STIVARGA 40 mg 84 film kaplı tablet Kısa Ürün Bilgisi
{ Regorafenib }
1. BEŞERİ TIBBİ ÜRÜNÜN ADI
STIVARGA® 40 mg film kaplı tablet Sitotoksik
2. KALİTATİF VE KANTİTATİF BİLEŞİM
Etkin madde
Regorafenib 40 mg (41,49 mg regorafenib monohidrat olarak)
Yardımcı maddeler
STIVARGA'nın günlük dozunda (4 tablet; 160 mg);
Sodyum 2,438 mmol (56,06 mg'a eşdeğer)
Lesitin (soyadan elde edilir) 1,68 mg Yardımcı maddeler için 6.1'e bakınız.
3. FARMASÖTİK FORMU
Film kaplı tablet.
Bir yüzünde “BAYER†logosu, diğer yüzünde “40†işareti bulunan, 16 mm uzunluğunda ve 7 mm kalınlığında, açık pembe renkte oval film kaplı tabletler.
4. KLİNİK ÖZELLİKLER
4.1. Terapötik endikasyonlar
STIVARGA, öncesinde floropirimidin-, oksaliplatin- ve irinotekan bazlı kemoterapi, anti- VEGF (anti vasküler endotelyal büyüme faktörü) tedavisi ve RAS-doğal tip ise anti-EGFR (anti epidermal büyüme faktörü reseptörü) ile tedavi görmüş ve progresyon göstermiş, ECOG performans skoru: 0-1 olan, yeterli organ fonksiyonu bulunan ve yaşam beklentisi üç aydan fazla olan, rezeke edilemeyen metastatik kolorektal kanserli (mKRK) hastalarda endikedir.
4.2. Pozoloji ve uygulama şekli
STIVARGA antikanser tedavisinin uygulanmasında deneyimli hekimler tarafından reçete
edilmelidir.
STIVARGA ile 4 haftalık bir tedavi kürü ilaç alınan 3 tedavi haftası ve ardından tedavisiz 1 haftadan oluşur. STIVARGA'nın önerilen dozu 3 tedavi haftası süresince günde bir kez oral yolla alınan 160 mg regorafenibdir (40 mg regorafenib içeren 4 adet STIVARGA tablet).
Tedaviye yarar sağlandığı sürece veya tedavi sırasında kabul edilemeyen toksisite ortaya
çıkana kadar devam edilmelidir (bkz. Bölüm 4.4).
Performans skoru (PS) 2 veya daha yüksek olan hastalar klinik çalışmaya dahil edilmemiştir. PS≥2'den büyük olan hastalar için kısıtlı veri bulunmaktadır.
Doz modifikasyonları
Bireysel güvenliliğe ve tolerabiliteye bağlı olarak ilacın kullanımına ara verilmesi ve/veya dozun azaltılması gerekebilir. Doz modifikasyonları 40 mg'lık (bir tablet) doz adımları şeklinde uygulanır. En düşük önerilen günlük doz 80 mg'dır. Maksimum günlük doz 160 mg'dır.
El-ayak deri reaksiyonları (EADR)/ palmar-plantar eritrodizestezi sendromu) durumunda
önerilen doz modifikasyonları ve önlemler Tablo 1'de özetlenmiştir.
Tablo 1: EADR durumunda önerilen doz modifikasyonları ve önlemler
Deri toksisitesi derecesi | Ortaya çıkış | Önerilen doz modifikasyonları ve önlemler |
Derece 1 | Herhangi bir zaman | Mevcut doz düzeyi korunur ve semptomların giderilmesi için derhal destekleyici önlemler alınır. |
Derece 2 | İlk olay | Doz 40 mg (bir tablet) azaltılır ve derhal destekleyici önlemler alınır.
Dozun azaltılmasına karşın iyileşme görülmezse, en az 7 gün süreyle, toksisite Derece 0-1'e gerileyene kadar tedaviye ara verilir.
Tedaviden sorumlu hekimin kararı doğrultusunda dozun yeniden artırılmasına izin verilir. |
7 gün içinde iyileşme olmaması veya ikinci kez ortaya çıkış | Toksisite Derece 0-1'e gerileyene kadar tedaviye ara verilir.
Aradan sonra tedaviye yeniden başlarken, doz 40 mg (bir tablet) azaltılır.
Tedaviden sorumlu hekimin kararı doğrultusunda dozun yeniden artırılmasına izin verilir. | |
Üçüncü kez ortaya çıkış | Toksisite Derece 0-1'e gerileyene kadar tedaviye ara verilir.
Aradan sonra tedaviye yeniden başlarken, doz 40 mg (bir tablet) azaltılır.
Tedaviden sorumlu hekimin kararı doğrultusunda dozun yeniden artırılmasına izin verilir. | |
Dördüncü kez ortaya çıkış | STIVARGA ile tedavi tamamen sonlandırılır. | |
Derece 3
| İlk olay
| Derhal destekleyici önlemler alınır. En az 7 gün süreyle, toksisite Derece 0-1'e gerileyene kadar tedaviye ara verilir. |
|
| (bir tablet) azaltılır.
Tedaviden sorumlu hekimin kararı doğrultusunda dozun yeniden artırılmasına izin verilir. |
İkinci kez ortaya çıkış | Derhal destekleyici önlemler alınır. En az 7 gün süreyle, toksisite Derece 0-1'e gerileyene kadar tedaviye ara verilir.
Aradan sonra tedaviye yeniden başlarken, doz 40 mg (bir tablet) azaltılır. | |
Üçüncü kez ortaya çıkış | STIVARGA ile tedavi tamamen sonlandırılır. |
Karaciğer fonksiyon testlerinde, STIVARGA tedavisi ile ilişkili olduğu düşünülen kötüleşmenin gözlendiği durumlarda önerilen önlemler ve doz modifikasyonları Tablo 2'de özetlenmiştir (ayrıca bkz. Bölüm 4.4).
Tablo 2: İlaçla ilişkili karaciğer fonksiyon testi anormallikleri durumunda önerilen önlemler ve doz modifikasyonları
ALT ve/veya AST değerlerinde gözlenen artışlar | Ortaya çıkış | Önerilen önlemler ve doz modifikasyonları |
≤5 x Normalin üst sınırı (NÜS) (en fazla Derece 2) | Herhangi bir zamanda ortaya çıkış | STIVARGA tedavisine devam edilir. Transaminazlar <3 x NÜS (Derece 1) veya başlangıç değerine dönene kadar haftada bir kez karaciğer fonksiyonları takip edilir. |
>5 x NÜS ila ≤20 x NÜS (Derece 3) | İlk olay | STIVARGA tedavisine ara verilir. Transaminazlar <3 x NÜS veya başlangıç değerine dönene kadar haftada bir kez takip edilir.
Yeniden başlama: Potansiyel yarar hepatotoksisite riskine ağır basıyorsa, STIVARGA tedavisi yeniden başlatılır. Aradan sonra tedaviye yeniden başlarken, doz 40 mg (bir tablet) azaltılır ve en az 4 hafta boyunca haftada bir kez karaciğer fonksiyonları takip edilir. |
Nüks | STIVARGA ile tedavi tamamen sonlandırılır. | |
>20 x NÜS (Derece 4) | Herhangi bir zamanda ortaya çıkış | STIVARGA ile tedavi tamamen sonlandırılır. |
>2 x NÜS bilirubin ile | Herhangi bir | STIVARGA ile tedavi tamamen sonlandırılır. |
eş zamanlı olarak | zamanda ortaya |
|
>3 x NÜS | çıkış | Karaciğer fonksiyonları düzelene veya |
(Derece 2 veya daha |
| başlangıçtaki duruma dönene kadar haftada |
yüksek) |
| bir kez takip edilir. |
|
| İstisna: Gilbert sendromu olan ve transaminaz |
| düzeyleriyükselen hastalar, ALT ve/veya ASTdeğerlerindeartışlargözlendiğinde |
|
| yukarıda özetlenen önerilere göre tedavi edilmelidir. |
ALT: Alanin aminotransferaz AST: Aspartat aminotransferaz
Uygulama şekli:
STIVARGA, oral kullanım içindir.
STIVARGA her gün aynı saatte alınmalıdır. Tabletler %30'dan düşük oranda yağ içeren hafif bir yemeğin ardından bir bütün halinde su ile yutulmalıdır (bkz. Bölüm 5.2). Hafif (düşük yağ içeren) bir öğüne örnek olarak; bir fincan tahıl (yaklaşık 30 g), bir bardak yağsız süt, bir dilim reçelli kızarmış ekmek, 1 bardak elma suyu ve bir fincan kahve veya çay verilebilir (520 kalori, 2 g yağ).
Hasta STIVARGA'nın bir dozunu almayı unutursa, unuttuğu dozu aynı gün içinde hatırlar hatırlamaz almalıdır. Hasta unuttuğu dozu dengelemek için aynı gün çift doz almamalıdır.
Regorafenib uygulanmasından sonra hastada kusma gözlenirse, hasta ilave tablet
almamalıdır.
Özel popülasyonlara ilişkin ek bilgiler:
Böbrek yetmezliği:
Mevcut klinik veriler hafif, orta veya şiddetli böbrek yetmezliği bulunan hastalarda regorafenib ve metabolitleri M-2 ile M-5 için maruziyetin böbrek fonksiyonu normal olan hastalardaki maruziyete benzer olduğunu göstermektedir. Hafif, orta veya şiddetli böbrek yetmezliği olan hastalarda doz ayarlaması gerekli değildir (Bkz. Bölüm 5.2 Farmakodinamik özellikler).
Karaciğer yetmezliği:
Regorafenib başlıca karaciğer yoluyla atılmaktadır.
Klinik çalışmalarda, hafif karaciğer yetmezliği (Child-Pugh A) olan hastalar ve karaciğer fonksiyonları normal olan hastalar arasında maruziyet, güvenlilik veya etkililik açısından anlamlı farklılıklar gözlenmemiştir. Hafif karaciğer yetmezliği olan hastalar için doz ayarlaması gerekli değildir. Orta derecede (Child-Pugh B) karaciğer yetmezliği olan hastalarda sadece kısıtlı veri bulunduğundan, doz önerisi sağlanamamaktadır. Bu hastalarda genel güvenliliğin yakından takip edilmesi önerilmektedir (ayrıca bkz. Bölüm 4.4 ve Bölüm 5.2).
