |
-1 | 600 mg günlük doz | (12 saat arayla 400 mg ve 200 mg, 12 saat arayla 2 tablet ve 1 tablet-herhangi bir doz önce alınabilir) |
-2 | 400 mg günlük doz | (Günde iki kez 200 mg, günde iki kez 1 tablet) |
-3 | 200 mg günlük doz | (Günde bir kez bir 200 mg, günde bir kez 1 tablet) |
Tablo 3: Diferansiye Tiroid Kanserli Hastalarda Deri Toksisitesi Durumunda Önerilen Doz Modifikasyonları
Derece | Ortaya çıkış | SOFEXAN doz modifikasyonu* |
Derece 1 | Herhangi bir zaman | Derhal destekleyici önlemler alınır ve SOFEXAN tedavisine devam edilir. |
Derece 2 | İlk olay | Derhal destekleyici önlemler alınır ve SOFEXAN dozu günde 600 mg'a düşürülür (12 saat arayla 400 mg ve 200 mg). Eğer 7 gün içinde iyileşme görülmezse, tablonun devamını inceleyiniz. |
7 gün içinde iyileşmezse veya ikinci kez ortaya çıkarsa | Derece 0-1'e gerileyinceye kadar, SOFEXAN'a ara verilir. SOFEXAN yeniden başlatılırken, doz bir doz düzeyi azaltılır. | |
Üçüncü kez | Derece 0-1'e gerileyinceye kadar, SOFEXAN'a ara verilir. SOFEXAN yeniden başlatılırken, doz iki doz düzeyi azaltılır. | |
Dördüncü kez | SOFEXAN tedavisi tamamen sonlandırılır. | |
Derece 3 | İlk olay | Derece 0-1'e gerileyinceye kadar, SOFEXAN'a ara verilir. SOFEXAN yeniden başlatılırken, doz bir doz düzeyi azaltılır. |
İkinci kez | Derece 0-1'e gerileyinceye kadar, SOFEXAN'a ara verilir. SOFEXAN yeniden başlatılırken, doz iki doz düzeyi azaltılır. | |
Üçüncü kez | SOFEXAN tedavisi tamamen sonlandırılır. |
* Derece 2 veya 3 deri toksisitesi için doz azaltımı gereken hastalarda, azaltılmış SOFEXAN dozu ile en az 28 gün tedaviden sonra deri toksisitesi derece 0-1'e gerilerse, SOFEXAN dozu artırılabilir.
Özel popülasyonlara ilişkin ek bilgiler:
Böbrek/ karaciğer yetmezliği:
Hafif, orta dereceli ya da diyaliz gerektirmeyen şiddetli böbrek yetmezliği olan hastalarda doz ayarlaması gerekli değildir. Sorafenib diyalize girmekte olan hastalarda incelenmemiştir (bkz. Bölüm 5.2).
Böbrek fonksiyon bozukluğu riski olan hastalarda sıvı dengesinin ve elektrolitlerin izlenmesi önerilir.
Child-Pugh A veya B karaciğer yetmezliği olan hastalarda doz ayarlaması gerekli değildir. Sorafenib, Child-Pugh C karaciğer yetmezliği olan hastalarda incelenmemiştir (bkz. Bölüm 5.2).
Pediyatrik popülasyon:
Sorafenibin çocuklardaki ve 18 yaş altındaki adölesanlarda güvenliliği ve etkililiği belirlenmemiştir.
Geriyatrik popülasyon:
Yaşlı hastalarda (65 yaş üzeri) doz ayarlaması gerekli değildir.
Diğer:
Hastanın cinsiyeti ya da vücut ağırlığı temelinde doz ayarlaması gerekli değildir.
4.3. Kontrendikasyonlar
SOFEXAN, sorafenibe ya da SOFEXAN'ın diğer bileşenlerinden (bkz. Bölüm 6.1) herhangi birine karşı şiddetli aşırı duyarlılığı bilinen hastalarda kontrendikedir.
4.4. Özel kullanım uyarıları ve önlemleri
Anevrizmalar ve arter diseksiyonları:
VEGF yolak inhibitörlerinin, hipertansiyonu olan veya olmayan hastalarda kullanılması, anevrizmalar ve/veya arter diseksiyonları oluşumunu kolaylaştırabilir. SOFEXAN başlamadan önce hipertansiyon veya anevrizma öyküsü gibi risk faktörleri olan hastalarda bu risk dikkatle değerlendirilmelidir.
Dermatolojik toksisiteler:
El-ayak deri reaksiyonu (palmar-plantar eritrodisestezi) ve döküntü sorafenibe bağlı en yaygın advers ilaç reaksiyonlarını temsil etmektedir. Döküntü ve el-ayak deri reaksiyonu sıklıkla CTC (National Cancer Institute Common Toxicity Criteria) derece 1 ve 2'dir ve genel olarak sorafenib tedavisinin ilk altı haftası içinde ortaya çıkmaktadır.
Dermatolojik toksisitelerin tedavisinde semptomatik iyileşme amaçlı topikal tedaviler, tedavinin geçici olarak durdurulması ve/veya sorafenib doz modifikasyonu ya da şiddetli veya ısrarlı durumlarda sorafenib tedavisine tamamen son verilmesi gibi yöntemler uygulanabilir (bkz. Bölüm 4.8).
Hipertansiyon riski:
Sorafenib tedavisi alan hastalarda hipertansiyon insidansında artış gözlenmiştir.
SOFEXAN uygulanan ilk 6 hafta boyunca kan basıncı haftalık olarak takip edilmelidir. Bu süreden sonra, kan basıncı izlenmeli ve hipertansiyon, gerekirse, standart tıbbi uygulama doğrultusunda tedavi edilmelidir. HSK çalışmasında, hipertansiyon sorafenib ile tedavi edilen hastaların yaklaşık %9,4'ünde ve plasebo ile tedavi edilen gruptaki hastaların %4,3'ünde bildirilmiştir. RHK Çalışması 1'de, hipertansiyon sorafenib ile tedavi edilen hastaların yaklaşık %16,9'unda ve plasebo ile tedavi edilen gruptaki hastaların %1,8'inde bildirilmiştir. DTK çalışmasında, hipertansiyon sorafenib ile tedavi edilen hastaların yaklaşık %40,6'sında ve plasebo ile tedavi edilen hastaların %12,4'ünde bildirilmiştir. Hipertansiyon sıklıkla hafif ile orta derecelidir, tedavinin erken dönemlerinde ortaya çıkar ve standart antihipertansif rejimlerle tedavi edilebilir niteliktedir. Kan basıncı düzenli olarak izlenmeli ve gerekli olduğunda standart tıbbi uygulama doğrultusunda tedavi edilmelidir. Uygun ve yeterli bir antihipertansif tedaviye karşın şiddetli ya da ısrarlı hipertansiyon veya hipertansif kriz olgularında, sorafenibin tamamen kesilmesi gündeme getirilmelidir (bkz. Bölüm 4.8). HSK çalışmasında sorafenib ile tedavi edilen 297 hastadan 1'inde, RHK Çalışması 1'de sorafenib ile tedavi edilen 451 hastadan 1'inde ve DTK çalışmasında sorafenib ile tedavi edilen 207 hastadan 1'inde hipertansiyon nedeniyle tedavinin kalıcı olarak kesilmesi gerekmiştir.
Hipoglisemi:
Sorafenib tedavisi sırasında, klinik açıdan semptomatik ve bilinç kaybı nedeniyle hastaneye yatış gerektiren bazı olgular dahil olmak üzere, kan glukoz değerinde azalmalar bildirilmiştir. Semptomatik hipoglisemi geliştiği takdirde, sorafenib geçici olarak kesilmelidir. Diyabetik hastalardaki anti-diyabetik tıbbi ürün dozajının ayarlanmasının gerekip gerekmediğinin değerlendirilmesi için kan glukoz düzeyleri düzenli olarak kontrol edilmelidir.
Hemoraji riski:
Sorafenib uygulamasını takiben kanama riskinde artış ortaya çıkabilir. Şiddetli kanama olaylarının insidansı seyrektir. HSK çalışmasında, nedensellikten bağımsız olarak aşırı kanama belirgin olarak izlenmemiş ve özofagus varislerinde kanama oranı sorafenib ile tedavi edilen hastalarda %2,4 ve plasebo ile tedavi edilen hastalarda %4 olarak kaydedilmiştir. Herhangi bir bölgede ölümle sonuçlanan kanama, sorafenib ile tedavi edilen hastaların
%2,4'ünde ve plasebo ile tedavi edilen hastaların %4'ünde bildirilmiştir. RHK Çalışması 1'de, nedensellikten bağımsız olarak kanama sorafenib ile tedavi edilen gruptaki hastaların
%15,3'ünde ve plasebo ile tedavi edilen gruptaki hastaların %8,2'sinde bildirilmiştir. CTCAE Derece 3 ve 4 kanama, sorafenib ile tedavi edilen hastalarda sırasıyla %2 ve %0 olarak kaydedilirken, plasebo ile tedavi edilen hastalarda sırasıyla %1,3 ve %0,2 olarak bildirilmiştir. RHK Çalışmasında her tedavi grubunda bir ölümcül hemoraji gerçekleşmiştir. DTK çalışmasında, kanama sorafenib ile tedavi edilen hastaların %17,4'ünde ve plasebo ile tedavi edilen hastaların %9,6'sında bildirilmiş ancak CTCAE Derece 3 kanama insidansı sorafenib ile tedavi edilen hastalarda %1 ve plasebo ile tedavi edilen hastalarda %1,4 olmuştur. Derece 4 kanama hiç bildirilmezken, plasebo edilen bir hastada ölümcül hemoraji meydana gelmiştir. Eğer herhangi bir kanama olayı tıbbi girişim gerektirirse sorafenib tedavisinin tamamen kesilmesi gündeme getirilmelidir (bkz. Bölüm 4.8). Potansiyel kanama riski nedeniyle diferansiye tiroid kanserli hastalarda sorafenib uygulanmadan önce trakeal, bronşiyal ve özefageal infiltrasyon lokalize terapi ile tedavi edilmelidir.
Varfarin:
Sorafenib tedavisinde iken varfarin alan bazı hastalarda, yaygın olmayan kanama olayları ya da Uluslararası Normalize Oran (INR) değerlerinde yükselmeler bildirilmiştir. Eş zamanlı olarak varfarin almakta olan hastalar, protrombin zamanında uzama, INR ve klinik kanama epizodları için düzenli şekilde izlenmelidir (bkz. Bölüm 4.8).
