SUTENT 37.5 mg kaps�l (28 kaps�l) K�sa �r�n Bilgisi
{ Sunitinib Maleat }
1. BE�ER� TIBB� �R�N�N ADI
SUTENT® 37,5 mg Kaps�l Sitotoksik
2. KAL�TAT�F VE KANT�TAT�F B�LE��M
Etkin madde
Her bir kaps�l 37,5 mg sunitinibe e�de�er 50,1 mg sunitinib malat i�erir.
Yard�mc� maddeler
Yard�mc� maddeler i�in 6.1'e bak�n�z.
3. FARMAS�T�K FORMU
Sert jelatin kaps�l
�ki par�al�, opak bask�l�, standart sar�, sert jelatin boyut 3 kaps�ller, sar� ile turuncu aras� renkte gran�l i�erir.
4. KL�N�K �ZELL�KLER
4.1. Terap�tik endikasyonlar
Gastrointestinal Stromal T�m�r (G�ST)
SUTENT imatinib mesilat tedavisine diren�li veya intoleran anrezektabl ve/veya
metastatik gastrointestinal stromal t�m�rlerin (G�ST) tedavisinde endikedir.
Metastatik Renal H�creli Karsinom (mRHK)
SUTENT ilerlemi� ve/veya metastatik renal h�creli karsinom (mRHK) tedavisinde endikedir.
Pankreatik N�roendokrin T�m�r (pNET)
SUTENT, metastatik veya lokal ileri evrede olup cerrahi tedavisi m�mk�n olmayan, somatostatin analoglar� tedavisi sonu progresyon geli�en iyi differansiye pankreatik n�roendokrin t�m�rlerin (pNET) tedavisinde endikedir.
4.2. Pozoloji ve uygulama �ekli
Pozoloji/uygulama s�kl��� ve s�resi:
Tedavi kanser ila�lar�n� uygulama konusunda tecr�beli bir hekim taraf�ndan ba�lat�lmal�d�r.
G�ST ve mRHK i�in; �nerilen SUTENT dozu 4 hafta kesintisiz g�nde 1 defa 50 mg a��zdan al�narak ve daha sonra 2 hafta ara vermek suretiyle 6 haftal�k k�r� tamamlayacak �ekildedir.
pNET i�in SUTENT'in �nerilen dozu; planl� bir ara verme d�nemi olmaks�z�n g�nde bir
kez oral yolla 37,5 mg'd�r.
G�ST ve mRHK i�in, 12,5 mg'l�k art�� veya azaltmalarla doz modifikasyonlar� bireysel g�venlilik ve tolerabiliteye ba�l� olarak uygulanabilir. G�nl�k dozlar 25 mg'�n alt�na d��memeli ve 75 mg'� ge�memelidir.
Bireysel g�venlilik ve tolere edilebilirlik g�z �n�ne al�narak, pNET i�in 12,5 mg'l�k ad�mlarla doz ayarlamas� yap�labilir. Faz III pNET �al��mas�nda uygulanan maksimum doz g�nde 50 mg olmu�tur.
Bireysel g�venlilik ve tolerabiliteye ba�l� olarak doza ara vermek gerekebilir.
Rifampisin gibi kuvvetli CYP3A4 ind�kleyici ile birlikte SUTENT kullan�m�ndan sak�n�lmal�d�r. E�er kullan�lmas� gerekiyorsa, kullanan hastalarda dozun 12,5 mg'l�k miktarlarla (G�ST ve mRHK i�in g�nde 87,5 mg'a kadar veya pNET i�in g�nde 62,5 mg'a kadar) art�r�lmas� gerekebilir. Klinik yan�t ve tolerabilite dikkatle izlenmelidir. Ketokonazol gibi CYP3A4 inhibit�r� ile birlikte SUTENT kullan�m�ndan sak�n�lmal�d�r. E�er birlikte kullan�lmas� mutlaka laz�msa kullanan hastalarda tolerabilite ve/veya klinik yan�ta ba�l� olarak SUTENT dozu 12,5 mg'l�k kademelerle G�ST ve mRHK i�in g�nl�k minimum 37,5 mg'a veya pNET i�in g�nde 25 mg'a d���r�lebilir. E�zamanl� ila� se�iminde CYP3A4'� ind�kleyici veya inhibe edici potansiyeli �ok d���k olan veya hi� olmayan alternatif ila�lar d���n�lmelidir. (Bkz. B�l�m 4.4)
Uygulama �ekli:
SUTENT, a� karn�na veya yemekle beraber, oral olarak al�nabilir. Bir doz atland�ysa hastaya ilave doz verilmemelidir. Hasta bir sonraki g�n �nerilen normal doz ile devam etmelidir.