STIVARGA şiddetli karaciğer yetmezliği (Child-Pugh C) olan hasta popülasyonunda araştırılmadığından, STIVARGA'nın bu hastalarda kullanılması önerilmemektedir.
Pediyatrik popülasyon:
STIVARGA'nın metastatik kolorektal kanser endikasyonunda pediyatrik popülasyonda
kullanımı bulunmamaktadır.
STIVARGA'nın gastrointestinal stromal tümör (GİST) endikasyonunda çocuklarda ve 18 yaşın altındaki ergenlerde güvenliliği ve etkililiği belirlenmemiştir. Veri bulunmamaktadır.
STIVARGA'nın hepatoselüler karsinom endikasyonunda pediyatrik popülasyonda kullanımı
bulunmamaktadır.
Geriyatrik popülasyon:
Klinik çalışmalarda, yaşlılar (65 yaş ve üzeri) ve daha genç hastalar arasında maruziyet, güvenlilik veya etkililik açısından anlamlı farklılıklar gözlenmemiştir. Yaşlı hastalarda doz ayarlaması gerekli değildir (ayrıcabkz.Bölüm5.2).
Cinsiyet:
Klinik çalışmalarda, erkek ve kadın hastalar arasında maruziyet, güvenlilik veya etkililik açısından anlamlı farklılıklar gözlenmemiştir. Cinsiyete göre doz ayarlaması gerekli değildir (ayrıca bkz. Bölüm 5.2).
Etnik farklılıklar:
Klinik çalışmalarda, farklı etnik gruplara ait hastalar arasında maruziyet, güvenlilik veya etkililik açısından anlamlı farklılıklar gözlenmemiştir. Etnik kökene göre doz ayarlaması gerekli değildir (ayrıca bkz. Bölüm 5.2). El ayak deri reaksiyonu (EADR), karaciğer fonksiyon testlerinde ciddi anormallikler ve karaciğer fonksiyon bozukluğu insidansının STIVARGA tedavisi alan Asyalı (özellikle Japon) hastalarda beyaz ırka kıyasla daha yüksek olduğu gözlenmiştir. Klinik çalışmalarda STIVARGA tedavisi alan Asyalı hastaların primer olarak Doğu Asya kökenli olduğu kaydedilmiştir.
4.3. Kontrendikasyonlar
STIVARGA, etkin madde ya da yardımcı maddelerin herhangi birine (bkz.Bölüm 6.1) karşı aşırı duyarlılığı olan kişilerde kontrendikedir.
Eğer fıstık ya da soya alerjisi varsa STIVARGA kullanılmamalıdır.
4.4. Özel kullanım uyarıları ve önlemleri
Anevrizmalar ve arter diseksiyonları
VEGF yolak inhibitörlerinin, hipertansiyonu olan veya olmayan hastalarda kullanılması, anevrizmalar ve/veya arter diseksiyonları oluşumunu kolaylaştırabilir. STIVARGA'ya başlamadan önce hipertansiyon veya anevrizma öyküsü gibi risk faktörleri olan hastalarda bu risk dikkatle değerlendirilmelidir.
Karaciğer üzerine etkiler:
STIVARGA ile tedavi edilen hastalarda sıklıkla karaciğer fonksiyon testlerinde (alanin amino transferaz [ALT], aspartat aminotransferaz [AST] ve bilirubin) anormallikler gözlenmiştir. Küçük bir hasta grubunda şiddetli karaciğer fonksiyon testi anormallikleri (Derece 3 ila 4) ve klinik seyir gösteren karaciğer fonksiyon bozukluğu (ölümcül sonuçlar dahil) bildirilmiştir (bkz. Bölüm 4.8). Klinik çalışmalarda karaciğer fonksiyon testlerinde ciddi anormallik ve karaciğer fonksiyon bozukluğu insidansının STIVARGA tedavisi alan Asyalı (özellikle Japon) hastalarda beyaz ırka kıyasla daha yüksek olduğu gözlenmiştir (bkz. Bölüm 4.2).
STIVARGA ile tedaviye başlamadan önce karaciğer fonksiyon testlerinin (ALT, AST ve bilirubin) yaptırılması ve bu değerlerin tedavinin ilk 2 ayı boyunca yakından takip edilmesi (en az iki haftada bir) önerilmektedir. Bu dönemin ardından ise, periyodik takibe en az ayda bir ve klinik olarak gerekli olduğunda devam edilmelidir.
Regorafenib bir üridin difosfat glukuronozil transferaz (UGT) 1A1 inhibitörüdür (bkz. Bölüm 4.5). Gilbert sendromu olan hastalarda hafif, indirekt (konjuge olmayan) hiperbilirubinemi görülebilir.
Karaciğer fonksiyon testlerinde, STIVARGA tedavisi ile ilişkili olduğu düşünülen kötüleşmenin gözlendiği hastalar için (post-hepatik kolestaz veya hastalık progresyonu gibi alternatif bir nedenin olmadığı durumlarda) Tablo 2'de verilen doz modifikasyonları ve takip önerileri izlenmelidir (bkz. Bölüm 4.2).
Regorafenib, başlıca karaciğeryoluylaelimineedilmektedir. Hafif veya orta derecede
önerilmektedir (bkz. Bölüm 4.2 Bölüm 5.2). STIVARGA, şiddetli karaciğer yetmezli
(Child-Pugh C) olan hasta popülasyonunda araştırılmadığından ve bu hastalarda maruziyet daha yüksek olabileceğinden STIVARGA'nın bu hastalarda kullanılması önerilmemektedir.
Enfeksiyonlar:
STIVARGA enfeksiyon olaylarının insidansında artış ile ilişkilendirilmiştir ve bu olayların bir kısmı ölümcül olmuştur (bkz. Bölüm 4.8).
Enfeksiyon olaylarının kötüleşmesi halinde, STIVARGA tedavisine ara verilmesi
düşünülmelidir.
Hemoraji:
STIVARGA, bazıları ölümcül olan hemorajik olayların insidansında artış ile ilişkilendirilmiştir (bkz. Bölüm 4.8). Kanamaya yatkınlık yaratan durumların görüldüğü, antikoagülanlar (örn. varfarin ve fenprokumon) veya kanama riskini artıran diğer tıbbi ürünlerle eşzamanlı tedavi edilen hastaların kan sayımları ve koagülasyon parametreleri takip edilmelidir. Karaciğer sirozu bulunan hastalarda, STIVARGA tedavisine başlanmadan önce standart olarak özofageal varisler için tarama ve ardından tedavi uygulanmalıdır. Acil tıbbi girişim gerektiren şiddetli kanama durumunda, STIVARGA'nın tamamen kesilmesi düşünülmelidir.
Gastrointestinal perforasyon ve fistül:
STIVARGA ile tedavi edilen hastalarda gastrointestinal perforasyon (fatal sonuçları olanlar dahil) ve fistül oluşumu bildirilmiştir (bkz. Bölüm 4.8). Bu olayların aynı zamanda intra- abdominal maligniteleri olan hastalarda hastalıkla ilişkili yaygın komplikasyonlar oldukları bilinmektedir. Gastrointestinal perforasyon veya fistül oluşan hastalarda STIVARGA'nın kesilmesi önerilmektedir.
Kardiyak iskemi ve infarktüs:
STIVARGA, miyokard iskemisi ve infarktüsü insidansında artışla ilişkilendirilmiştir (bkz. Bölüm 4.8). Stabil olmayan anjina veya yeni başlangıçlı anjina (STIVARGA tedavisine başlamadan önceki 3 ay içinde), yakın zamanda miyokard enfarktüsü geçirmiş (STIVARGA tedavisine başlamadan önceki 6 ay içinde) ve New York Kalp Derneği (NYHA) sınıflamasına göre kalp yetmezliği 2 veya daha yüksek olan hastalar klinik çalışmalara dahil edilmemiştir.
İskemik kalp hastalığı öyküsü olan hastalar miyokard iskemisinin klinik bulgu ve semptomları açısından takip edilmelidir. Kardiyak iskemi ve/veya infarktüsü gelişen hastalarda iyileşme görülene kadar STIVARGA tedavisine ara verilmelidir.
STIVARGA ile tedaviye yeniden başlama kararı alınırken, her bir hasta için potansiyel yarar ve risklere ilişkin dikkatli bir inceleme yapılmalıdır. Hastada düzelme görülmezse STIVARGA tamamen kesilmelidir.
Geri Dönüşümlü Posterior Lökoensefalopati Sendromu (PRES):
STIVARGA tedavisi ile ilişkili olarak Geri dönüşümlü posterior lökoensefalopati sendromu
(GPLS) bildirilmiştir (bkz. Bölüm 4.8).
GPLS'nin bulgu ve semptomları arasında nöbet, baş ağrısı, mental durumda değişiklikler, konfüzyon, hipertansiyon ile ilişkili olan ya da olmayan görme bozuklukları veya kortikal körlük yer almaktadır. GPLS tanısının beyin görüntülemesi ile doğrulanması gerekmektedir. GPLS gelişen hastalarda, hipertansiyon kontrolü ve diğer semptomlar için destekleyici tıbbi kontrol ile birlikte STIVARGA'nın kesilmesi önerilmektedir.
Arteriyel hipertansiyon:
STIVARGA arteriyel hipertansiyon insidansında artışla ilişkilendirilmiştir (bkz. Bölüm 4.8). Hipertansiyon gelişen çoğu hastada, hipertansiyon başlangıcı STIVARGA tedavisinin ilk siklusunda ortaya çıkmıştır. Kan basıncı STIVARGA ile tedaviye başlamadan önce kontrol edilmelidir. Kan basıncı izlenmeli ve standart tıbbi uygulamalar doğrultusunda hipertansiyon tedavi edilmelidir. Yeterli tıbbi tedaviye karşın şiddetli veya dirençli hipertansiyon vakalarında, tedaviden sorumlu hekimin kararına bağlı olarak STIVARGA tedavisine geçici olarak ara verilmeli ve/veya doz azaltılmalıdır (bkz. Bölüm 4.2). Hipertansif kriz durumunda STIVARGA kullanımı sonlandırılmalıdır.