Yara iyileşmesi komplikasyonları:
Sorafenibin yara iyileşmesi üzerindeki etkisi konusunda randomize bir çalışma yürütülmemiştir. Majör cerrahi girişim geçirecek hastalarda bir önlem olarak sorafenib tedavisinin geçici olarak durdurulması önerilir. Majör cerrahi girişim sonrasında tedaviye yeniden başlama zamanı konusundaki klinik deneyim kısıtlıdır. Bu nedenle, majör cerrahi girişim sonrasında sorafenib tedavisine tekrar başlama kararı yara iyileşmesinin yeterliliğine yönelik klinik yargıya dayandırılmalıdır.
Yaşlı popülasyon:
Böbrek yetmezliği olguları bildirilmiştir. Böbrek fonksiyonunun izlenmesi düşünülmelidir.
QT aralığının uzaması:
Sorafenibin ventriküler aritmiler açısından riskin artmasına neden olabilecek bir durum olan QT/QTc aralığını uzattığı gösterilmiştir (bkz. Bölüm 5.1). Konjenital uzun QT sendromu olan hastalarda SOFEXAN kullanımından kaçınınız. Sorafenib, yüksek kümülatif dozda antrasiklin tedavisi gören, bazı antiaritmik ilaçları veya QT uzamasına neden olan diğer tıbbi
ürünleri kullanan hastalar gibi QTc uzaması olan veya gelişebilecek hastalar ile hipokalemi, hipokalsemi veya hipomagnezemi gibi elektrolit bozuklukları olan hastalarda dikkatli kullanılmalıdır. Bu hastalarda SOFEXAN kullanımı sırasında elektrokardiyogram ve elektrolit ölçümü (magnezyum, kalsiyum, potasyum) ile periyodik izlem göz önünde bulundurulmalıdır. QTc aralığı 500 milisaniyenin üzerinde olduğu veya başlangıca göre 60 milisaniye veya üzeri artış gösterdiği takdirde SOFEXAN kullanımı durdurulmalıdır.
Kardiyak iskemi ve/veya enfarktüs:
Randomize, plasebo kontrollü, çift kör bir çalışmada (çalışma 1, bkz. Bölüm 5.1), tedaviye bağlı kardiyak iskemi/enfarktüs olayları sorafenib grubunda (%4,9), plasebo grubuna (%0,4) kıyasla daha yüksektir. Çalışma 3'te (bkz. Bölüm 5.1), tedaviye bağlı kardiyak iskemi/enfarktüs olaylarının insidansı sorafenib grubunda %2,7 iken, plasebo grubunda
%1,3'tür. Stabil olmayan koroner arter hastalığı öyküsü olan ya da yakın zamanda miyokard enfarktüsü geçiren hastalar bu çalışmanın dışında bırakılmıştır. Kardiyak iskemi ve/veya enfarktüs geçiren hastalarda sorafenibe geçici olarak ya da tamamen son verilmesi gündeme getirilmelidir (bkz. Bölüm 4.8, Bölüm 5.1).
Gastrointestinal perforasyon:
Gastrointestinal perforasyon, sorafenib almakta olan hastalarda seyrek görülen bir advers olaydır. Hastaların %1'inden daha azında bildirilmiştir. Olguların bazılarında bu durum, görünür bir intraabdominal tümör ile ilişkilendirilememiştir. Gastrointestinal perforasyon durumunda sorafenib tedavisine son verilmelidir (bkz. Bölüm 4.8).
Tümör lizis sendromu (TLS):
Sorafenib ile tedavi edilen hastalarda pazarlama sonrası gözetimde bazıları ölümcül olan tümör lizis sendromu (TLS) vakaları bildirilmiştir. TLS için risk faktörleri arasında yüksek tümör yükü, önceden var olan kronik böbrek yetmezliği, oligüri, dehidratasyon, hipotansiyon ve asidik idrar bulunur. Klinik olarak tespit edildiğinde, bu hastalar yakından izlenmeli, derhal tedavi edilmeli ve profilaktik hidrasyon düşünülmelidir.
Karaciğer yetmezliği:
Child-Pugh C (şiddetli) karaciğer yetmezliği olan hastalara ilişkin veri bulunmamaktadır. Sorafenib başlıca karaciğer yoluyla elimine edildiği için şiddetli karaciğer yetmezliği olan hastalarda ilaca sistemik maruziyet artabilir (bkz. Bölüm 5.2).
İlaç indüklü hepatit:
Sorafenib indüklü hepatit, karaciğer yetmezliği ve ölümle sonuçlanabilecek şekilde, transaminazlarda anlamlı artışlarla birlikte seyreden hepatosellüler paternde karaciğer hasarıyla karakterizedir. Bilirubin ve INR'de de artış görülebilir. Global monoterapi veritabanında, normalin üst sınırının 20 kat üzerine çıkan transaminaz düzeyleri veya anlamlı klinik sekel bırakan transaminaz artışları (örneğin INR artışı, assit, ölümcül sonuç veya transplantasyon) olarak tanımlanan şiddetli ilaç indüklü karaciğer hasarı insidansı 3.357 hastada iki hasta (%0,06) olarak kayıtlıdır. Karaciğer fonksiyon testleri düzenli olarak izlenmelidir. Viral hepatit veya altta yatan malignitede progresyon gibi alternatif bir açıklaması olmayan anlamlı transaminaz artışı izlendiği takdirde SOFEXAN kesilmelidir.
Hipokalsemi:
Diferansiye tiroid kanserli hastalarda sorafenib kullanılıyorken, kan kalsiyum düzeyinin yakından izlenmesi önerilmektedir. Klinik çalışmalarda hipokalsemi; diferansiye tiroid kanserli hastalarda (özellikle hipoparatiroidizm öyküsü olanlarda), renal hücreli veya
hepatoselüler kanserli hastalar ile karşılaştırıldığında daha sık ve daha şiddetli görülmüştür (bkz. Bölüm 4.8).
DTK'de tiroid stimüle edici hormon (TSH) supresyonu:
Sorafenib eksojen tiroid supresyonunu olumsuz etkiler. DTK çalışmasında, hastaların
%99'unun başlangıçtaki tiroid stimüle edici hormon (TSH) düzeyi 0,5 mU/L'nin altındadır. 0,5 mU/L'nin üzerine çıkan TSH düzeyleri, sorafenib ile tedavi edilen hastaların %41'inde, plasebo ile tedavi edilen hastaların %16'sında gözlemlenmiştir. Sorafenib kullanırken TSH supresyonunda bozulma görülen hastalarda, medyan maksimum TSH 1,6 mU/L olup, hastaların %25'inde TSH düzeyi 4,4 mU/L'nin üzerinde kaydedilmiştir.
Diferansiye tiroid kanserli hastalarda sorafenib kullanılıyorken, TSH düzeyinin yakından izlenmesi ve gerekirse tiroid replasman tedavisinde ayarlama yapılması önerilmektedir.
İlaç etkileşimleri:
UGT1A yolu: Sorafenib, esas olarak UGT1A1 yolu veya UGT1A9 yolu aracılığıyla metabolize veya elimine edilen bileşikler (örn. irinotekan) ile birlikte uygulanırken dikkatli olunması önerilmektedir (bkz. Bölüm 4.5).
Dosetaksel: Dosetaksel (75 ya da 100 mg/m) ile birlikte sorafenib (günde iki kez 200 ya da 400 mg) uygulaması (dosetaksel uygulaması sırasında sorafenibe 3 gün ara verme şeklinde), dosetaksel EAA değerinde %36-80 artış ile sonuçlanmıştır. Sorafenib dosetaksel ile birlikte uygulanırken dikkatli olunması önerilmektedir (bkz. Bölüm 4.5).
Neomisin: Eş zamanlı neomisin uygulaması ve bağırsak florasını bozan diğer antibiyotiklerin uygulanması sorafenibin biyoyararlanımında azalmaya neden olabilir (bkz. Diğer tıbbi ürünler ile etkileşimler). Antibiyotiklerle bir tedavi kürüne başlanmadan önce, plazma sorafenib konsantrasyonlarının azalma riski değerlendirilmelidir.
Akciğer karsinomunda platin-bazlı kemoterapi sırasında sorafenib kullanılması yüksek mortaliteye neden olur. Küçük hücreli dışı akciğer kanseri tanılı hastaların değerlendirildiği iki randomize çalışmada, skuamöz karsinomlu hastaların oluşturduğu alt grupta paklitaksel/karboplatine ek olarak sorafenib tedavisi alanlarda genel sağkalım için HR 1,81 (%95 GA 1,19; 2,74) olarak belirlenirken, gemsitabin/sisplatine ek tedavi için 1,22 (%95 GA 0,82; 1,8) olmuştur. Tekli ölüm nedenlerinden hiçbiri baskın olmamış; ancak, platin bazlı kemoterapilere ek olarak sorafenib uygulanan hastalarda solunum yetmezliği, kanamalar ve enfeksiyöz advers olaylar bakımından daha yüksek bir insidans kaydedilmiştir.
Hastalığa özel uyarılar
Diferansiye tiroid kanseri (DTK):
Hekimlere, tedaviyi başlatmadan önce maksimum lezyon boyutu (bkz. Bölüm 5.1), hastalıkla ilgili semptomlar (bkz. Bölüm 5.1) ve progresyon hızını dikkate alarak hastadaki prognozu dikkatli bir şekilde değerlendirmesi önerilmektedir.
Şüpheli advers ilaç reaksiyonlarının yönetimi sorafenib tedavisinin geçici olarak kesilmesini veya dozunun azaltılmasını gerektirebilir. Çalışma 5'de (bkz. Bölüm 5.1), sorafenib tedavisinin henüz 1. siklusunda gönüllülerin %37'sinde doza ara verilmesi ve %35'inde dozun azaltılması gerekmiştir.
Doz azaltımı, advers reaksiyonların hafifletilmesinde yalnızca kısmen başarılı olmuştur. Bu
nedenle, anti-tümör aktivitesi ve tolerabilite de dikkate alınarak tekrarlanan fayda/risk değerlendirmeleri önerilmektedir.
DTK'de hemoraji:
Potansiyel kanama riski nedeniyle, DTK hastalarına sorafenib uygulanmadan önce trakeal, bronşiyal ve özefageal infiltrasyon için lokal tedavi uygulanmalıdır.
DTK'de hipokalsemi:
DTK hastalarında sorafenib kullanılırken kan kalsiyum düzeyinin yakından izlenmesi önerilir.
Klinik çalışmalarda, özellikle hipoparatiroidizm öyküsü olanlar olmak üzere DTK hastalarında hipokalsemi, renal hücreli veya hepatosellüler karsinomu olan hastalara göre daha sık ve daha şiddetli olmuştur. Derece 3 ve 4 hipokalsemi, sorafenib ile tedavi edilen DTK hastalarının %6,8'i ve %3,4'ünde gelişmiştir (bkz. Bölüm 4.8). QT uzaması veya Torsade de pointes gibi komplikasyonların önlenmesi için (bkz. Bölüm 4.4.) şiddetli hipokalsemi düzeltilmelidir.