Yara iyileşme komplikasyonları :
Anti-anjiyojenik özellikleri bulunan tıbbi ürünler yara iyileşmesini geciktirebileceği veya engelleyebileceği için, büyük bir ameliyat geçirecek olan hastalarda STIVARGA kullanımının önlem olarak geçici şekilde kesilmesi önerilir. Büyük bir ameliyattan sonra STIVARGA ile tedaviye devam etme kararı, yeterli yara iyileşmesine dair klinik değerlendirmelere dayanmalıdır.
Dermatolojik toksisite:
El-ayak deri reaksiyonu (EADR /palmar-plantar eritrodizestezi sendromu) ve döküntü STIVARGA ile gözlenen en yaygın dermatolojik advers ilaç reaksiyonlarını temsil etmektedir (bkz. Bölüm 4.8). Klinik çalışmalarda STIVARGA ile tedavi edilen Beyaz hastalara kıyasla Asyalı hastalarda (özellikle Japon) EADR insidansının daha yüksek olduğu gözlenmiştir (bkz. Bölüm 4.2). EADR'yi önlemek için alınacak önlemler arasında nasır kontrolü ve ayak tabanlarındaki ve avuç içlerindeki basıncı azaltmak üzere ayakkabı yastıklarının ve eldivenlerin kullanılması yer almaktadır.
EADR tedavisi, semptomların giderilmesi için keratolitik kremlerin (örn., sadece etkilenmiş alanlara az miktarda uygulanan üre-, salisilik asit- veya alfa hidroksil asit bazlı kremler) ve nemlendirici kremlerin (istenildiği kadar uygulanan) kullanılmasını içerebilir. STIVARGA dozunun azaltılması ve/veya tedaviye geçici olarak ara verilmesi ya da şiddetli veya dirençli durumlarda STIVARGA'nın tamamen kesilmesi düşünülmelidir (bkz. Bölüm 4.2).
Biyokimyasal ve metabolik laboratuvar testi anormallikleri:
STIVARGA, elektrolit anormalliklerinin (hipofosfatemi, hipokalsemi, hiponatremi ve hipokalemi) ve metabolik anormalliklerinin (tiroid stimülan hormon, lipaz ve amilazda artışlar dahil) insidansında artışla ilişkilendirilmiştir. Anormallikler genellikle hafif ila orta şiddette olup, klinik seyir göstermez ve genellikle dozun kesilmesini veya azaltılmasını gerektirmez. STIVARGA tedavisi sırasında biyokimyasal ve metabolik parametrelerin takip edilmesi ve gerekirse standart klinik uygulamaya göre uygun replasman tedavisinin başlatılması önerilmektedir. Dirençli veya tekrar eden anlamlı anormalliklerin görülmesi durumunda dozun kesilmesi veya azaltılması ya da STIVARGA'nın tamamen bırakılması düşünülmelidir (bkz. Bölüm 4.2).
Trombotik mikroanjiyopati (TMA):
Trombotik trombositopenik purpura (TTP) dahil olmak üzere trombotik mikroanjiyopati (TMA), regorafenib kullanımıyla ilişkilendirilmiştir (bkz. bölüm 4.8). Hemolitik anemi, trombositopeni, yorgunluk, değişken nörolojik belirtiler, böbrek yetmezliği ve ateş ile başvuran hastalarda TMA tanısı düşünülmelidir. TMA gelişen hastalarda regorafenib tedavisi kesilmelidir ve acil tedavi gereklidir. Tedavinin kesilmesinden sonra TMA'nın etkilerinin tersine döndüğü gözlemlenmiştir.
Hastalığa özgü önlemler - Hepatoselüler karsinom (HSK):
Plasebo kontrollü pivot faz III çalışmada, hastalar önceden sorafenib tedavisi almıştır.
Sodyum:
STIVARGA'nın önerilen günlük dozu (4 tablet; 160 mg) 2,438 mmol (56,06 mg'a eşdeğer) sodyum ihtiva eder. Bu durum, kontrollü sodyum diyetinde olan hastalar için göz önünde bulundurulmalıdır.
Lesitin:
STIVARGA'nın önerilen günlük dozu (4 tablet; 160 mg), 1,68 mg lesitin (soyadan elde edilir) ihtiva eder. Eğer fıstık ya da soya alerjisi varsa STIVARGA kullanılmamalıdır.
4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri
CYP3A4 ve UGT1A9 inhibitörleri/CYP3A4 indükleyicileri:
İn vitro veriler regorafenibin, sitokrom CYP3A4 ve üridin difosfat glukuronozil transferaz
UGT1A9 ile metabolize olduğunu göstermektedir.
Güçlü bir CYP3A4 inhibitörü olan ketokonazolün (18 gün boyunca 400 mg) tek doz regorafenib (5. günde 160 mg) ile birlikte uygulanması, regorafenibe ortalama maruziyette (EAA) yaklaşık %33'lük bir artış ve aktif metabolitler, M-2 (N-oksit) ve M-5'e (N-oksit ve N-destemil) ortalama maruziyette yaklaşık %90 azalma ile sonuçlanmıştır. Regorafenibin ve metabolitlerinin (M-2 ve M-5) kararlı durum maruziyetleri üzerine etkileri araştırılmamış olduğundan, CYP3A4 aktivitesinin güçlü inhibitörlerinin (örn., klaritromisin, greyfurt suyu, itrakonazol, ketokonazol, posakonazol, nefazodon, telitromisin ve vorikonazol) STIVARGA ile birlikte kullanılmasından kaçınılması önerilmektedir.
Regorafenib ve onun metabolitlerinin kararlı durum maruziyetine etkileri çalışılmadığından; regorafenib tedavisi süresince, güçlü UGT1A9 inhibitörlerinin (örn. mefenamik asit, diflunisal ve niflumik asit) birlikte kullanımından kaçınılmalıdır.
Güçlü bir CYP3A4 indükleyicisi olan rifampisinin (9 gün boyunca 600 mg) tek doz regorafenib (7. günde 160 mg) ile birlikte uygulanması, regorafenibe ortalama maruziyette (EAA) yaklaşık %50 azalma ve aktif metabolit M-5'e ortalama maruziyette 3- ila 4- kat artış ile sonuçlanırken, aktif metabolit M-2'ye maruziyette herhangi bir değişiklik gözlenmemiştir. Diğer güçlü CYP3A4 indükleyicileri de (örn., fenitoin, karbamazepin, fenobarbital, St.John's Wort) regorafenibin metabolizmasını artırabilirler. Regorafenibin plazma konsantrasyonundaki düşüş etkililiğin azalmasına neden olabileceğinden, güçlü CYP3A4 indükleyicilerinin kullanımından kaçınılmalı veya CYP3A4'ü indükleme potansiyeli olmayan ya da çok düşük bir potansiyele sahip eşzamanlı kullanılacak alternatif bir tıbbi ürünün seçilmesi düşünülmelidir.
Meme Kanseri Direnç Proteini (BCRP) ve P-glikoprotein substratları:
BCRP substratı olan rosuvastatin tek doz (5 mg) uygulamasından önce regorafenib (14 gün 160 mg) uygulaması, ortalama rosuvastatin maruziyetinde (EAA) 3,8 kat artış ile Cdeğerinde 4,6 kat artışa sebep olmuştur.
Klinik veriler, regorafenibin digoksinin farmakokinetik özellikleri üzerinde etkisi olmadığını bu sebeple klinik olarak anlamlı ilaç etkileşimleri olmadan digoksin gibi p-glikoprotein substratları ile eşzamanlı uygulanabileceğini göstermiştir.
UGT1A1 ve UGT1A9 substratları:
İn vitro veriler, gerek regorafenibin gerekse aktif metaboliti olan M-2'nin, in vivo kararlı durumda elde edilen konsantrasyonlarda, üridin difosfat glukuronil transferazlar UGT1A1 ve UGT1A9'un aracılık ettiği glukuronidasyonu inhibe ettiğini göstermektedir. Diğer yandan M- 5 sadece UGT1A1'i inhibe etmektedir.
İrinotekan uygulanmasından önce 5 günlük bir ara ile regorafenib uygulanması, UGT1A1
substratı ve irinotekan aktif metaboliti olan SN-38'e ortalama maruziyette (EAA) yaklaşık
%44'lük artışla sonuçlanmıştır. Aynı zamanda irinotekanın EAA'da yaklaşık %28'lik artışı gözlenmiştir. Bu durum, regorafenibin UGT1A1 ve UGT1A9 substratları ile birlikte uygulanmasının sistemik maruziyeti artırabileceğini göstermektedir. Bu bulguların klinik anlamı bilinmemektedir.
Meme Kanseri Direnç Proteini (BCRP) ve P-glikoprotein substratları inhibitör/indükleyicileri:
İn vitro veriler M-2 ve M-5 aktif metabolitlerinin BCRP ve P-glikoproteinin substratları olduğunu göstermektedir. BCRP ve P-glikoproteinin inhibitörleri ve indükleyicilerinin M-2 ve M-5 maruziyetini etkileyebilir. Bu bulguların klinik önemi bilinmemektedir (bkz. Bölüm 5.2)
CYP izoform-selektif substratlar:
İn vitro veriler regorafenibin, in vivo kararlı durumda elde edilen konsantrasyonlarda (8,1 mikromolar doruk plazma konsantrasyonu) sitokrom CYP2C8 (Kdeğeri 0,6 mikromolar), CYP2C9 (Kdeğeri 4,7 mikromolar), CYP2B6 (Kdeğeri 5,2 mikromolar)'nın yarışmalı inhibitörü olduğunu göstermektedir. CYP3A4 (Kdeğeri 11,1 mikromolar) ve CYP2C19 (16,4 mikromolar Kdeğeri)'a karşı in vitro inhibe edici potens daha az belirgindir.
160 mg regorafenibin 14 gün süreyle uygulanmasının CYP2C8 (rosiglitazon), CYP2C9 (S- varfarin), CYP2C19 (omeprazol) ve CYP3A4 (midazolam) prob substratlarının farmakokinetiği üzerindeki etkisini değerlendirmek üzere klinik prob substrat çalışması yapılmıştır.
Farmakokinetik veriler, regorafenibin, klinik olarak anlamlı bir ilaç etkileşimi olmaksızın CYP2C8, CYP2C9, CYP3A4 ve CYP2C19 substratları ile eşzamanlı verilebileceğini göstermektedir (ayrıca bkz. Bölüm 4.4).
Antibiyotikler:
Konsantrasyon-zaman profili, regorafenib ve metabolitlerinin enterohepatik dolaşıma girebildiğini göstermektedir (bkz. Bölüm 5.2). Gastrointestinal mikroflora eradikasyonu için kullanılan ve zayıf emilime uğrayan antimikrobiyal bir ilaç olan neomisin (regorafenibin enterohepatik dolaşımıyla etkileşebilir) ile birlikte uygulama regorafenib maruziyetini etkilememiş ancak in vitro ve in vivo koşullarda regorafenibin farmakolojik aktivitesine benzer aktivite gösteren M-2 ve M-5 metabolitlerinin maruziyetinde yaklaşık %80 azalmaya neden olmuştur. Neomisin etkileşiminin klinik açıdan anlamlılığı bilinmemekle birlikte,
regorafenibin etkililiğini azalmaola sılığıvardır.Diğerantibiyotiklerle farmakokinetik
Safra asidi bağlayıcı ajanlar
Regorafenib, M-2 ve M-5 enterohepatik dolaşımına geçmesi muhtemeldir (bkz. Bölüm 5.2). Kolestiramin ve kolosevelam gibi safra tuzu bağlayıcı ajanlar, absorpsiyonu (veya reabsorpsiyonu) etkileyebilen ve bu nedenle potansiyel azalmış maruziyet ile sonuçlanan çözünmeyen kompleksler oluşturarak, regorafenib ile etkileşime girebilirler. Bu potansiyel etkileşimlerin klinik önemi bilinmemektedir, ancak regorafenibin etkililiğinde azalmaya neden olabilir.
4.6. Gebelik ve laktasyon
Gebelik kategorisi D'dir.
Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon)
Çocuk doğurma potansiyeli bulunan kadınlar regorafenibin, fetüs üzerinde zararlı etkilere neden olabileceği konusunda bilgilendirilmelidir.
Çocuk doğurma potansiyeli olan kadınlar ve ayrıca erkekler tedavi süresince ve tedavinin tamamlanmasını takiben 8 haftaya kadar etkili doğum kontrol yöntemi kullanmalıdırlar.
Gebelik dönemi
Regorafenibin gebe kadınlarda kullanımına ilişkin bilgi mevcut değildir. Regorafenibin etki mekanizmasına dayalı olarak, gebelik döneminde uygulandığında fetüs üzerinde zararlı etkilere neden olması beklenmektedir.
Hayvanlar üzerinde yapılan çalışmalar regorafenibin üreme toksisitesine sahip olduğunu göstermiştir (bkz. Bölüm 5.3).
STIVARGA, kesinlikle gerekli olmadıkça ve anne için sağlayacağı yararları ile fetüs üzerindeki riskleri dikkatle değerlendirilmeden gebelik döneminde kullanılmamalıdır.
Laktasyon dönemi
Regorafenib veya metabolitlerinin insan sütüyle atılıp atılmadığı bilinmemektedir. Bununla birlikte sıçanlarda, regorafenib veya metabolitleri süte geçmektedir.
Emzirilen çocuk için risk göz ardı edilemez. Regorafenib bebeğin büyümesine ve gelişimine zarar verebilir (bkz. Bölüm 5.3).
STIVARGA ile tedavi sırasında emzirme durdurulmalıdır.
Üreme yeteneği/Fertilite
STIVARGA'nın insan fertilitesi üzerindeki etkisine dair bilgi bulunmamaktadır. Hayvanlar üzerinde yapılan çalışmalardan elde edilen bulgular, regorafenibin erkek ve dişi fertilitesini azaltabileceğini göstermektedir (bkz. Bölüm 5.3).
4.7. Araç ve makine kullanımı üzerindeki etkiler
STIVARGA'nın araç veya makine kullanma yeteneği üzerindeki etkisine dair herhangi bir çalışma yürütülmemiştir. Hastaların, STIVARGA tedavisi esnasında konsantrasyon ya da reaksiyon yeteneklerini etkileyen belirtiler yaşaması durumunda, bu etkiler geçene kadar araç ve makine kullanmamaları tavsiye edilir.
4.8. İstenmeyen etkiler
Güvenlilik profilinin özeti
dahil olmak üzere klinik çalışmalarda tedavi edilen 4800'ün üzerinde hastadan elde edilen verilere dayanmaktadır.
Regorafenibin bu çalışmalardaki güvenlilik profili, standart tedavilerin ardından hastalığı progresyon gösteren metastatik kolorektal kanserli 2872 hasta ile yürütülen faz III B çalışmanın güvenlilik bulguları ile uyumludur.
STIVARGA kullanan hastalarda gözlenen en yaygın advers ilaç reaksiyonları (≥%30) ağrı, el-ayak deri reaksiyonu, asteni/yorgunluk, diyare, iştah kaybı ve besin alımının azalması, hipertansiyon ve enfeksiyondur.
STIVARGA kullanan hastalarda gözlenen en ciddi advers ilaç reaksiyonları; şiddetli karaciğer hasarı, hemoraji, gastrointestinal perforasyon ve enfeksiyondur.
Advers reaksiyonların listesi
STIVARGA ile tedavi edilen hastalar ile yapılan klinik çalışmalarda bildirilen advers ilaç
reaksiyonları aşağıda verilmektedir.
Advers ilaç reaksiyonları, aşağıda sistem-organ sınıfı (MedDRA versiyon 14.1) ve sıklık derecesine göre listelenmektedir. Belirli bir reaksiyonu, onun eşanlamlısını ve ilişkili durumları tanımlamak için en uygun MedDRA terimi kullanılmıştır. Şu terimler ve sıklık dereceleri kullanılmıştır: Çok yaygın (≥1/10); yaygın (≥1/100 ila <1/10); yaygın olmayan (≥1/1000 ila <1/100); seyrek (≥1/10,000 ila <1/1000); çok seyrek (<1/10,000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor).
Her sıklık grubu içinde, istenmeyen etkiler azalan ciddiyet derecesine göre sıralanmıştır.
Enfeksiyonlar ve enfestasyonlar
Çok yaygın: Enfeksiyon*
İyi huylu ve kötü huylu neoplazmalar (Kist ve polipler dahil olmak üzere)
Seyrek: Keratoakantoma/skuamöz hücreli deri karsinomu
Kan ve lenf sistemi hastalıkları Çok yaygın: Trombositopeni, anemi Yaygın: Lökopeni
Seyrek: Trombotik mikroanjiyopati (TMA)
İmmun sistem hastalıkları
Yaygın olmayan: Hipersensitivite reaksiyonu
Endokrin hastalıkları
Yaygın: Hipotiroidizm
Metabolizma ve beslenme hastalıkları
Çok yaygın: İştah kaybı ve besin alımının azalması
Yaygın: Hipokalemi, hipofosfatemi, hipokalsemi, hiponatremi, hipomagnezemi, hiperürisemi, dehidratasyon
Sinir sistemi hastalıkları
Seyrek: Geri dönüşümlü posterior lökoensefalopati sendromu
Kardiyak hastalıklar
Yaygın olmayan: Miyokard infarktüsü, miyokard iskemisi
Vasküler hastalıklar
Çok yaygın: Hemoraji*, hipertansiyon
Yaygın olmayan: Hipertansif kriz
Bilinmiyor: Anevrizmalar ve arter diseksiyonları
Solunum, göğüs bozuklukları ve mediastinal hastalıklar
Çok yaygın: Disfoni
Gastrointestinal hastalıklar
Çok yaygın: Diyare, stomatit, kusma, bulantı, kabızlık
Yaygın: Tat alma bozuklukları, ağız kuruluğu, gastroözofageal reflü, gastroenterit Yaygın olmayan: Gastrointestinal perforasyonlar*, gastrointestinal fistül, pankreatit
Hepato-bilier hastalıklar
Çok yaygın: Hiperbilirubinemi, transaminazlarda artış
Yaygın olmayan: Şiddetli karaciğer hasarı (karaciğer yetmezliği dahil)*#
Deri ve deri altı doku hastalıkları
Çok yaygın: El-ayak deri reaksiyonu**, döküntü Yaygın: Deri kuruluğu, eksfoliyatif döküntü, alopesi
Yaygın olmayan: Tırnaklarda bozukluk, eritema multiforme Seyrek: Stevens-Johnson sendromu, toksik epidermal nekroliz
Kas-iskelet bozuklukları, bağ doku ve kemik hastalıkları
Yaygın: Kas spazmları
Böbrek ve idrar yolu hastalıkları
Yaygın: Proteinüri
Genel bozukluklar ve uygulama bölgesine ilişkin hastalıklar
Çok yaygın: Asteni/yorgunluk, ağrı***, ateş, mukozal inflamasyon
Araştırmalar
Çok yaygın: Kilo kaybı
Yaygın: Amilaz artışı, lipaz artışı, anormal uluslararası normalizasyon oranı (anormal INR)
ölümcül vakalar bildirilmiştir.
** MedDRA terminolojisinde palmar-plantar eritrodizestezi sendromu.
*** En sık bildirilen ağrı türleri (≥%10) karın ağrısı ve sırt ağrısıdır.
Seçili advers reaksiyonların tanımı Şiddetli karaciğer hasarı
Klinik çalışmalarda fatal sonuçlu şiddetli karaciğer hasarı vakaları bildirilmiştir. Bu hastalarda karaciğer fonksiyon bozukluğu tedavinin ilk 2 ayı içinde başlamış ve > 20x ULN (Normalin Üst Sınırı) düzeyinde transaminaz artışını bilirubindeki artışın takip ettiği hepatoselüler hasar paterniylekarakterizeolmuştur.Klinikçalışmalarda ölümcül sonuçlanan
Hemoraji
Plasebo kontrollü faz III çalışmalarda, STIVARGA ile tedavi edilen hastalarda hemorajinin genel insidansı %18,2 ve plasebo alan hastalarda %9,5'tir. STIVARGA ile tedavi edilen hastalarda görülen kanama olayı vakalarının çoğu hafif ila orta şiddette olup (Derece 1 ve 2:
%15,2), en belirgin olanı epistaksistir (%6,1). STIVARGA ile tedavi edilen hastalarda ölümcül sonuçlar yaygın olmayıp (%0,7), bu olaylara serebral, solunum ile ilgili, gastrointestinal ve genito-üriner olaylar dahildir.