DTK'de TSH supresyonu:
Çalışma 5'de (bkz. Bölüm 5.1), sorafenib ile tedavi edilen hastaların TSH düzeylerinde 0,5 mU/L'nin üzerinde artışlar gözlemlenmiştir. DTK hastalarında sorafenib kullanılırken TSH düzeyinin yakından izlenmesi önerilir.
Renal hücreli karsinom:
MSKCC (Memorial Sloan Kettering Cancer Center) prognostik grubuna göre Yüksek Riskli Hastalar, renal hücreli karsinoma ilişkin faz III klinik çalışmaya dahil edilmemiş (bkz. Bölüm 5.1), dolayısıyla bu hastalarda fayda-risk değerlendirmesi yapılmamıştır.
4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri
CYP3A4 indükleyicileri:
Tek doz sorafenib uygulanmadan önce 5 gün süreyle rifampisin uygulanması sorafenib EAA değerinde ortalama %37'lik bir azalmayla sonuçlanmıştır. CYP3A4 aktivitesinin ve/veya glukuronidasyonunun diğer indükleyicileri de (örn. St. John's wort Hypericum perforatum bitkisi, fenitoin, karbamazepin, fenobarbital ve deksametazon), sorafenib metabolizmasını artırabilir ve böylelikle sorafenib konsantrasyonlarını azaltabilirler.
CYP3A4 inhibitörleri:
Güçlü bir CYP3A4 inhibitörü olan ketokonazol, sağlıklı erkek gönüllülere 7 gün süreyle günde bir kez uygulandığında, tek doz 50 mg sorafenibin ortalama EAA değerini değiştirmemiştir. Bu nedenle sorafenib ve CYP3A4 inhibitörleri arasında klinik farmakokinetik etkileşim olasılığı pek bulunmamaktadır.
CYP2B6, CYP2C8 ve CYP2C9 substratları:
Sorafenib, in vitro koşullarda CYP2B6, CYP2C8 ve CYP2C9'u benzer potensle inhibe etmiştir. Bununla birlikte, klinik farmakokinetik çalışmalarında günde iki kez sorafenib 400 mg'ın bir CYP2B6 substratı olan siklofosfamid veya bir CYP2C8 substratı olan paklitaksel ile birlikte uygulanması klinik olarak anlamlı inhibisyonla sonuçlanmamıştır. Bu veriler, önerilen doz olan günde iki kez 400 mg sorafenibin CYP2B6 veya CYP2C8'in in vivo inhibitörü olmayabileceğini düşündürmektedir.
Sorafenib in vitro koşullarda CYP2B6 ve CYP2C8'i sırasıyla 6 ve 1-2 µM Ki değerleri ile inhibe etmektedir.
Ayrı bir klinik çalışmada, sorafenibin paklitaksel ile eş zamanlı uygulaması, CYP2C8 tarafından oluşturulan etkin paklitaksel metaboliti 6-OH paklitaksel maruziyetinde azalma yerine artışa neden olmuştur. Bu veriler sorafenibin in vivo olarak CYP2C8 inhibitörü olmayabileceğini ileri sürmektedir. Farklı bir klinik çalışmada sorafenibin siklofosfamid ile eşzamanlı kullanımı siklofosfamid maruziyetinde küçük bir azalma ile sonuçlanmıştır. Ancak CYP2B6 tarafından oluşturulan siklofosfamidin etkin metaboliti 4-OH siklofosfamidin sistemik maruziyetinde bir azalma meydana gelmemiştir. Bu veriler sorafenibin, CYP2B6'nın in vivo inhibitörü olmayabileceğini işaret etmektedir.
İnsan karaciğer mikrozomları ile çalışmalar sorafenibin 7-8 µM Ki değeri ile CYP2C9'un kompetitif inbibitörü olduğunu göstermiştir. Ayrıca sorafenib ve bir CYP2C9 substratı olan varfarin ile eş zamanlı tedavi, ortalama PT-INR'de plaseboya göre değişiklik oluşturmamıştır. Dolayısıyla, CYP2C9'un sorafenib tarafından klinik olarak ilgili in vivo inhibisyonu riskinin de düşük olması beklenebilir. Ancak, varfarin veya fenprokumon kullanan hastaların INR'leri düzenli olarak kontrol edilmelidir (bkz. Bölüm 4.4).
CYP3A4, CYP2D6 ve CYP2C19 substratları:
Sırasıyla CYP3A4, CYP2D6 ve CYP2C19 sitokromlarının substratları olan sorafenib ve midazolam, dekstrometorfan veya omeprazolün eş zamanlı uygulaması bu ajanlara maruz kalmayı değiştirmemiştir. Bu durum, sorafenibin bu sitokrom P450 izoenzimlerinin inhibitörü veya indükleyicisi olmadığını göstermektedir. Bu nedenle, sorafenibin bu enzimlerin substratları ile klinik farmakokinetik etkileşimi öngörülmemektedir.
UGT1A1 ve UGT1A9 substratları:
İn vitro koşullarda, sorafenib UGT1A1 ve UGT1A9 aracılığıyla glukuronidasyonu inhibe etmiştir. Bu bulgunun klinik ilgisi bilinmemektedir (bkz. Bölüm 4.4). Sorafenibin irinotekan ile birlikte klinik olarak kullanımı sonucu SN-38 olan aktif metaboliti UGT1A1 aracılığıyla tekrardan metabolize edilir ve sonucunda SN-38'in EAA'ında %67-120 artış gözlenir.
CYP enzim indüksiyonuna ilişkin in vitro koşullarda yapılmış çalışmalar:
CYP1A2 ve CYP3A4'ün aktiviteleri, kültürü yapılan insan hepatositlerinin sorafenib ile tedavisinden sonra değişmemiştir, bu da sorafenibin CYP1A2 ve CYP3A4'ün indükleyicisi olma olasılığının düşük olduğunu göstermektedir.
P-gp substratları:
İn vitro, sorafenibin transport protein p-glikoproteini (P-gp) inhibe ettiği gösterilmiştir. Sorafenib ile eş zamanlı tedavi sonucunda digoksin gibi P-gp substratlarının plazma konsantrasyonlarında artış olasılığı dışlanamaz.
Diğer antineoplastik ajanlar ile kombinasyon:
Sorafenib klinik çalışmalarda, gemsitabin, sisplatin, oksaliplatin, paklitaksel, karboplatin, kapesitabin, doksorubisin, irinotekan, dosetaksel ve siklofosfamid gibi çeşitli diğer anti- neoplastik ajanlar ile birlikte, bu ajanların sıklıkla kullanılan dozaj rejimleriyle kullanılmıştır.
Sorafenib gemsitabin, sisplatin, karboplatin, oksaliplatin veya siklofosfamid farmakokinetiği üzerinde klinik olarak anlamlı bir etki göstermemiştir.
Paklitaksel/Karboplatin
Paklitaksel/karboplatin uygulaması sırasında sorafenib dozunda 3 günlük bir ara verilerek, paklitaksel (225 mg/m) ve karboplatinin (EAA=6) sorafenible (günde iki kez ≤400 mg) birlikte uygulanması, paklitaksel farmakokinetik değerlerinde anlamlı herhangi bir etki oluşturmamıştır.
Paklitaksel (225 mg/m, her 3 haftada bir kez) ve karboplatinin (EAA=6) sorafenible (günde iki kez 400 mg, sorafenib dozunda bir ara olmaksızın) birlikte uygulanması, sorafenib maruziyetinde %47'lik, paklitaksel maruziyetinde %29'luk ve 6-OH paklitaksel maruziyetinde ise %50'lik bir artış oluşturmuştur. Karboplatin farmakokinetik değerleri etkilenmemiştir.
Bu veriler, paklitaksel ve karboplatinin sorafenib dozunda 3 günlük bir ara dikkate alınarak sorafenible birlikte uygulanması durumunda doz ayarlamasına gerek olmadığını ortaya koymaktadır. Ara olmaksızın verilen eş zamanlı sorafenibi takiben sorafenib ve paklitaksel maruziyetlerinde gözlenen artışın klinik anlamı bilinmemektedir.
Kapesitabin
Eş zamanlı kapesitabin (750-1050 mg/m-günde iki kez, her 21 günlük süre içinde 1-14. günlerde) ve sorafenib (günde iki kez 200 ya da 400 mg, sürekli-kesintisiz uygulama) uygulaması, sorafenib maruziyetinde anlamlı bir değişiklik oluşturmamış; ancak kapesitabin maruziyetinde %15-50'lik, 5-FU maruziyetinde ise %0-52'lik bir artışla sonuçlanmıştır. Sorafenible eşzamanlı uygulandığında kapesitabin ve 5-FU maruziyetlerinde gözlenen bu hafif ila orta düzeyli artışların klinik anlamı bilinmemektedir.
Doksorubisin/İrinotekan
Sorafenib ile eş zamanlı uygulama, doksorubisinin EAA değerinde %21'lik bir artışla sonuçlanmıştır. Aktif metaboliti SN-38'in daha sonra UGT1A1 yoluyla metabolize olduğu irinotekan ile birlikte uygulandığında, SN-38'in EAA değerinde %67-120, irinotekanın EAA değerinde ise %26-42 artış vardır. Bu bulguların klinik önemi bilinmemektedir (bkz. Bölüm 4.4).
Dosetaksel
Dosetaksel (21 günde bir uygulanan 75 ya da 100 mg/m) ile sorafenibin (21 günlük siklusun
2. gününden 19. gününe kadar, günde iki kez 200 ya da 400 mg; dosetaksel uygulaması sırasında sorafenibe 3 gün ara verme şeklinde) birlikte uygulanması, dosetakselin EAA değerinde %36-80 ve dosetakselin Cdüzeyinde %16-32 artış ile sonuçlanmıştır. Sorafenib ile dosetakselin birlikte uygulanması sırasında dikkatli olunması önerilmektedir (bkz. Bölüm 4.4).
Antibiyotikler ile kombinasyon
Neomisin
Gastrointestinal florayı eradike etmek için kullanılan sistemik olmayan bir antimikrobiyal ajan olan neomisinin eş zamanlı uygulaması, sorafenibin enterohepatik geri dönüşümünü engeller (bkz. Bölüm 5.2) ve bunun sonucunda sorafenib maruziyetinde düşüşe neden olur. 5 gün süreli neomisin rejimiyle tedavi gören sağlıklı gönüllülerde ortalama sorafenib maruziyeti
%54 azalmıştır. Bu bulguların klinik anlamı bilinmemektedir. Diğer antibiyotiklerin etkileri araştırılmamıştır ancak büyük olasılıkla glukuronidaz aktivitesini azaltma yetenekleriyle ilişkilendirileceklerdir.