Enfeksiyon
Plasebo kontrollü faz III çalışmalarda, STIVARGA ile tedavi edilen hastalarda plasebo kullanan hastalar ile karşılaştırıldığında enfeksiyonlar daha yaygın gözlenmiştir (tüm dereceler: %31,6'ya karşı %17,2). STIVARGA ile tedavi edilen hastalarda en sık görülen enfeksiyonlar hafif ila orta şiddette olup (Derece 1 ve 2: %23,0), idrar yolu enfeksiyonları (%5,7), nazofarenjit (4,0%), mukokutanöz ve sistemik mantar enfeksiyonlarının (%3,3) yanı sıra pnömoni (%2,6) de bunlara dahildir. Enfeksiyonla ilişkili ölümcül sonuçlar, STIVARGA ile tedavi edilen hastalarda (%1,0) plasebo alan hastalara (%0,3) kıyasla daha sık gözlenmiştir ve bunlar ağırlıklı olarak solunum sistemi olaylarıdır.
El-ayak deri reaksiyonu
Plasebo kontrollü faz III çalışmalarda, el ayak deri reaksiyonu (EADR) STIVARGA tedavisi alan hastalarda plasebo alanlara kıyasla daha sık görülmüştür (tüm dereceler: %51,4 ve %6,5, KRK; %66,7 ve %15,2 , GİST ve %51,6 ve %7,3 HSK). STIVARGA ile tedavi edilen hastalarda görülen el-ayak deri reaksiyonu vakalarının çoğu tedavinin ilk siklusu sırasında ortaya çıkmış olup, hafif ila orta şiddettedir (Derece 1 ve 2: %34,3, mKRK, %44,7, GİST ve
% 39,3 HSK). Derece 3 el-ayak deri reaksiyonu insidansı %17,1 (mKRK), %22,0 (GİST) ve
%12,3 (HSK)'tür. EADR insidansının STIVARGA tedavisi alan Asyalı hastalarda daha yüksek olduğu gözlenmiştir (tüm dereceler: %74,8, KRK, %88,2 GİST ve % 67,1 HSK; 3. Derece: %20,5, KRK, %23,5 GİST ve %13,5 HSK) (ayrıca bkz. Bölüm 4.2.).
Hipertansiyon
Plasebo kontrollü faz III çalışmalarda, hipertansiyonun genel insidansı, STIVARGA ile tedavi edilen hastalarda, plasebo alanlara kıyasla, daha yüksektir (% 29,6'ya karşı % 7,5 KRK, % 60,6'ya karşı % 25,8 GIST, % 31,0'a karşı % 6,2 HSK).
STIVARGA ile tedavi edilen hastalarda görülen hipertansiyon vakalarının çoğu tedavinin ilk
siklusu sırasında ortaya çıkmış olup, hafif ila orta şiddettedir (Derece 1 ve 2: %20,9, KRK,
% 31,8, GİST ve %15,8 HSK). Derece 3 hipertansiyon insidansı %8,7 (KRK) % 28,0 (GİST) ve %15,2 (HSK)'dir. GİST çalışmasında bir kez Derece 4 hipertansiyon vakası bildirilmiştir.
Plasebo kontrollü faz III çalışmalarda, tedaviye bağlı proteinürinin genel insidansı, plasebo uygulanan hastaların %1,9'una kıyasla STIVARGA ile tedavi edilen hastalarda %9,1 bulunmuştur.
STIVARGA kolunda bildirilen olayların %35,6'sı ile plasebo kolunda bildirilen olayların
%54,5'nn iyileşmediği/düzelmediği bildirilmiştir.
Tüm klinik çalışmalarda, kardiyak bozukluk olayları (tüm derecelerde) STIVARGA ile tedavi edilen 75 yaş altı hastalara (N=4108) kıyasla STIVARGA ile tedavi edilen 75 yaş ve üstü hastalarda (N=410) daha sık bildirilmiştir (%6,5 ve %13,7).
Laboratuvar testi anormallikleri
anormallikleri Tablo 3 ve Tablo 4'te yer almaktadır (ayrıca bkz. Bölüm 4.4).
Tablo 3: Plasebo kontrollü faz III çalışmalarda metastatik CRC (CORRECT), GIST (GRID) ve HSK'lı (RESORCE) hastalarda bildirilen tedaviye bağlı laboratuvar testi anormallikleri
mCRC (CORRECT) | |||||
Laboratuvar parametreleri | Stivarga artı EİDT (n=500) | Plasebo artı EİDT (n=253) | Stivarga artı EİDT (n=500) | Plasebo artı EİDT (n=253) | |
Derece | |||||
Tüm dereceler (%) | 3./4. Derece (%) | ||||
Kan ve lenf sistemi hastalıkları Hemoglobin sayısında azalma Trombositopeni Nötropeni Lenfopeni |
78,5 40,5 2,8 54,1 |
66,3 16,8 0 34,8 |
5,3 2,8 0,6 9,3 |
2,8 0,4 0 4,0 | |
Metabolizma ve beslenme hastalıkları Hipokalsemi Hipokalemi Hipofasfatemi |
59,3 25,7 57,4 |
18,3 8,3 11,1 |
1,2 4,3 31,1 |
1,2 0,4 3,6 | |
Hepato-biliyer hastalıkları Hiperbilirubinemi AST artışı ALT artışı |
44,6 65,0 45,2 |
17,1 45,6 29,8 |
12,2 5,9 5,5 |
8,4 5,2 3,2 | |
Böbrek ve idrar yolu hastalıkları Proteinüri |
83,6 |
61,0 |
1,8 |
0,8 | |
Araştırmalar INR artışı* Lipaz artışı Amilaz artışı |
23,7 46,0 25,5 |
16,6 18,7 16,7 |
4,2 11,4 2,6 |
1,6 4,4 2,4 | |
GIST (GRID) | |||||
Laboratuvar parametreleri | Stivarga artı EİDT (n=132) | Plasebo artı EİDT (n=66) | Stivarga artı EİDT (n=132) | Plasebo artı EİDT (n=66) | |
Derece | |||||
Tüm dereceler (%) | 3./4. Derece (%) | ||||
Kan ve lenf sistemi hastalıkları Hemoglobin sayısında azalma Trombositopeni Nötropeni Lenfopeni |
75,0 12,9 15,9 29,9 |
72,7 1,5 12,1 24,2 |
3,0 0,8 3,1 7,6 |
1,5 1,5 3,0 3,0 | |
Metabolizma ve beslenme hastalıkları Hipokalsemi Hipokalemi Hipofasfatemi |
16,7 20,5 54,5 |
4,5 3,0 3,1 |
1,5 3,0 21,2 |
0 0 1,5 | |
Hepato-biliyer hastalıkları Hiperbilirubinemi | 58,3 | 47,0 | 3,8 | 1,5 3,0 | |
AST artışı ALT artışı | 39,4 | 39,4 | 4,6 | 1,5 |
Böbrek ve idrar hastalıkları Proteinüri |
59,2 |
52,5 |
3,1 |
3,4 |
Araştırmalar INR artışı* Lipaz artışı Amilaz artışı |
9,3 14,4 - |
12,5 4,6 - |
1,6 0,8 - |
4,7 0 - |
HSK (RESORCE) | ||||
Laboratuvar parametreleri | Stivarga artı EİDT (n=374) | Plasebo artı EİDT (n=193) | Stivarga artı EİDT (n=374) | Plasebo artı EİDT (n=193) |
Derece | ||||
Tüm dereceler (%) | 3./4. Derece (%) | |||
Kan ve lenf sistemi hastalıkları Hemoglobin sayısında azalma Trombositopeni Nötropeni Lenfopeni |
72,5 63,1 13,6 67,8 |
71,3 50,0 14,9 58,5 |
6,0 5,4 3,0 17,4 |
4,8 0 1,0 11,7 |
Metabolizma ve beslenme hastalıkları Hipokalsemi Hipokalemi Hipofasfatemi |
23,4 30,7 70,4 |
10,1 9,0 31,4 |
0,3 4,3 33,9 |
0 2,1 6,9 |
Hepato-biliyer hastalıkları Hiperbilirubinemi AST artışı ALT artışı |
78,2 92,7 70,4 |
54,5 84,3 58,6 |
15,9 17,8 6,2 |
15,7 19,9 4,7 |
Böbrek ve idrar hastalıkları Proteinüri |
51,0 |
36,5 |
16,7 |
3,1 |
Araştırmalar INR artışı* Lipaz artışı Amilaz artışı |
44,4 40,5 23,0 |
35,4 27,0 19,0 |
0,7 14,2 2,8 |
2,1 8,7 2,7 |
4.9. Doz aşımı ve tedavisi
5. FARMAKOLOJİK ÖZELLİKLER
5.1. Farmakodinamik özellikler
Farmakoterapötik grup: Diğer antineoplastik ajanlar, protein kinaz inhibitörleri
ATC kodu: L01EX05
Etki mekanizması ve farmakodinamik etkiler:
Regorafenib tümör anjiyogenezinde (VEGFR1, -2, -3, TIE2), onkogenezde (KIT, RET, RAF- 1, BRAF, BRAFV600E), metastazda (VEGFR3, PDGFR, FGFR) ve tümör immünitesinde (CSF1R) yer alan kinazlar dahil olmak üzere çoklu protein kinazları güçlü bir şekilde engelleyen bir oral tümör deaktivasyon ilacıdır. Regorafenib özellikle gastrointestinal stromal tümörlerde majör bir onkojenik etken olan mutasyona uğramış KIT'i inhibe eder ve
böylelikle tümör hücresi proliferasyonunu bloke eder. Klinik öncesi çalışmalarda, regorafenib
kolorektal, gastrointestinal stromal ve hepatoselüler tümör modelleri dahil olmak üzere
birçok tümör modelinde muhtemel antianjiyogenik ve antiproliferatif etkileri aracılığı ile güçlü anti-tümör aktivite göstermiştir. Ek olarak, regorafenib tümör ile ilişkili makrofajların seviyelerini düşürmüş ve in vivo anti-metastatik etki göstermiştir. Majör insan metabolitleri (M-2 ve M-5) in vitro ve in vivo modellerde regorafenib ile karşılaştırıldığında benzer etkililik sergilemişlerdir.
Klinik etkililik ve güvenlilik:
Metastatik kolorektal kanser (mKRK)
STIVARGA'nın klinik etkililiği ve güvenliliği, standart tedavi ile başarısızlık sonrasında progresyon göstermiş daha önce yoğun tedavi görmüş mKRK'li hastalar ile yapılan uluslararası, çok merkezli, randomize, çift kör, plasebo kontrollü bir faz III çalışmasında (CORRECT) değerlendirilmiştir.