Proton pompası inhibitörleri ile kombinasyon
Omeprazol
Omeprazol ile birlikte kullanımın sorafenibin farmakokinetik değerleri üzerinde herhangi bir etkisi yoktur. Sorafenib için bir doz ayarlaması yapılması gerekmez.
Özel popülasyonlara ilişkin ek bilgiler
Veri bulunmamaktadır.
Pediyatrik popülasyon:
Veri bulunmamaktadır.
4.6. Gebelik ve laktasyon
Gebelik kategorisi: D
Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon) Hayvanlar üzerinde yapılan araştırmalar sorafenibin teratojenik ve embriyotoksik olduğunu göstermiştir. Çocuk doğurma potansiyeli olan kadınlar tedavi süresince ve tedavinin ardından en az 2 haftaya kadar, etkili doğum kontrolü uygulamak zorundadırlar (bkz. Bölüm 4.4, Bölüm 5.3).
Kadınlar tedavi altında iken gebe kalmaktan kaçınmalıdır. Çocuk doğurma potansiyeline sahip kadınlara fetüs üzerindeki ağır malformasyon (teratojenisite), büyüme geriliği ve fetal ölüm (embriyotoksisite) gibi potansiyel tehlikeler bildirilmelidir.
Gebelik dönemi
Sorafenibin gebelik ve/veya fetüs/yenidoğan üzerinde zararlı farmakolojik etkileri bulunmaktadır. Sorafenib kullanan gebe kadınlar üzerinde yürütülmüş yeterli ve iyi kontrollü çalışmalar bulunmamaktadır. Hayvanlarda yürütülen çalışmalarda malformasyonları içeren reprodüktif toksisite gösterilmiştir (bkz. Bölüm 4.4). Sıçanlarda sorafenib ve metabolitlerinin plasentaya geçtikleri bulunmuştur ve sorafenibin fetusta anjiyojenezi inhibe etmesi beklenmektedir.
SOFEXAN gebelik sırasında kullanılmamalıdır. Doktorlar bu ilacın kullanımını, sadece potansiyel yararları fetüs üzerindeki potansiyel riskleri haklı çıkarıyorsa gündeme getirebilirler (bkz. Bölüm 4.4, Bölüm 5.3).
Laktasyon dönemi
Sorafenibin insan sütü ile atılıp atılmadığı bilinmemektedir.
Hayvanlar üzerinde yapılan çalışmalar sorafenib ve/veya metabolitlerinin süt ile atıldığını göstermektedir. Sorafenibin fetüsün büyümesine ve gelişmesine zararlı etkileri olabileceğinden, sorafenib tedavisi sırasında emzirme bırakılmalıdır.
Üreme yeteneği/Fertilite
Hayvan çalışmalarının sonuçları, sorafenibin erkek ve kadınlarda fertiliteyi bozabileceğine işaret etmektedir (bkz. Bölüm 5.3).
4.7. Araç ve makine kullanımı üzerindeki etkiler
Sorafenib ile araç ya da makine kullanma yetileri üzerindeki etkilerine yönelik çalışma yapılmamıştır. Sorafenibin araç ya da makine kullanma yetilerini etkilediğine ilişkin bir veri bulunmamaktadır.
4.8. İstenmeyen etkiler
En önemli ciddi advers reaksiyonlar, miyokard enfarktüsü/iskemisi, gastrointestinal perforasyon, ilaçla indüklenen hepatit, hemoraji ve hipertansiyon/hipertansif kriz olmuştur.
En yaygın advers reaksiyonlar diyare, yorgunluk, alopesi, enfeksiyon, el-ayak deri reaksiyonu (MedDRA'da palmar-plantar eritrodizestezi sendromuna karşılık gelir), döküntü, kilo kaybı, iştah kaybı, mide bulantısı, gastrointestinal ve abdominal ağrı, hipertansiyon ve hemoraji olmuştur.
Birden fazla klinik çalışma sırasında meydana gelen ya da pazarlama sonrası kullanım süresince saptanan tüm advers reaksiyonlar, aşağıda sistem-organ sınıfı (MedDRA) ve sıklık derecesine göre listelenmektedir. Sıklık dereceleri şu şekilde tanımlanmaktadır; Çok yaygın (≥1/10); yaygın (≥1/100 ila <1/10); yaygın olmayan (≥1/1.000 ila <1/100); seyrek (≥1/10.000 ila <1/1.000); çok seyrek (<1/10.000); bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor).
Her sıklık grubu içinde, istenmeyen etkiler azalan şiddet derecesine göre sıralanmıştır.
Enfeksiyonlar ve enfestasyonlar
Çok yaygın: Enfeksiyon Yaygın: Folikülit
Kan ve lenf sistemi hastalıkları
Çok yaygın: Lenfopeni
Yaygın: Lökopeni, nötropeni, anemi, trombositopeni
Bağışıklık sistemi hastalıkları
Yaygın olmayan: Anafilaktik reaksiyon, aşırı duyarlılık reaksiyonları (deri reaksiyonları ve ürtiker dahil)
Seyrek: Anjiyoödem
Endokrin hastalıkları
Yaygın: Hipotiroidizm
Yaygın olmayan: Hipertiroidizm
Metabolizma ve beslenme hastalıkları
Çok yaygın: Anoreksi, hipofosfatemi
Yaygın: Hipokalsemi, hipokalemi, hiponatremi, hipoglisemi Yaygın olmayan: Dehidrasyon
Bilinmiyor: Tümör lizis sendromu
Psikiyatrik hastalıklar
Yaygın: Depresyon
Sinir sistemi hastalıkları
Yaygın: Periferik duyusal nöropati, disguzi
Yaygın olmayan: Geri dönüşümlü posterior lökoensefalopati* Bilinmiyor: Ensefalopati
Kulak ve içkulak hastalıkları
Yaygın: Tinnitus
Kardiyak hastalıklar
Yaygın: Konjestif kalp yetmezliği*, miyokard iskemisi ve/veya enfarktüsü* Seyrek: QT uzaması
Vasküler hastalıklar
Çok yaygın: Hemoraji (gastrointestinal*, solunum yolu* ve serebral hemoraji* dahil), hipertansiyon
Yaygın: Sıcak basması (flushing) Yaygın olmayan: Hipertansif kriz*
Bilinmiyor: Anevrizmalar ve arter diseksiyonları
Solunum, göğüs bozuklukları ve mediyastinal hastalıklar
Yaygın: Rinore, disfoni
Yaygın olmayan: İnterstisyel akciğer hastalığı benzeri olaylar* (pnömonit, radyasyon pnömoniti, akut solunum güçlüğü, interstisyel pnömoni, pulmonit ve akciğer inflamasyonu bildirimleri de dahil olmak üzere)
Gastrointestinal hastalıklar
Çok yaygın: Diyare, bulantı, kusma, konstipasyon
Yaygın: Stomatit (ağız kuruluğu ve glossodini dahil), dispepsi, disfaji, gastroözofajiyal reflü hastalığı
Yaygın olmayan: Pankreatit, gastrit, gastrointestinal perforasyonlar*
Hepatobiliyer hastalıklar
Yaygın olmayan: Bilirübin artışı ve sarılık, kolesistit, kolanjit Seyrek: İlaca bağlı hepatit*
Deri ve derialtı dokusu hastalıkları
Çok yaygın: Deri kuruluğu, döküntü, alopesi, el-ayak deri reaksiyonu**, pruritus, eritem Yaygın: Deride keratoakantomalar/skuamöz hücreli kanser, eksfolyatif dermatit, akne, deri deskuamasyonu, hiperkeratoz
Yaygın olmayan: Egzama, eritema multiforme
Seyrek: Radyasyon dermatiti, lökositoklastik vaskülit, Stevens-Johnson sendromu, toksik epidermal nekroliz*
Kas-iskelet bozuklukları, bağ dokusu ve kemik hastalıkları
Çok yaygın: Artralji
Yaygın: Miyalji, kas spazmları Seyrek: Rabdomiyoliz
Böbrek ve idrar yolu hastalıkları
Yaygın: Böbrek yetmezliği, proteinüri
Seyrek: Nefrotik sendrom
Üreme sistemi ve meme hastalıkları
Yaygın: Erektil disfonksiyon Yaygın olmayan: Jinekomasti
Genel bozukluklar ve uygulama bölgesine ilişkin hastalıklar
Çok yaygın: Yorgunluk, ağrı (ağız, abdominal, kemik, tümör ağrısı ve baş ağrısı dahil), ateş Yaygın: Asteni, grip benzeri hastalık, mukozal inflamasyon
Araştırmalar
Çok yaygın: Kilo kaybı, amilaz artışı, lipaz artışı Yaygın: Transaminazlarda geçici artış
Yaygın olmayan: Kan alkalen fosfatazında geçici artış, anormal INR (Uluslararası Normalizasyon Oranı), protrombin düzeyinde anormallik
* Bu advers reaksiyonlar hayatı tehdit edici ya da ölümcül sonuç verebilir. Bu gibi olaylar ya yaygın olmayan ya da yaygın olmayandan daha az sıklıktadır.
**MedDRA'da palmar plantar eritrodisestezi sendromuna uygun düşmektedir.
Bazı advers ilaç reaksiyonları ile ilgili ilave bilgi
Konjestif Kalp Yetmezliği - Yürütülen klinik çalışmalarda, konjestif kalp yetmezliği sorafenib tedavisi gören hastaların (n=2.276) %1,9'unda advers olay olarak bildirilmiştir. Renal hücreli karsinoma (RHK) çalışması 11213'te (çalışma 1) konjestif kalp yetmezliğiyle uyumlu advers olaylar sorafenib tedavisi gören hastaların %1,7'sinde ve plasebo grubunun %0,7'sinde bildirilmiştir. Hepatoselüler karsinoma (HSK) çalışması 100554'de (çalışma 3), sorafenib tedavisi görenlerin %0,99'unda, plasebo kullananların ise %1,1'inde bu olaylar bildirilmiştir.
İleri küçük hücre dışı akciğer karsinomu (KHDAK) olan hastaların ilk basamak tedavisi olarak sorafenibin ikili platinyum-bazlı kemoterapilerle (karboplatin/paklitaksel ve ayrı ayrı gemsitabin/sisplatin) kombine kullanımına karşılık sadece ilgili ikili platinyum-bazlı kemoterapinin güvenlilik ve etkililiğinin karşılaştırıldığı iki randomize plasebo kontrollü çalışma, genel sağkalımın iyileşmesi olan birincil sonlanım noktasına ulaşamamıştır. Güvenlilik olayları genellikle daha önce bildirilen olaylarla uyumludur. Ancak, iki çalışmada da tek başına ikili platinyum-bazlı kemoterapi (karboplatin/paklitaksel ve ayrı ayrı gemsitabin/sisplatin) tedavisiyle karşılaştırıldığında, sorafenib ve ikili platinyum-bazlı kemoterapi tedavisi uygulanan skuamöz hücreli akciğer kanseri olan bir hasta alt grubunda daha yüksek mortalite izlenmiştir (HR 1,22, %95 GA 0,82-1,8). Bu bulgulara ilişkin kesin bir gerekçe tanımlanmamıştır.
Güvenlilik aynı zamanda bir faz II çalışma havuzunda da değerlendirilmiştir. Bu toplu veriler, sorafenib ile tedavi edilen 638 hastaya aittir ve bunların arasında 202 hastada renal hücreli karsinoma, 137 hastada hepatoselüler karsinoma ve 299 hastada başka kanser türleri bulunmaktadır. Bu toplu veriler içinde sorafenib tedavisindeki hastalarda en sık bildirilen ilaca bağlı advers olaylar; döküntü (%38), diyare (%37), el-ayak deri reaksiyonu (%35) ve yorgunluk (%33) olmuştur. Sorafenib tedavisindeki hastalarda ortak toksisite kriterleri (CTC) (v 2.0) Derece 3 ve 4 ilaca bağlı advers olaylar, sırasıyla %37 ve %3'dür.
Klinik çalışmalarda, renal hücreli karsinoma veya hepatoselüler karsinoma çalışmalarında yer alan hastalar ile karşılaştırıldığında diferansiye tiroid kanserli hastalarda el-ayak deri
reaksiyonu, diyare, alopesi, kilo kaybı, hipertansiyon, hipokalsemi ve deride keratoakantomalar/skuamöz hücreli kanser gibi belli advers ilaç reaksiyonları büyük ölçüde daha yüksek sıklıkta ortaya çıkmıştır.
RHK hastalarında laboratuvar test anormallikleri (Çalışma 11213) (Çalışma 1):
Lipaz ve amilaz düzeylerinde artış sık olarak bildirilmiştir. Çalışma 11213'te, Advers olaylar için ortak terminoloji kriterleri (CTCAE) derece 3 ya da 4 lipaz artışları, sorafenib grubundaki hastaların %12'sinde bildirilirken, plasebo grubundaki hastaların %7'sinde bildirilmiştir. CTCAE derece 3 ya da 4 amilaz artışları, sorafenib grubundaki hastaların %1'inde bildirilirken, plasebo grubundaki hastaların %3'ünde bildirilmiştir. Çalışma 1'de klinik pankreatit, sorafenib tedavisindeki 451 hastanın 2'sinde (CTCAE derece 4) ve plasebo grubundaki 451 hastanın 1'inde (CTCAE derece 2) bildirilmiştir.
Hipofosfatemi sık rastlanan laboratuvar anomalisidir, sorafenib tedavisindeki hastaların
%45'inde, plasebo hastalarının %11'inde gözlenmiştir. CTCAE derece 3 hipofosfatemi (1-2 mg/dL), sorafenib ile tedavi edilen hastaların %13'ünde ve plasebo grubundaki hastaların
%3'ünde ortaya çıkmıştır. CTCAE derece 4 hipofosfatemi (<1 mg/dL) olgusu sorafenib ve plasebo kollarında bildirilmemiştir. Sorafenib ile ilişkili hipofosfateminin etiyolojisi bilinmemektedir.
CTCAE derece 3 ya da 4 olaylar, lenfopeni için sorafenib tedavisindeki hastaların %13'ünde ve plasebo hastalarının %7'sinde, nötropeni için sorafenib tedavisindeki hastaların %5'inde ve plasebo hastalarının %2'sinde, anemi için sorafenib tedavisindeki hastaların %2'sinde ve plasebo hastalarının %4'ünde ve trombositopeni için sorafenib tedavisindeki hastaların %1 ve plasebo hastalarının %0'ında bildirilmiştir.
Sorafenib ile tedavi edilen hastaların %12'sinde hipokalsemi bildirilirken plasebo alan hastalarda bu oran %7,5 olmuştur. Bildirilen çoğu hipokalsemi düşük derecededir (CTCAE Derece 1 ve 2). CTCAE derece 3 hipokalsemi (6-7 mg/dL) sorafenib ile tedavi edilen hastaların %1,1'inde, plasebo grubundaki hastaların ise %0,2'sinde görülmüş; CTCAE derece
4 hipokalsemi de (<6 mg/dL) sorafenib ile tedavi edilen hastaların %1,1'inde, plasebo grubundaki hastaların ise %0,5'inde görülmüştür. Sorafenib ile ilişkili hipokalseminin etiyolojisi bilinmemektedir.
Sorafenib ile tedavi edilen hastaların %5,4'ünde hipokalemi bildirilirken, plasebo alan hastalarda bu oran %0,7 olmuştur. Bildirilen çoğu hipokalemi düşük derecededir (CTCAE derece 1). CTCAE derece 3 hipokalemi, sorafenib ile tedavi edilen hastaların %1,1'inde, plasebo grubundaki hastaların ise %0,2'sinde görülmüştür. Derece 4 hipokalemi bildirilmemiştir.
HSK hastalarında laboratuvar anormallikleri (Çalışma 100554):
Lipaz artışı sorafenib ile tedavi edilen hastaların %40'ında gözlenirken, plasebo grubundaki hastaların %37'sinde gözlenmiştir. CTCAE derece 3 ya da 4 lipaz artışları, her iki grupta da hastaların %9'unda ortaya çıkmıştır. Amilaz artışları, sorafenib ile tedavi edilen hastaların
%34'ünde gözlenirken, plasebo grubundaki hastaların %29'unda gözlenmiştir. CTCAE derece 3 ya da 4 amilaz artışları, her iki grupta da hastaların %2'sinde bildirilmiştir. Lipaz ve amilaz artışlarının çoğu geçicidir ve olguların büyük çoğunluğunda sorafenib tedavisine ara verilmemiştir. Klinik pankreatit sorafenib tedavisindeki 297 hastanın 1'inde bildirilmiştir (CTCAE derece 2).
Hipofosfatemi sık rastlanan bir laboratuvar bulgusudur, sorafenib ile tedavi edilen hastaların
%35'inde, plasebo hastalarının %11'inde gözlenmiştir. CTCAE derece 3 hipofosfatemi (1-2 mg/dL), sorafenib ile tedavi edilen hastaların %11'inde ve plasebo grubundaki hastaların
%2'sinde ortaya çıkmıştır; plasebo grubunda bildirilen 1 CTCAE derece 4 hipofosfatemi (<1 mg/dL) olgusu bulunmaktadır. Sorafenib ile ilişkili hipofosfateminin etiyolojisi bilinmemektedir.
Karaciğer fonksiyon testlerindeki artışlar, çalışmanın 2 kolu arasında karşılaştırılabilir niteliktedir. Aspartat aminotransferaz (AST) artışları sorafenib ile tedavi edilen hastaların
%94'ü ve plasebo hastalarının %91'inde gözlenmiştir. CTCAE derece 3 ya da 4 AST artışları, sorafenib ile tedavi edilen hastaların %16'sında ve plasebo grubundaki hastaların %17'sinde bildirilmiştir. ALT artışları sorafenib ile tedavi edilen hastaların %69'unda ve plasebo hastalarının %68'inde gözlenmiştir. CTCAE derece 3 ya da 4 alanin aminotransferaz (ALT) artışları, sorafenib ile tedavi edilen hastaların %3'ünde ve plasebo tedavisindeki hastaların
%8'inde bildirilmiştir. Bilirübin artışları, sorafenib ile tedavi edilen hastaların %47'sinde ve plasebo hastalarının %45'inde gözlenmiştir. CTCAE derece 3 ya da 4 bilirübin artışları sorafenib ile tedavi edilen hastaların %10'unda ve plasebo tedavisindeki hastaların %11'inde bildirilmiştir. Hipoalbüminemi, sorafenib ile tedavi edilen hastaların %59'unda ve plasebo hastalarının %47'sinde gözlenmiştir; her iki grupta da CTCAE derece 3 ya da 4 hipoalbüminemi gözlenmemiştir.
Alkalen fosfataz artışları, sorafenib ile tedavi edilen hastaların %82,2'sinde ve plasebo hastalarının %82,5'inde gözlenmiştir. CTCAE derece 3 alkalen fosfataz artışları, sorafenib ile tedavi edilen hastaların %6,2'sinde ve plasebo tedavisindeki hastaların %8,2'sinde bildirilmiştir; her iki grupta da hiç CTCAE derece 4 alkalen fosfataz artışı gözlenmemiştir.
INR artışları, sorafenib ile tedavi edilen hastaların %42'sinde ve plasebo hastalarının
%34'ünde gözlenmiştir. CTCAE derece 3 INR artışları sorafenib ile tedavi edilen hastaların
%4'ünde ve plasebo hastalarının %2'sinde bildirilmiştir; her iki grupta da CTCAE derece 4 INR yükselmesi bulunmamaktadır.
Lenfopeni, sorafenib ile tedavi edilen hastaların %47'sinde ve plasebo hastalarının %42'sinde gözlenmiştir. CTCAE derece 3 ya da 4 lenfopeni, her bir gruptaki hastaların %6'sında bildirilmiştir. Nötropeni sorafenib ile tedavi edilen hastaların %11'inde ve plasebo hastalarının %14'ünde gözlenmiştir. CTCAE derece 3 ya da 4 nötropeni, her bir gruptaki hastaların %1'inde bildirilmiştir.
Anemi, sorafenib ile tedavi edilen hastaların %59'unda ve plasebo hastalarının %64'ünde gözlenmiştir. CTCAE derece 3 ya da 4 anemi, her bir gruptaki hastaların %3'ünde bildirilmiştir.
Trombositopeni, sorafenib ile tedavi edilen hastaların %46'sında ve plasebo hastalarının
%41'inde gözlenmiştir. CTCAE derece 3 ya da 4 trombositopeni sorafenib ile tedavi edilen hastaların %4'ünde ve plasebo hastalarının %1'den daha azında bildirilmiştir.
Sorafenib ile tedavi edilen hastaların %26,5'inde hipokalsemi bildirilirken plasebo alan hastalarda bu oran %14,8 olmuştur. Bildirilen çoğu hipokalsemi düşük derecededir (CTCAE derece 1 ve 2). CTCAE derece 3 hipokalsemi (6-7 mg/dL) sorafenib ile tedavi edilen hastaların %1,8'inde, plasebo grubundaki hastaların ise %1,1'inde görülmüş; CTCAE derece
4 hipokalsemi de (<6 mg/dL) sorafenib ile tedavi edilen hastaların %0,4'ünde, plasebo
grubundaki hastaların ise %0'ında görülmüştür. Sorafenib ile ilişkili hipokalseminin etiyolojisi bilinmemektedir.