Bu çalışmanın birincil etkililik sonlanım noktası genel sağkalımdır (OS). İkincil sonlanım noktaları progresyonsuz sağkalım (PFS), objektif tümör yanıt oranı ve hastalık kontrol oranıdır.
Toplamda, 760 hasta 2:1 oranında 3 hafta boyunca günde bir kez oral yolla uygulanan 160 mg regorafenib (her biri 40 mg regorafenib içeren 4 adet STIVARGA tablet) ile birlikte En İyi Destek Tedavi (EİDT) (N=505) ya da denk plasebo (N=255) ile birlikte EİDT uygulanmasını takiben 1 hafta tedavisiz periyoda randomize edilmiştir. Kullanılan ortalama günlük regorafenib dozu 147 mg'dır.
Hastalar, hastalık progresyonu veya kabul edilemeyen toksisite ortaya çıkana kadar tedaviye devam etmiştir. 432 ölüm meydana geldiğinde etkililik için önceden planlanmış bir interim analiz gerçekleştirilmiştir.
OS için planlanmış bu interim analiz, önceden belirlenmiş etkililik sınırını geçtikten sonra, yani plasebo ile birlikte uygulanan EİDT ile karşılaştırıldığında STIVARGA ile birlikte EİDT uygulanmasının sağkalımda uzama sağladığının kanıtı gösterildiğinde çalışmanın körlüğü kaldırılmıştır.
Randomize edilen 760 hastanın medyan yaşı 61 olup, %61'i erkek, %78'i beyaz ırktandır ve hastaların tamamının başlangıç ECOG (Eastern Cooperative Oncology Group) performans skoru (PS) 0 veya 1'dir. Hastaların %11,4'ünde STIVARGA tedavisi sırasında PS en az 2 bildirilmiştir.
Doz modifikasyonu ve doz azaltımı oranlarının yanı sıra medyan tedavi süresi ve günlük doz, plasebo alıp PS'nin en az 2 olarak bildirildiği hastalarda (%8,3) gözlemlenenle benzerdir. PS'nin en az 2 olduğu hastaların çoğunda tedavi hastalığın progresyonu nedeniyle durdurulmuştur. PS2 hastalar ve başlangıçtaki dehidratasyonu ≥1 olan hastalar, pivot çalışmanın dışında tutulmuştur. Birincil hastalık bölgesi kolon (%65), rektum (%29) veya her ikisidir (%6). Çalışmanın başlangıcında hastaların %57'sinde KRAS mutasyonu bildirilmiştir.
Hastaların çoğu (%52) metastatik hastalığın tedavisi için daha önce 3 veya daha az basamak tedavi almıştır. Bu tedaviler, floropirimidin içeren kemoterapi, anti-VEGF tedavisi, hasta KRAS doğal tip ise, anti-EGFR tedavisini içermiştir.
STIVARGA'nın EİDT'ye eklenmesi, plasebonun EİDT'ye eklenmesine kıyasla anlamlı olarak daha uzun sağkalım lasonuçlanmıştır; p değeri gruplanmış log-sıra testine göre
anlamlı olarak daha uzundur (tehlike oranı: 0,494, p<0,000001, bkz., Tablo 5 ve Şekil 2). Yanıt oranı (tam yanıt veya kısmi yanıt), STIVARGA ve plasebo ile tedavi edilen hastalarda sırasıyla (p= 0,188432, tek yönlü) %1 ve %0,4 olmuştur. Hastalık kontrol oranı (tam yanıt veya kısmi yanıt ya da stabil hastalık), STIVARGA ile tedavi edilen hastalarda anlamlı düzeyde daha yüksek olmuştur (%41,0 karşısında %14,9, p<0,000001, tek yönlü).
Tablo 5: CORRECT çalışmasından elde edilen etkililik sonuçları
Etkililik parametresi | Tehlike Oranı* (%95 GA) | P-değeri (tek taraflı) | Medyan (%95 GA) | |
STIVARGA + EİDT (N=505) | Plasebo + EİDT (N=255) | |||
Genel Sağkalım | 0,774 (0,636, 0,942) | 0,005178 | 6,4 ay (5,9, 7,3) | 5,0 ay (4,4, 5,8) |
Progresyonsuz Sağkalım** | 0,494 (0,419, 0,582) | <0,000001 | 1,9 ay (1,9, 2,1) | 1,7 ay (1,7, 1,7) |
* Tehlike oranı <1 STIVARGA lehine.
** Araştırmacının tümör yanıtını değerlendirmesine dayanır.
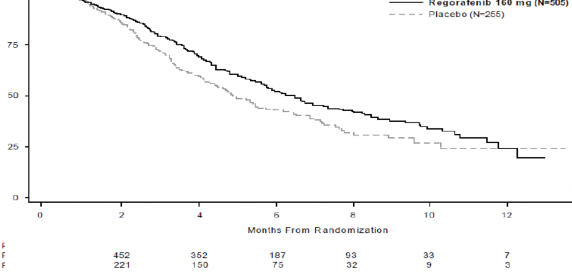
Randomizasyondan sonraki aylar
Şekil 1: Genel Sağkalım için Kaplan-Meier eğrileri
Genel sağkalım ve progresyonsuz sağkalım için yaş (<65; ≥65), cinsiyet, ECOG PS, primer hastalık yeri, metastatik hastalığın ilk tanısına kadar geçen süre, önceki antikanser tedavisi, metastatik hastalık için önceki tedavi basamakları ve KRAS mutasyon durumuna göre alt grup analizi, tedavi etkisinin plasebo rejimine kıyasla regorafenib rejimi lehine olduğunu göstermiştir.
Önceki KRAS mutasyon durumuna göre alt grup analizinin sonuçları, KRAS doğal tip tümörlü hastalarda GS açısından plaseboya kıyasla regorafenib lehine tedavi etkisi olduğunu göstermektedir. KRAS mutant tümörlü hastalarda ise sayıca daha az etki bildirilmiştir. PFS açısından KRAS mutasyon tipinden bağımsız olarak regorafenib lehine tedavi etkisi gözlemlenmiştir. Genel sağkalım tehlike oranı (%95 GA), KRAS doğal tip tümörlü hastalar için 0,653 (0,476 ila 0,895) şeklindeyken KRAS mutant tümörlü hastalarda 0,867 (0,670 ila 1,123) bulunmuştur. Tedavi etkisinde heterojenite kanıtı görülmemiştir (anlamlı olmayan
etkileşim testi). Progresyonsuz sağkalım tehlike oranı (%95 GA), KRAS doğal tip tümörlü
hastalar için 0,475 (0,362 ila 0,623) şeklindeyken KRAS mutant tümörlü hastalarda 0,525
(0,425 ila 0,649) bulunmuştur
İkinci bir faz III, uluslararası, çok merkezli, randomize, çift kör, plasebo kontrollü çalışmada (CONCUR) STIVARGA'nın etkililik ve güvenliliği önceden tedavi almış metastatik kolorektal kanseri olan ve floropirimidin bazlı kemoterapi başarısızlığından sonra progresyon görülen 204 Asyalı hastada (>%90 Doğu Asyalı) değerlendirilmiştir. CONCUR çalışmasındaki hastaların sadece %59,5'u öncesinde VEGF veya EGFR hedefli tedaviler ile tedavi edilmiştir.
Primer etkililik sonlanım noktası olarak OS değerlendirilmiştir. EİDT'ye STIVARGA eklenmesi plasebo artı EİDT'ye kıyasla anlamlı derecede daha uzun sağkalımla sonuçlanmış, tehlike oranının 0,550 (p = 0,000159 gruplanmış log-sıra testi) ve medyan GS'nin 8,8 aya kıyasla 6,3 ay olduğu [%95 GA 0,395, 0,765] belirlenmiştir.
STIVARGA artı EİDT alan hastalarda PFS'nin de anlamlı derecede daha uzun olduğu belirlenmiştir, medyan PFS STIVARGA ile 3,2 ayken plasebo ile 1,7 aydır. (tehlike oranı: 0,311, p<0,000001). CONCUR çalışmasında STIVARGA artı EİDT'nin güvenlilik profilinin CORRECT çalışmasında gözlenen güvenlilik profiliyle tutarlı olduğu görülmüştür.
Gastrointestinal stromal tümörler (GİST)
STIVARGA'nın klinik etkililiği ve güvenliliği, öncesinde 2 tirozin kinaz inhibitörü (imatinib ve sunitinib) ile tedavi görmüş GİST'li hastalar ile yapılan uluslararası, çok merkezli, randomize, çift kör, plasebo kontrollü bir faz III çalışmasında değerlendirilmiştir.
Progresyonsuz sağkalım (PFS) birincil etkililik sonlanım noktası analizi, 144 PFS vakasından sonra gerçekleştirilmiştir (merkezi körlenmiş değerlendirme). Progresyona kadar geçen süre (TTP) ve genel sağkalımdan (OS) oluşan ikincil sonlanım noktaları da değerlendirilmiştir (interim analiz).
Toplamda 199 GİST hastası, 2:1 oranında 3 hafta boyunca ya günde bir kez oral yolla uygulanan 160 mg regorafenib ile birlikte EİDT (n=133) ya da denk plasebo ile birlikte EİDT (n=66) uygulanmasını takiben 1 hafta tedavisiz periyoda randomize edilmiştir. Kullanılan ortalama günlük regorafenib dozu 140 mg'dır.
Hastalar, hastalık progresyonu veya kabul edilemeyen toksisite ortaya çıkana kadar tedaviye devam etmiştir. Plasebo kullanan ve hastalığında progresyon görülen hastalara açık etiketli regorafenib tedavisi (çapraz geçiş seçeneği) olanağı sunulmuştur. Regorafenib kullanan ve hastalığında progresyon görülen ve araştırmacının kanaatine göre regorafenib ile tedavinin klinik fayda sağlamakta olduğu hastalara açık etiketli regorafenibe devam etme imkanı verilmiştir.
Randomize edilen 199 hastanın ortalama yaşı 58 olup %64'ü erkek, %68'i beyaz ırktandır ve hastaların tamamının başlangıç ECOG Performans Skoru 0 veya 1'dir. En yakın progresyon veya relapstan bu yana geçen genel medyan süre 6 hafta olarak belirlenmiştir.