Sorafenib ile tedavi edilen hastaların %9,5'inde hipokalemi bildirilirken, plasebo alan hastalarda bu oran %5,9 olmuştur. Bildirilen çoğu hipokalemi düşük derecededir (CTCAE derece 1). CTCAE derece 3 hipokalemi, sorafenib ile tedavi edilen hastaların %0,4'ünde, plasebo grubundaki hastaların ise %0,7'sinde görülmüştür. Derece 4 hipokalemi bildirilmemiştir.
Tiroid karsinoma hastalarında laboratuvar test anormallikleri (Çalışma 5):
Sorafenib ile tedavi edilen hastaların %35,7'sinde hipokalsemi bildirilirken, plasebo grubundaki hastalarda bu oran %11 olmuştur. Bildirilen çoğu hipokalsemi düşük derecededir. CTCAE derece 3 hipokalsemi sorafenib ile tedavi edilen hastaların %6,8'inde, plasebo grubundaki hastaların %1,9'unda; CTCAE derece 4 hipokalsemi ise sorafenib ile tedavi edilen hastaların %3,4'ünde, plasebo grubundaki hastaların %1'inde görülmüştür.
Çalışmada gözlemlenen diğer klinik olarak anlamlı laboratuvar anormallikleri, Tablo 4' de gösterilmektedir.
Tablo 4: Diferansiye Tiroid Kanserli Hastalarda Bildirilen Tedaviye Bağlı Laboratuvar Testi Anormallikleri_Çift Kör Dönemi (Çalışma 5)
Laboratuvar parametresi, (% araştırılan numuneler) | Sorafenib, n=207 | Plasebo, n=209 | ||||
Tüm Dereceler* | Derece 3* | Derece 4* | Tüm Dereceler * | Derece 3* | Derece 4* | |
Kan ve lenf sistemi hastalıkları | ||||||
Anemi | 30,9 | 0,5 | 0 | 23,4 | 0,5 | 0 |
Trombositopeni | 18,4 | 0 | 0 | 9,6 | 0 | 0 |
Nötropeni | 19,8 | 0,5 | 0,5 | 12 | 0 | 0 |
Lenfopeni | 42 | 9,7 | 0,5 | 25,8 | 5,3 | 0 |
Metabolizma ve beslenme hastalıkları | ||||||
Hipokalemi | 17,9 | 1,9 | 0 | 2,4 | 0 | 0 |
Hipofosfatemi** | 19,3 | 12,6 | 0 | 2,4 | 1,4 | 0 |
Hepatobiliyer hastalıklar | ||||||
Bilirubin artışı | 8,7 | 0 | 0 | 4,8 | 0 | 0 |
ALT artışı | 58,9 | 3,4 | 1 | 24,4 | 0 | 0 |
AST artışı | 53,6 | 1 | 1 | 14,8 | 0 | 0 |
Araştırmalar | ||||||
Amilaz artışı | 12,6 | 2,4 | 1,4 | 6,2 | 0 | 1 |
Lipaz artışı | 11,1 | 2,4 | 0 | 2,9 | 0,5 | 0 |
* Advers Olaylar için Ortak Terminoloji Kriteri (CTCAE), Versiyon 3.0
** Sorafenib ile ilişkili hipofosfateminin etiyolojisi bilinmemektedir.
Şüpheli advers reaksiyonların raporlanması
Ruhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar/risk dengesinin sürekli olarak izlenmesine olanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu Türkiye Farmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e-
posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks: 0 312 218 35 99).
4.9. Doz aşımı ve tedavisi
SOFEXAN doz aşımı için spesifik bir tedavi bulunmamaktadır.
Klinik olarak incelenen en yüksek sorafenib dozu günde iki kez 800 mg'dır. Bu dozda gözlenen advers reaksiyonlar başlıca diyare ve dermatolojik olaylar olmuştur.
Bir doz aşımı kuşkusu durumunda, SOFEXAN uygulaması durdurulmalı ve destekleyici tedavi başlatılmalıdır.
5. FARMAKOLOJİK ÖZELLİKLER
5.1. Farmakodinamik özellikler
Farmakoterapötik grup: Antineoplastik ilaçlar, protein kinaz inhibitörü ATC kodu: L01EX02
Sorafenib, in vitro ve in vivo anti-proliferatif ve anti-anjiyojenik özellikler sergileyen bir multikinaz inhibitörüdür.
Etki mekanizması ve farmakodinamik etkiler
Sorafenib, in vitro koşullarda tümör hücresi proliferasyonunu azaltan bir multikinaz inhibitörüdür. Sorafenib, atimik farelerde tümör anjiyogenezinde azalma ile birlikte, insan tümörü ksenograftlarının geniş bir spektrumunda tümör büyümesini inhibe etmektedir. Sorafenib tümör hücresinde (CRAF, BRAF, V600E BRAF, c-KIT ve FLT-3) ve tümör vaskülatüründe (CRAF, VEGFR-2, VEGFR-3 ve PDGFR-ß) bulunan hedeflerin aktivitesini inhibe etmektedir. Serin/treonin kinazlar RAF kinazları iken c-KIT, FLT-3, VEGFR-2, VEGFR-3 ve PDGFR-ß reseptör tirozin kinazlarıdır.
Klinik etkililik ve güvenlilik
Sorafenibin klinik etkililiği ve güvenliliği hepatoselüler karsinomalı (HSK), metastatik renal hücreli karsinomalı (RHK) ve diferansiye tiroid karsinomalı (DTK) hastalarda çalışılmıştır.
Hepatoselüler karsinoma
Çalışma 3 (çalışma 100554), 602 hepatosellüler karsinom hastasıyla yapılan uluslararası, çok merkezli, randomize, çift körlü, plasebo kontrollü bir Faz III çalışmadır. Sorafenib ve plasebo grubunda ECOG skoru açısından demografik ve başlangıçtaki hastalık özellikleri benzerdir (skor 0: %54'e karşı %54; skor 1: %38'e karşı %39; skor 2: %8'e karşı %7), TNM evresi (evre I: <%1'e karşı <%1; evre II: %10,4'e karşı %8,3; evre III: %37,8'e karşı %43,6; evre IV: %50,8'e karşı %46,9) ve BCLC evresi (evre B: %18,1'e karşı %16,8; evre C: %81,6'ya karşı %83,2; evre D: <%1'e karşı %0).
Çalışma, planlanan ara OS analizi önceden belirlenen etkililik sınırını geçtikten sonra durdurulmuştur. Bu OS analizi, sorafenibin OS açısından plaseboya göre istatistiksel olarak anlamlı ölçüde avantajlı olduğunu göstermiştir (HR: 0,69, p = 0,00058, bkz. Tablo 5).
Bu çalışmada Child Pugh B karaciğer yetmezliği olan hastalara ilişkin veriler sınırlıdır ve Child Pugh C olan yalnızca bir hasta çalışmaya dahil edilmiştir.
Tablo 5: Hepatosellüler Karsinomda Çalışma 3 (Çalışma 100554) Etkililik Sonuçları
Etkililik Parametresi | Sorafenib (n=299) | Plasebo (n=303) | P değeri | HR (%95 GA) |
Genel Sağkalım (OS) [medyan, hafta (%95 GA)] | 46,3
(40,9; 57,9) | 34,4
(29,4; 39,4) | 0,00058* | 0,69
(0,55; 0,87) |
İlerlemeye Kadar Geçen Zaman (TTP) [medyan, hafta (%95 GA)]** | 24
(18; 30) | 12,3
(11,7; 17,1) |
0,000007 | 0,58
(0,45; 0,74) |
GA=Güven aralığı, HR=Tehlike oranı (plaseboya göre Sorafenib)
*p değeri, önceden belirlenen O'Brien Fleming durma sınırı olan 0,0077'in altında olduğundan istatistiksel olarak anlamlı
**Bağımsız radyolojik inceleme
İkinci Faz III, uluslararası, çok merkezli, randomize, çift körlü, plasebo kontrollü çalışmada (Çalışma 4, 11849) ilerlemiş hepatosellüler karsinomu olan 226 hastada sorafenibin klinik faydası değerlendirilmiştir. Çin, Kore ve Tayvan'da yürütülen bu çalışma, sorafenibin olumlu fayda-risk profili açısından Çalışma 3'ün bulgularını doğrulamıştır (HR (OS): 0,68, p = 0,01414).
Çalışma 3 ve 4'te önceden belirlenen katmanlandırma faktörlerinde (ECOG skoru, makroskopik vasküler invazyon ve/veya ekstrahepatik tümör yayılması varlığı veya yokluğu) tehlike oranı tutarlı bir şekilde plaseboya göre sorafenib lehine olmuştur. Araştırma amaçlı alt grup analizleri, başlangıçta uzak metastazları olan hastalarda tedavi etkisinin daha az belirgin olduğunu göstermiştir.
Renal hücreli karsinoma
Sorafenibin ilerlemiş renal hücreli karsinoma (RHK) tedavisindeki güvenliliği ve etkililiği 2 klinik çalışmada incelenmiştir:
Çalışma 1 (çalışma 11213), 903 hastayla yapılan çok merkezli, randomize, çift körlü, plasebo kontrollü bir Faz III çalışmadır. Yalnızca berrak hücreli renal karsinomu ve düşük ve orta MSKCC (Memorial Sloan Kettering Kanser Merkezi) riski olan hastalar çalışmaya dahil edilmiştir. Primer sonlanım noktaları genel sağkalım ve progresyonsuz sağkalım (PFS) olmuştur.
Hastaların yaklaşık yarısında ECOG performans skoru 0'dır ve hastaların yarısı düşük risk MSKCC prognostik grubundadır.
PFS, RECIST kriterleri kullanılarak körleştirilmiş bağımsız radyolojik inceleme ile değerlendirilmiştir. PFS analizi 769 hastada 342 olayla yürütülmüştür. Medyan PFS, sorafenibe randomize edilen hastalarda 167 gün olarak bulunurken, plasebo hastalarında 84 gün olmuştur (HR=0,44; %95 GA: 0,35-0,55; p<0,000001). Yaş, MSKCC prognostik grubu, ECOG PS ve önceki tedavi, tedavinin etki boyutunu etkilememiştir.
Genel sağkalıma yönelik ara analiz (ikinci ara analiz) 903 hastada 367 ölümle yapılmıştır. Bu analiz için nominal alfa değeri 0,0094'tür. Medyan sağkalım, sorafenibe randomize edilen hastalarda 19,3 ay olarak bulunurken, plasebo hastalarında 15,9 ay olmuştur (HR= 0,77; %95 GA: 0,63, 0,95; p = 0,015). Bu analiz sırasında yaklaşık 200 hasta plasebo grubundan
sorafenib grubuna geçmiştir.
Çalışma 2, RHK'yı da içeren metastatik maligniteleri olan hastalarla yapılan bir Faz II, kesme çalışmasıdır. Sorafenib tedavisinde hastalığı stabil olan hastalar plaseboya randomize edilmiş veya sorafenib tedavisine devam etmiştir. RHK'sı olan hastalarda progresyonsuz sağkalım (PFS), sorafenib grubunda (163 gün) plasebo grubuna (41 gün) göre anlamlı ölçüde daha uzun olmuştur (p= 0,0001, HR= 0,29).
Diferansiye tiroid karsinoma
Çalışma 5 (çalışma 14295), radyoaktif iyoda dirençli lokal olarak ilerlemiş veya metastatik diferansiye tiroid kanserli 417 hasta üzerinde yürütülen, uluslararası, çok-merkezli, randomize, çift-kör, plasebo-kontrollü bir Faz III araştırmadır.
Körlenmiş bir bağımsız radyolojik inceleme ile RECIST kriterleri kullanılarak değerlendirilen PFS, çalışmanın birincil sonlanım noktasıdır. Sekonder sonlanım noktaları arasında genel sağkalım (OS), tümör yanıt oranı ve yanıt süresi yer almıştır. Progresyon sonrasında, hastaların açık etiketli sorafenib kullanmalarına izin verilmiştir.
Kayıttan önce 14 ay içinde hastalarda progresyon görüldüyse ve Radyoaktif iyota (RAİ) dirençli DTK varsa hastalar çalışmaya dahil edilmiştir. RAİ'ye dirençli DTK RAİ taramasında iyot tutulumu olmayan bir lezyonu olan veya kümülatif RAİ>600 mCi alan veya kayıttan önceki 16 ay içinde bir RAİ tedavisi ya da 16 aylık süre içinde yapılmış iki RAİ tedavisinden sonra progresyon gösteren olarak tanımlanmıştır.
Başlangıç dönemi demografik özellikleri ve hasta özellikleri her iki tedavi grubu arasında iyi bir dengelenme göstermiştir. Hastaların %86'sında akciğerlerde, %51'inde lenf nodunda ve
%27'sinde kemikte metastaz vardır. Hastaların hemen hemen tamamına tiroidektomi (%99,5) uygulanmış ve ortanca yaklaşık 400 mCi kümülatif radyoaktif aktivite almıştır. Hastaların çoğunda papiller karsinomayı (%56,8) takiben foliküler (%25,4) ve daha az diferansiye karsinoma (%9,6) vardır.
Tüm analiz kümesinde, 207 hasta günde iki kez 400 mg sorafenibe ve 210 hasta plaseboya randomize edilmiştir. PFS, körlemeli bağımsız radyolojik inceleme yoluyla, RECIST kriterleri kullanılarak değerlendirilmiştir.
Ortanca PFS süresi, sorafenib grubunda 329 gün (10,8 ay) ve plasebo grubunda 175 gündür (5,8 ay). PFS için bağıl risk (hastalık progresyonu veya ölüm), sorafenib ile tedavi edilen hastalarda, plasebo ile tedavi edilen bireyler ile karşılaştırıldığında tehlike oranıyla yaklaşık
%41 azalmıştır (Tehlike Oranı (TO): 0,587; %95 GA: 0,454; 0,758; tek yönlü p<0,0001)
(Tablo 6, Şekil 2).
Sorafenibin PFS üzerindeki etkisi coğrafik bölge, 60 yaş üzeri veya altı yaş, cinsiyet, histolojik alt tip, tümör yükü ve kemik metastazının varlığı veya yokluğu dahil tüm alt kümelerde tutarlıdır.
Genel sağkalımda (OS) tedavi grupları arasında istatistiksel bir fark bulunmamaktadır (TO: 0,802; %95 GA: 0,539; 1,194, tek yönlü p değeri 0,138, Tablo 6). Ortanca OS'ye her iki kolda da erişilmemiştir. Plasebo koluna randomize edilen 150 (%71,4) ve sorafenib koluna randomize edilen 55 (%26,6) hasta açık etiketli olarak sorafenib aldılar.
RECIST'e göre tam yanıt (CR) gözlenmemiştir. Bağımsız radyolojik değerlendirmeye göre
genel yanıt oranı (CR + parsiyel yanıt (PR)) sorafenib grubunda (24 hasta, %11,6), plasebo grubuna (1 hasta, %0,5) kıyasla daha yüksektir (tek yönlü p<0,0001). PR veren, sorafenib ile tedavi edilen hastalarda ortalama yanıt süresi 309 gündür (%95 GA: 226, 505 gün).
Tablo 6: Çalışma 5: Diferansiye Tiroid Karsinomada Etkililik Sonuçları
Etkinlik Parametresi | Sorafenib (n=207) Olay (ölüm) ile deneklerin (%) oranı | Plasebo (n=210) Olay (ölüm) ile deneklerin (%) oranı | Sorafenib (n=207) [ortanca, gün (%95 GA)]* | Plasebo (n=210) [ortanca, gün (%95 GA)] | p-değeri | TO (%95 GA) |
Progresyonsuz Sağkalım (PFS) (12.08.2012) | 113 (%54,6) | 137 (%65,2) | 329 (278; 393) | 175 (160; 238) | <0,0001 | 0,587 (0,454-0,758) |
Genel Sağkalım (OS) (31.05.2013) | 66 (%31,9) | 72 (%34,3) | Erişilmemiştir | 1.110 (26-1.110) | 0,2359 | 0,884 (0,663; 1,236) |
GA=Güven aralığı, TO=Tehlike oranı (plaseboya karşı sorafenib)
* Bağımsız radyolojik inceleme
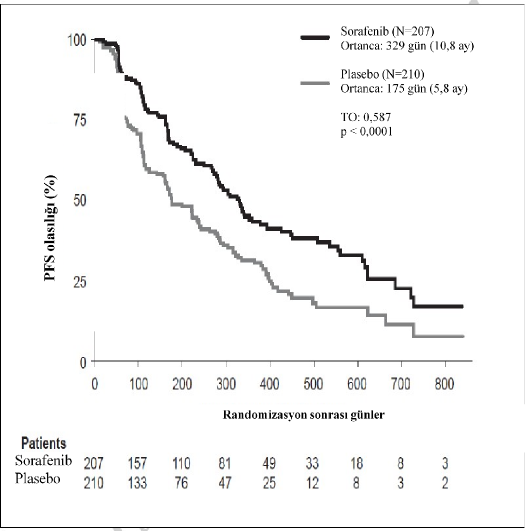
Riskli hastalar
Şekil 1: Çalışma 5: Tiroid Karsinomada Progresyonsuz Sağkalımın (PFS) Kaplan-Meier eğrisi (tüm analiz kümesi)
QT aralığının uzaması
Bir klinik farmakoloji çalışmasında, 31 hastada başlangıçta (tedavi öncesi) ve tedavi sonrası QT/QTc ölçümleri kaydedilmiştir. 28 günlük tek bir tedavi küründen sonra, maksimum sorafenib konsantrasyonunun izlendiği zaman noktasında, plasebo tedavisinin başlangıç değerleriyle karşılaştırıldığında QTcB 4±19 msn, QTcF ise 9±18 msn uzamıştır. Tedavi sonrası EKG izlemi sırasında hiçbir hastada QTcB veya QTcF>500 msn izlenmemiştir (bkz. Bölüm 4.4).
5.2. Farmakokinetik özellikler
Genel özelliklerEmilim:
Sorafenib tablet uygulamasından sonra, oral çözeltiye kıyasla ortalama bağıl biyoyararlanım
%38-49'dur. Oral uygulamayı izleyerek, sorafenib doruk plazma düzeylerine yaklaşık 3 saatte ulaşır. Orta dereceli yağ içeren bir yemek ile verildiğinde, biyoyararlanımı aç karına olduğu gibidir. Yağdan zengin bir yemek ile verildiğinde, sorafenib biyoyararlanımı aç karına uygulamaya kıyasla %30 azalmaktadır. Yedi gün süreyle çoklu sorafenib dozlarının uygulanması, tek doz uygulamaya kıyasla 2,5 ile 7 katlık bir birikim ile sonuçlanır. Kararlı durum plazma sorafenib konsantrasyonları 7 gün içinde elde edilir ve ortalama konsantrasyonların tepe-vadi oranı 2'den düşüktür. Günde iki kez 400 mg sorafenib uygulamasının kararlı durum farmakokinetiği, tiroid karsinoma, RHK ve HSK hastalarında değerlendirilmiştir. Tüm tümör tipleri için maruz kalmadaki değişkenlik yüksek olmasına rağmen, en yüksek ortalama maruziyet tiroid kanserli hastalarda görülmüştür. Tiroid kanserli hastalarda EAA'daki artışın klinik anlamı bilinmemektedir.
Dağılım:
Sorafenibin insan plazma proteinlerine in vitro bağlanması %99,5 düzeyindedir.
Biyotransformasyon:
Sorafenib esas olarak karaciğerde metabolize olur, CYP3A4'ün aracılık ettiği oksidatif metabolizmaya girerken, aynı zamanda UGT1A9 aracılığıyla glukuronidasyona uğrar. Sorafenib konjugatları gastrointestinal kanalda bakteriyel glukuronidaz aktivitesi tarafından parçalanabilirler, bu da konjuge olmayan ilacın tekrar emilimini sağlar. Eş zamanlı neomisin uygulaması bu süreci engelleyerek sorafenibin ortalama biyoyararlanımını %54 oranında düşürür.
Sorafenib kararlı durumda, plazmada dolaşan metabolitlerin yaklaşık %70-85'ini oluşturur. Sorafenibin sekiz metaboliti tanımlanmıştır, bunlardan beşi plazmada saptanmıştır. Sorafenibin plazmada dolaşan esas metaboliti, piridin N-oksit, sorafenibe benzer bir in vitro potens gösterir. Bu metabolit kararlı durumda dolaşımdaki metabolitlerin yaklaşık %9-16'sını oluşturur.
Eliminasyon:
Çözelti şeklinde bir sorafenib formülasyonu oral yoldan 100 mg dozda uygulandıktan sonra dozun %96'sı 14 gün içinde atılmıştır, bu miktarın %77'si feçes ile, %19'u idrarda glukuronize metabolitler şeklinde atılmıştır. Dozun %51'ini oluşturan değişmemiş haldeki sorafenib, feçeste bulunmakta, ama idrarda bulunmamaktadır. Bu da, değişmemiş haldeki ilacın eliminasyonuna safra ile atılımın katkıda bulunduğunu göstermektedir.