EİDT ile regorafenib kullanılması, plasebo ile birlikte uygulanan EİDT'ye kıyasla anlamlı derecede daha uzun PFS ile sonuçlanmış olup, tehlike oranı 0,268 [%95 GA 0,185, 0,388] ve medyan PFS 0,9 aya karşın 4,8 aydır (p<0,000001). Hastalık progresyonu veya ölüm için
bağıl risk regorafenib ile tedaviedilenhastalarda, plasebo ile tedavi edilen hastalar ile
yaş, cinsiyet, coğrafik bölge, önceki tedavi basamakları ve ECOG performans skorundan bağımsız olarak tutarlı olmuştur.
TTP, EİDT ile birlikte regorafenib kullanan hastalarda, EİDT ile birlikte plasebo uygulanan hastalara kıyasla anlamlı derecede daha uzun olmuş olup, tehlike oranı 0,248 [%95 GA 0,170, 0,364] ve medyan TTP 0,9 aya karşın 5,4 aydır (p<0,000001) (bkz., Tablo 6).
Başlangıçta plasebo koluna randomize edilmiş hastaların %85'inde progresyon sonrası çapraz geçiş gerçekleşmiş olmakla birlikte, OS analizinin tehlike oranı, pozitif tedavi etkisi yönünde eğilim göstermiştir (tehlike oranı=0,772 [%95 GA, 0,423, 1,408]; p=0,199; iki kolda da medyan OS'ye ulaşılamamıştır) (bkz., Tablo 6, Şekil 4).
Tablo 6 : GRID çalışmasından elde edilen etkililik sonuçları
Etkililik | Tehlike Oranı* | P-değeri | Medyan (%95 GA) | |
parametresi | (%95 GA) | (tek taraflı) | STIVARGA + EİDT (N=133) | Plasebo + EİDT (N=66) |
Progresyonsuz Sağkalım | 0,268 (0,185, 0,388) | <0,000001 | 4,8 ay (4,0, 5,7) | 0,9 ay (0,9, 1,1) |
Progresyona Kadar Geçen Süre | 0,248 (0,170, 0,364) | <0,000001 | 5,4 ay (4,1, 5,7) | 0,9 ay (0,9, 1,1) |
Genel Sağkalım | 0,772 (0,423, 1,408) | 0,199 | Ulaşılamamıştır | Ulaşılamamıştır |
GA: Güven aralığı
* Tehlike oranı <1 STIVARGA lehine.
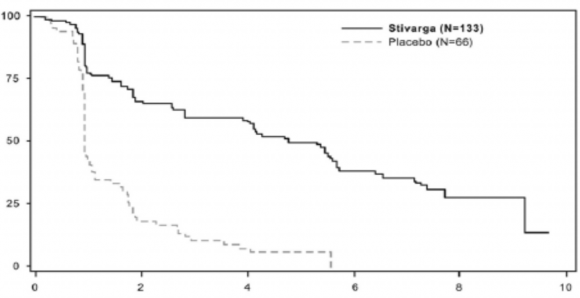
Şekil 2: Progresyonsuz Sağkalım için Kaplan-Meier eğrileri
Progresyonsuz Sağkalım Olasılığı (%)
Randomizasyondan sonraki aylar
![]()
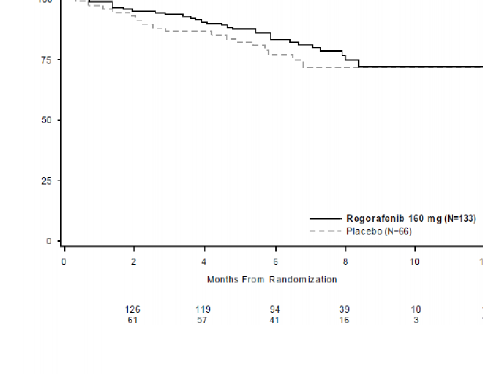
Genel Sağkalım Olasılığı (%)
Şekil 3: Genel sağkalım için Kaplan-Meier eğrileri
Ek olarak, EİDT ile plasebo uygulanan 56 hasta, hastalık progresyonunu takiben çapraz geçişten sonra açık etiketli STIVARGA kullanmıştır ve EİDT ile STIVARGA uygulanan toplam 41 hasta, hastalık progresyonunu takiben STIVARGA tedavisine devam etmiştir. Medyan ikincil PFS (araştırmacı değerlendirmelerine göre ölçüldüğünde) sırasıyla 5,0 ve 4,5 aydır.
Hepatoselüler karsinom (HSK)
STIVARGA'nın klinik etkililik ve güvenliliği, daha önce sorafenib tedavisi almış hepatoselüler karsinomlu hastalarla yürütülen uluslararası, çok merkezli, randomize, çift kör, plasebo kontrollü bir faz III çalışmada (RESORCE) değerlendirilmiştir.
Primer etkililik sonlanım noktası Genel Sağkalım (GS) olmuştur. Sekonder sonlanım noktaları Progresyonsuz Sağkalım (PFS), Progresyona Kadar Geçen Süre (TTP), Objektif Tümör Yanıt Oranı (ORR) ve Hastalık Kontrol Oranıdır (DCR).
Demografik özellikler ve başlangıç özellikleri STIVARGA ve plasebo tedavisi alan gruplarda
benzerdir ve randomize edilen 573 hasta için aşağıdaki tabloda gösterilmiştir:
Medyan yaş: 63 yaş
5.2. Farmakokinetik özellikler
Genel ÖzelliklerEmilim:
Her biri 40 mg içeren 4 tablet olarak verilen 160 mg'lık tek bir oral regorafenib dozundan yaklaşık 3 ila 4 saat sonra, regorafenib yaklaşık 2,5 mg/L'lik ortalama doruk plazma düzeyine ulaşır. Tabletlerin ortalama bağıl biyoyararlanımı oral çözeltiye kıyasla %69-83'tür. Regorafenib ve farmakolojik olarak aktif majör metabolitlerinin M-2 (N-oksit) ve M-5 (N- oksit ve N-desmetil) konsantrasyonları, ilacın az yağlı (hafif) bir kahvaltının ardından verilmesi durumunda yağdan zengin bir kahvaltı sonrasında ya da açlık koşullarında verilmesine kıyasla en yüksektir. Regorafenibe maruziyet açlık koşullarına kıyasla, ilacın yağdan zengin bir kahvaltı ile verilmesi durumunda %48, az yağlı bir kahvaltı ile verilmesi durumunda ise %36 artmıştır. M-2 ve M-5 metabolitlerine maruziyet, regorafenibin az yağlı bir kahvaltı ile verilmesi durumunda açlık durumuna kıyasla daha yüksek, yağdan zengin bir yemek ile verilmesi durumunda ise açlık koşullarında verilmesine kıyasla daha düşük olmuştur.
Dağılım:
Regorafenibin yanı sıra dolaşımdaki majör metabolitler için plazma konsantrasyon-zaman profilleri, 24 saatlik doz uygulama aralığında enterohepatik dolaşıma bağlı olarak çok sayıda pik göstermiştir.
Regorafenibin insan plazma proteinlerine in vitro protein bağlanması yüksektir (%99,5). Biyotransformasyon:
Regorafenib esas olarak karaciğerde metabolize olur, CYP3A4'ün aracılık ettiği oksidatif metabolizmaya girerken, aynı zamanda UGT1A9 aracılığıyla glukuronidasyona da uğrar. Plazmada iki majör ve altı minör regorafenib metaboliti tanımlanmıştır. Regorafenibin plazmada dolaşan esas metabolitleri, farmakolojik olarak aktif olan ve kararlı durumda regorafenib ile benzer konsantrasyonlara sahip M-2 (N-oksit) ve M-5 (N-oksit ve N- desmetil)'tir.
M-2 ve M-5'in in vitro proteine bağlanmaları regorafenibe göre daha yüksektir (sırasıyla
%99,8 ve %99,95).
Metabolitleri, gastrointestinal sistemde mikrobiyal flora tarafından indirgenebilir veya hidrolize olabilir ve bu da konjuge olmayan ilaç ve metabolitlerin geri emilmelerini sağlayabilir (enterohepatik dolaşım).
Eliminasyon:
Oral uygulamayı takiben, plazmadaki regorafenib ve metabolit M-2 için ortalama eliminasyon yarılanma ömrü farklı çalışmalarda 20 ila 30 saat arasında değişmektedir. Metabolit M-5 için ortalama eliminasyon yarılanma ömrü yaklaşık 60 saattir (40 ila 100 saat arasında).
Radyoaktif dozun yaklaşık %90'ı uygulamayı takiben 12 gün içinde geri kazanılmış ve dozun yaklaşık %71'i feçes ile (%47 ana bileşen, %24 metabolit olarak) ve dozun yaklaşık %19'u idrarda glukuronize metabolitler şeklinde atılmıştır. Kararlı durum koşullarında glukuronidlerin üriner itrahı %10'un altına düşmüştür. Feçeste bulunan ana bileşik, glukuronidlerin intestinal degradasyonundan veya M-2 metabolitinin (N-oksit) ve emilime uğramamış regorafenibin redüksiyonundan elde edilebilmiştir. Gastrointestinal kanalda mikrobiyal flora M-5 metabolitini M-4'e indirgeyerek M-4 için geri emilime (enterohepatik dolaşım) imkan verebilir. M-5 sonuç olarak feçeste M-4 üzerinden M-6 şeklinde atılmaktadır.
Doğrusallık/Doğrusal olmayan durum:
Kararlı durumda regorafenibin sistemik maruziyeti 60 mg'a kadar dozla orantılı olarak ve
60 mg'dan daha yüksek dozlarda dozla orantılı değerden daha düşük bir değerde artış göstermektedir. Kararlı durumda regorafenib birikimi plazma konsantrasyonlarında yaklaşık 2 katlık bir artışla sonuçlanır ve bu, eliminasyon yarılanma ömrü ve doz uygulama sıklığı ile tutarlıdır. Kararlı durumda, 160 mg regorafenibin oral uygulamasından sonra regorafenib yaklaşık 3,9 mg/L'lik (8,1 mikromolar) ortalama doruk plazma düzeylerine erişir ve ortalama plazma konsantrasyonlarının tepe:vadi oranı 2'den düşüktür.
Her iki metabolit de (M-2 ve M-5) doğrusal olmayan birikim göstermektedir. Tek doz regorafenibin verilmesinden sonra M-2 ve M-5'in plazma konsantrasyonları, ana bileşene oranla çok daha düşük olmakla birlikte, kararlı durumda plazma konsantrasyonları regorafenib ile benzerdir.
Hastalardaki karakteristik özellikler
Böbrek yetmezliği olan hastalar:
Mevcut klinik verilere ve fizyoloji bazlı farmakokinetik modellemeye göre, regorafenibin ve metabolitleri M-2 ve M-5'in kararlı durum maruziyeti hafif ve orta derecede böbrek yetmezliği olan hastalar ve böbrek fonksiyonları normal olan hastalarda benzerdir.
Şiddetli böbrek yetmezliği olan hastalar ile normal böbrek fonksiyonu olan hastalar karşılaştırıldığında, regorafenib maruziyeti benzerken, klinik olarak anlamlı kabul edilmeyen kararlı durum koşullarında M-2 ve M-5 maruziyeti yaklaşık %30 oranında azalmıştır.
Regorafenibin farmakokinetiği son evre böbrek yetersizliği olan hastalarda araştırılmamıştır. Bununla birlikte, fizyoloji bazlı farmakokinetik modelleme, bu hastalarda maruziyette görülebilecek herhangi bir anlamlı değişikliği öngörmemektedir.
Karaciğer yetmezliği olan hastalar:
Regorafenibin ve metabolitleri olan M-2 ve M-5'in maruziyeti, hafif derecede karaciğer yetmezliği olan (Child-Pugh A) hastalarda ve karaciğer fonksiyonları normal olan hastalarda benzerdir. 100 mg tek doz regorafenib uygulanmasından sonra orta derecede karaciğer yetmezliği (Child-Pugh B) olan hastalardaki maruziyetinin, karaciğer fonksiyonları normal olan hastalardaki farmakokinetiği ile benzer olduğuna ilişkin sınırlı veri bulunmaktadır. Child-Pugh C (şiddetli) karaciğer yetmezliği olan hastalara ilişkin bilgi bulunmamaktadır. Regorafenib başlıca karaciğer yoluyla atılmaktadır ve bu hasta popülasyonunda maruziyet artabilmektedir.
Geriyatrik hastalar:
Regorafenibin farmakokinetiği, çalışılan yaş aralığında (29 – 85 yaş), yaştan etkilenmemiştir.
Cinsiyet:
Regorafenibin farmakokinetiği cinsiyetten etkilenmemektedir.
Etnik farklılıklar:
Farklı Asya popülasyonlarındaki (Çinli, Japon, Koreli) regorafenib maruziyeti beyaz ırkta görülen ile aynı aralıktadır.
Kardiyak Elektrofizyoloji/QT uzaması:
Erkek ve kadın kanser hastalarında yapılan özel bir QT çalışmasında, kararlı durumda 160
5.3. Klinik öncesi güvenlilik verileri
Sistemik toksisite
Farelere, sıçanlara ve köpeklere tekrarlanan doz uygulanmasından sonra esas olarak böbreklerde, karaciğerde, sindirim sisteminde, kalpte, tiroid bezinde, kan ve lenf sisteminde, endokrin sistemde, üreme sisteminde ve deride olmak üzere bir dizi organda advers etkiler gözlenmiştir. Sıçanlarda 26 haftalık tekrar dozlu toksisite çalışmasında kalpte atrioventriküler damarlarda kalınlaşma insidansında hafif artış görülmüştür. Nedeni yaşa bağlı fizyolojik proseslerin hızlanması olabilir. Bu etkiler, öngörülen insan maruziyeti aralığında veya altındaki sistemik maruziyetlerde meydana gelmiştir (EAA karşılaştırmasına dayalı olarak).
Genç ve büyüme dönemindeki hayvanların ve yavru sıçanların dişlerinde ve kemiklerinde değişiklikler ve üreme sistemindeki advers etkiler daha belirgin olup, bu durum çocuklar ve ergenler için potansiyel bir riske işaret etmektedir.
Genotoksisite ve karsinojenisite
Regorafenibin karsinojenik potansiyeline ilişkin çalışma yürütülmemiştir.
Fareler üzerinde yürütülen in vitro ve in vivo standart analizlerde, regorafenibin genotoksik potansiyeline ilişkin bulgu gözlenmemiştir.
Üreme ve gelişim toksisitesi
Fertiliteye özgü çalışmalar yürütülmemiştir. Bununla birlikte, sıçanlarda ve köpeklerde öngörülen insan maruziyeti altındaki maruziyetlerde (EAA karşılaştırmasına dayalı olarak) tekrarlanan doz uygulamasını takiben testislerde, overlerde ve uterusta gözlenen morfolojik değişikliklere dayalı olarak regorafenibin erkek ve dişi üreme sistemini advers olarak etkileme potansiyelinin olduğu dikkate alınmalıdır. Gözlenen değişiklikler sadece kısmen geri dönüşümlüdür.
Tavşanlarda, öngörülen insan maruziyetinin altındaki maruziyetlerde (EAA karşılaştırmasına dayalı olarak) regorafenibin intrauterin gelişim üzerindeki etkisi gösterilmiştir. Başlıca bulgular üriner sistem, kalp ve majör damarlar ve iskelette malformasyonları içermiştir.
Çevresel Risk Değerlendirme (ÇRD)
Çevresel risk değerlendirme çalışmaları, regorafenibin çevreye karşı kalıcı, biyoakümülatif ve toksik olma potansiyeline sahip olduğunu; yerüstü suyu ve çökelti kısımlarına risk oluşturabildiğini göstermiştir.
6. FARMASÖTİK ÖZELLİKLER
6.1. Yardımcı maddelerin listesi
Mikrokristalin selüloz Kroskarmelloz sodyum Magnezyum stearatPovidon
Silika, kolloidal susuz Kırmızı demir oksit – E172 Sarı demir oksit – E172 Lesitin (soyadan elde edilir) Makrogol
Polivinil alkol, kısmen hidrolize
Talk
Titanyum dioksit – E171
6.2. Geçimsizlikler
Bilinen herhangi bir geçimsizliği bulunmamaktadır.
6.3. Raf ömrü
36 ay
Şişe ilk açıldıktan sonra sıkıca kapatılmalıdır. Şişe bir kez açıldıktan sonra tıbbi ürünün nem tutucu yokluğunda dahi 7 hafta (49 gün) stabil olduğu gösterilmiştir. Ardından tıbbi ürün imha edilmelidir.
6.4. Saklamaya yönelik özel tedbirler
30°C'nin altındaki oda sıcaklığında saklayınız. Nemden korumak için orijinal ambalajında saklayınız. Nem tutucu kapsülü şişeden çıkarmayınız.
6.5. Ambalajın niteliği ve içeriği
STIVARGA karton kutulara yerleştirilen ve her birinde 28 tablet ve 1 adet nem tutucu kapsül içeren çocuk emniyetli, sızdırmaz polipropilen kapaklı, beyaz opak 3 adet HDPE şişede ambalajlanmaktadır (bir kutuda toplam 28 tablet x 3 şişe = 84 tablet).
6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler
Üzerindeki talimatlara uygun şekilde kapağı bastırarak sola doğru çeviriniz. Şişe ilk açıldıktan sonra sıkıca kapatılmalıdır. Nem tutucu kapsül yutulmamalıdır.
Bu tıbbi ürün, çevreye karşı risk oluşturabilmektedir (bkz. Bölüm 5.3).
Regorafenibin kullanımı, yerüstü suyu ve çökelti kısımlarında risk ile sonuçlanabilmektedir.
Bu nedenle STIVARGA, atık sular (kanalizasyon) veya ev atıkları ile birlikte atılmamalıdır.
Kullanılmamış olan ürünler ya da atık materyaller “Tıbbi Atıkların Kontrolü Yönetmeliği†ve “Ambalaj Atıklarının Kontrolü Yönetmelikâ€lerine uygun olarak imha edilmelidir.
 HIV ve Aids
HIV, Human Immunodeficiency Virus’dür (İnsanlarda Bağışıklık Sistemini Bozan
Virüsdür). Bu virüs AIDS hastalığına sebep olur.
HIV ve Aids
HIV, Human Immunodeficiency Virus’dür (İnsanlarda Bağışıklık Sistemini Bozan
Virüsdür). Bu virüs AIDS hastalığına sebep olur. |
 Artrit
Artrit, oldukça yaygın bir hastalıktır ancak iyi anlaşılamamıştır. Aslında “artrit” tek bir hastalığın adı değildir; eklem ağrısı veya eklem hastalıklarını adlandırmanın gayri resmi yoludur.
Artrit
Artrit, oldukça yaygın bir hastalıktır ancak iyi anlaşılamamıştır. Aslında “artrit” tek bir hastalığın adı değildir; eklem ağrısı veya eklem hastalıklarını adlandırmanın gayri resmi yoludur. |
 |
Tiroid Kanseri En sık görülen tiroid kanseri türü olan papiller tiroid kanseri, tüm tiroid kanserlerinin yaklaşık %70'ini oluşturur. |
 |
Kolon, Rektum yada Bağırsak Kanseri Bağırsak kanseri kolon veya rektumda (arka geçit) herhangi bir bölgede ortaya çıkabilir.Kolorektal kanser erken safhalarda teşhis edilmesi halinde daha kolay ve daha başarılı bir şekilde tedavi edilir. |
 |
Mide Kanseri Mide kanseri genellikle mideyi tümüyle kaplayan ve mukus üretmekle görevli hücrelerde başlar. Bu kanser tipine adenokarsinom denir. |
İLAÇ GENEL BİLGİLERİ
Bayer Türk Kimya San. Tic. Ltd. Şti.
| Geri Ödeme Kodu | A15618 |
| Satış Fiyatı | 55250.21 TL [ 24 Mar 2025 ] |
| Önceki Satış Fiyatı | 55250.21 TL [ 17 Mar 2025 ] |
| Original / Jenerik | Original İlaç |
| Reçete Durumu | Normal Reçeteli bir ilaçdır. |
| Barkodu | 8699546090303 |
| Etkin Madde | Regorafenib |
| ATC Kodu | L01EX05 |
| Birim Miktar | 40 |
| Birim Cinsi | MG |
| Ambalaj Miktarı | 84 |
| Antineoplastik ve İmmünomodülatör Ajanlar |
| İthal ( ref. ülke : Fransa ) ve Beşeri bir ilaçdır. |