Sorafenibin eliminasyon yarı-ömrü 25-48 saat civarındadır.
Doğrusallık/Doğrusal olmayan durum:
Sorafenib doğrusal farmakokinetik özellik gösterir.
Hastalardaki karakteristik özellikler
Irk:
Beyaz ırk ile Asya ırkına mensup gönüllülerde farmakokinetik özellikler bakımından klinik olarak anlamlı farklılıklar bulunmamaktadır.
Cinsiyet:
Demografik verilerin analizleri, cinsiyete göre farmakokinetik açısından fark olmadığını göstermektedir.
Karaciğer yetmezliği:
Sorafenib esas olarak karaciğer tarafından elimine edilmektedir.
Hafif (Child-Pugh A) ya da orta derecede (Child-Pugh B) karaciğer yetmezliği olan hepatoselüler karsinomu hastalarda sistemik maruz kalma düzeyleri, karaciğer bozukluğu olmayan hastalarda gözlenen aralık içindedir. Child-Pugh A ve Child-Pugh B olup hepatoselüler karsinomu olmayan hastalarda sorafenibin farmakokinetiği, sağlıklı gönüllülerin farmakokinetiği ile benzerdir.
Sorafenib farmakokinetiği şiddetli karaciğer yetmezliği (Child-Pugh C) olan hastalarda incelenmemiştir (bkz. Özel kullanım uyarıları ve önlemleri ve Pozoloji ve uygulama şekli). Başlıca karaciğer aracılığıyla atıldığından bu hasta popülasyonunda maruziyet artabilir.
Böbrek yetmezliği:
Bir klinik farmakoloji çalışmasında sorafenibin farmakokinetiği, böbrek fonksiyonları normal olan olgulara ve hafif (CrCl 50-80 mL/dk), orta dereceli (CrCl 30 ile <50 mL/dk) ya da diyaliz gerektirmeyen şiddetli böbrek yetmezliği (CrCl <30 mL/dk) olan olgulara tek doz 400 mg uygulamasından sonra değerlendirilmiştir. Sistemik sorafenibe maruz kalma ve renal fonksiyon arasında ilişki gözlenmemiştir. Hafif, orta dereceli ya da diyaliz gerektirmeyen şiddetli böbrek yetmezliği nedeniyle doz ayarlaması gerekli değildir (bkz. Pozoloji ve uygulama şekli). Diyaliz gereken hastalardaki durum incelenmemiştir.
Yaşlılar (65 yaş üzeri):
Demografik verilerin analizleri, yaşa göre doz ayarlaması yapılmasının gerekli olmadığını göstermektedir.
Pediyatrik hastalar:
Pediyatrik hastalara ilişkin farmakokinetik veri bulunmamaktadır.
5.3. Klinik öncesi güvenlilik verileri
Karsinojenez, Mutajenez, Fertilite bozukluğu
Sorafenibin klinik öncesi güvenlilik profili fareler, sıçanlar, köpekler ve tavşanlarda değerlendirilmiştir.
Tekrarlı doz toksisitesi çalışmalarında, öngörülen klinik maruziyetin altındaki maruziyetlerde (EAA karşılaştırmaları doğrultusunda) çeşitli organlarda değişiklikler (dejenerasyon ve rejenerasyon) görülmüştür.
Genç ve büyümekte olan köpeklere tekrarlı doz uygulamasından sonra, kemik ve dişler üzerinde etkiler gözlenmiştir. Bu değişiklikler, günlük 600 mg/m vücut yüzey alanı sorafenib dozunda (vücut yüzey alanı temelinde önerilen klinik doz olan 500 mg/m'nin 1,2 katına eşdeğer) femoral büyüme plağında düzensiz kalınlaşmalar, 200 mg/m/gün düzeyinde değişen büyüme plağına komşu kemik iliğinde hiposelülarite ve 600 mg/m/gün düzeyinde dentin bileşiminde değişikliklerden oluşmaktaydı. Erişkin köpeklerde benzeri etkiler indüklenmemiştir.
Bir in vitro memeli hücresi çalışmasında (Çin hamsteri overleri), metabolik aktivasyon varlığında klastojenisite (kromozomal aberrasyonlar) için pozitif genotoksik etkiler elde edilmiştir. Üretim prosesi sırasında oluşan ve aynı zamanda bitmiş ilaç hammaddesinde de bulunan (<%0,15) bir ara madde, bir in vitro bakteriyel hücre incelemesinde (Ames testi) mutajenez açısından pozitiftir. Sorafenib, Ames testinde (%0,34 düzeyinde ara madde) ve bir in vivo koşullarda fare mikronukleus incelemesinde genotoksik bulunmamıştır.
Sorafenib ile karsinogenesite çalışmaları yürütülmemiştir.
Sorafenib ile hayvanlarda fertilite üzerindeki etkiyi değerlendirme amaçlı spesifik çalışmalar yapılmamıştır. Ancak erkek ve dişi fertilitesi üzerinde bir advers etki beklenebilir, çünkü hayvanlarda yürütülen tekrarlı doz çalışmaları, erkek ve dişi üreme organlarında değişimler olduğunu göstermiştir. Tipik değişimler, sıçanların testis, epididim, prostat ve seminal veziküllerinde dejenerasyon ve retardasyon bulgularından oluşmaktadır ve bu etkiler günlük 150 mg/m vücut yüzey alanı sorafenib dozunda açık bir şekilde belirmiştir (vücut yüzey alanı temelinde önerilen 500 mg/m'lik klinik dozun yaklaşık üçte biri). Dişi sıçanlarda korpus luteumda santral nekroz ve overlerde foliküler gelişim duraklaması görülmüştür ve gözlenen en düşük etki düzeyi 30 mg/m/gün olmuştur. Köpeklerde 600 mg/m/gün dozunda testislerde tübüler dejenerasyon ve 1200 mg/m/gün dozunda oligospermi görülmüştür.
Sorafenibin sıçanlar ve tavşanlara uygulandığında embriyotoksik ve teratojen olduğu gösterilmiştir. Gözlenen etkiler maternal ve fetal vücut ağırlığında azalma, fetal rezorpsiyon sayısında artış ve eksternal ve viseral malformasyon sayısında artıştan oluşmaktadır. Sıçanlarda oral 6 mg/m/gün dozunda ve tavşanlarda 36 mg/m/gün dozunda advers fetal sonlanımlar gözlenmiştir (bkz. Bölüm 4.4, Bölüm 4.6).
Çevresel Risk değerlendirme çalışmaları, sorafenib tosilatın çevre açısından persistan, biyoakümülatif ve toksik olma potansiyeli olduğunu göstermiştir.
6. FARMASÖTİK ÖZELLİKLER
6.1. Yardımcı maddelerin listesi
Mikrokristalin selüloz PH101 Mikrokristalin selüloz PH102 Kroskarmelloz sodyum
Hidroksi propil metilselüloz E5 LV premium Sodyum lauril sülfat
Magnezyum stearat Polietilen glikol 6000
Opadry complete film coating system 03B240041 pink (hipromelloz, titanyum dioksit, makrogol/PEG, kırmızı demiroksit)
6.2. Geçimsizlikler
Bilinen bir geçimsizliği bulunmamaktadır.
6.3. Raf ömrü
24 ay
6.4. Saklamaya yönelik özel tedbirler
SOFEXAN 25°C'nin altındaki oda sıcaklığında ambalajında saklanmalıdır.
6.5. Ambalajın niteliği ve içeriği
Şeffaf PVC-PE-PVDC-Alüminyum folyo blister ambalajlarda 112 film kaplı tablet
6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler
SOFEXAN isimli tıbbi ürün çevre için potansiyel risk oluşturabilir.
Kullanılmamış olan ürünler ya da atık materyaller, “Tıbbi Atıkların Kontrolü Yönetmeliği†ve “Ambalaj Atıklarının Kontrolü Yönetmeliğiâ€ne uygun olarak imha edilmelidir.
 Depresyonu Anlamak
Depresyon farklı kişileri farklı biçimlerde etkiler. Duygusal veya fiziksel
olmak üzere geniş alanda belirtilere sebep olabilir.Depresyona neler sebep olur?
Depresyonu Anlamak
Depresyon farklı kişileri farklı biçimlerde etkiler. Duygusal veya fiziksel
olmak üzere geniş alanda belirtilere sebep olabilir.Depresyona neler sebep olur? |
 Gıda Alerjisi
Her yıl milyonlarca insan yiyeceklere alerji gösteriyor.
Gıda Alerjisi
Her yıl milyonlarca insan yiyeceklere alerji gösteriyor. |
İLAÇ EŞDEĞERLERİ
| Eşdeğer İlaç Adı | Barkodu | İlaç Fiyatı |
|---|---|---|
| PROKINIB | 8699702099027 | 31,286.88TL |
| SOFEXAN | 8699540033658 | 31,286.88TL |
| Diğer Eşdeğer İlaçlar |
 |
Grip, Soğuk Algınlığı ve Öksürük Grip ve soğuk algınlığı (nezle) semptomları arasındaki farkı bilmek önemlidir. Soğuk algınlığı gripten daha hafif belirtiler gösteren bir solunum yolu hastalığıdır. |
 |
Pankreas Kanseri Pankreas karnın alt kısmında yatay şekilde bulunan bir organdır. Sindirime yardımcı olan enzimleri ve kan şekerini yönetmeye yardımcı olan hormonları vücuda dağıtmakla görevlidir. |
 |
Dış Gebelik Dış gebelik, her 100 gebelikten birini etkileyen, sık görülen ve ölüme sebep olabilecek bir durumdur. Bu, döllenen yumurta, rahimin dışına yerleşirse, oluşan bir durumdur. Gebelik ilerledikçe, ağrıya ve kanamalara sebep olur. |
İLAÇ GENEL BİLGİLERİ
Nobel İlaç Sanayii ve Tic. Anomim Şirketi
| Satış Fiyatı | 31286.88 TL [ 24 Mar 2025 ] |
| Önceki Satış Fiyatı | 31286.88 TL [ 17 Mar 2025 ] |
| Original / Jenerik | Original İlaç |
| Reçete Durumu | Normal Reçeteli bir ilaçdır. |
| Barkodu | 8699540033658 |
| Etkin Madde | Sorafenib |
| ATC Kodu | L01EX02 |
| Birim Miktar | 200 |
| Birim Cinsi | MG |
| Ambalaj Miktarı | 112 |
| Antineoplastik ve İmmünomodülatör Ajanlar |
| Yerli ve Beşeri bir ilaçdır. |