�zel pop�lasyonlara ili�kin ek bilgiler:
B�brek/Karaci�er yetmezli�i:
Hafif (Child-Pugh S�n�f A) ve orta (Child-Pugh S�n�f B) karaci�er yetmezli�i olan hastalarda ba�lang�� dozunun ayarlamas�na gerek yoktur. Ciddi (Child-Pugh S�n�f C) karaci�er yetersizli�i olan hastalarda �al��ma yap�lmam��t�r. Bu nedenle �iddetli karaci�er yetmezli�i olan hastalarda sunitinib kullan�m� ile ilgili bir �neride bulunulamamaktad�r. (Bkz. B�l�m 5.2)
Orta-ciddi b�brek yetmezli�i olan hastalarda veya hemodiyalize giren son d�nem b�brek yetmezli�i hastalar�na sunitinib verilirken ba�lang�� doz ayarlamas� gerekli de�ildir. Takip eden dozlar g�venlilik ve tolerabiliteye g�re ayarlanmal�d�r. (Bkz. B�l�m 5.2)
Pediyatrik pop�lasyon:
SUTENT'in 18 ya��n alt�ndaki hastalardaki g�venlilik ve etkilili�i de�erlendirilmemi�tir. �u an var olan veriler B�l�m 4.8, 5.1 ve 5.2'de anlat�lm��t�r, fakat bu pop�lasyonda SUTENT kullan�m� �nerilmemektedir.
Geriyatrik pop�lasyon:
SUTENT'e ait klinik �al��malardaki hastalar�n yakla��k %34'�n�n ya�� 65 veya �zeridir. Gen� ve ya�l� hastalar aras�nda g�venlilik ve etkililik a��s�ndan herhangi bir anlaml� fark g�zlenmemi�tir.
4.3. Kontrendikasyonlar
Sunitinib malat veya SUTENT kaps�lleri bile�enlerinden herhangi birine a��r� duyarl�l��� olan hastalarda kontrendikedir (Bkz. B�l�m 6.1).
4.4. �zel kullan�m uyar�lar� ve �nlemleri
Potent CYP3A4 ind�kleyicileri sunitinibin plazma konsantrasyonunu azaltabilece�inden
birlikte kullan�m�ndan ka��n�lmal�d�r (Bkz. B�l�m 4.2 ve 4.5).
Cilt ve doku bozukluklar�
Hastalar ayn� zamanda SUTENT ile tedavi boyunca sa� veya ciltte depigmentasyon olabilece�i konusunda uyar�lmal�d�r. Ciltte kuruluk, kal�nl�k veya �atlama, ayak tabanlar� veya avu� i�lerinde nadiren k�zar�kl�k veya kabarc�klar di�er olas� dermatolojik etkiler aras�nda say�labilir.
Yukar�da ad� ge�en olaylar k�m�latif (birikimli) de�ildir; tipik olarak reversibl olup genellikle tedaviye son vermeyi gerektirmemi�tir. Piyoderma gangrenosum (genellikle sunitinib kullan�m� b�rak�ld�ktan sonra geri d�n��l� olan a�r�l� deri �lseri) vakalar� bildirilmi�tir. Baz�lar� �l�mle sonu�lanm�� eritema multiforme (EM), Steven-Johnson sendromu (SJS) ve toksik epidermal nekroliz (TEN) dahil ciddi kutan�z reaksiyonlar bildirilmi�tir. SJS, TEN veya EM belirtileri veya semptomlar� (�rn. genellikle su toplamas� ve mukozal lezyonlar ile seyreden ilerlemi� deri d�k�nt�leri) mevcutsa, sunitinib tedavisi sonland�r�lmal�d�r. SJS veya TEN te�hisi do�rulanm��sa, tedaviye tekrar ba�lanmamal�d�r. EM'den ��phelenilen baz� olgularda, reaksiyonun sona ermesinin ard�ndan daha d���k dozda sunitinib tedavisine yeniden ba�lanmas� hastalar taraf�ndan tolere edilmi�tir. Bu hastalardan baz�lar� e� zamanl� olarak kortikosteroid ve antihistaminik tedavisi alm��t�r (Bkz. B�l�m 4.8).
Hemoraji ve t�m�rlerin kanamas�
SUTENT ile yap�lan klinik �al��malarda ve pazarlama sonras� deneyimlerde, gastrointestinal sistem, solunum, �riner sistem ve beyin hemorajisi gibi baz�lar� �l�mc�l olabilecek hemorajik olaylar bildirilmi�tir (Bkz. B�l�m 4.8).
Kanama olaylar�n�n rutin de�erlendirmesi, tam kan say�m� ve fizik muayene ile
yap�lmal�d�r.
Hemorajik olaylar ya�am�� solid t�m�rl� hastalar�n, yakla��k yar�s�nda en �ok g�r�len hemorajik advers olay burun kanamas�d�r. Burun kanamas� olaylar�ndan baz�lar� ciddi olup, �ok seyrek olarak �l�me yol a�m��t�r.
T�m�rlerin kanama olaylar� bazen t�m�r nekrozuna e�lik eder, bu kanamalar fatal olabilir.
T�m�r hemorajisi aniden olu�abilir ve akci�er t�m�rleri olgular�nda, ciddi ve hayat� tehdit edici hemoptizi veya akci�er kanamas� olarak g�r�lebilir. mRHK, akci�er kanseri ve G�ST tedavisi i�in SUTENT kullanan hastalarda pazarlama sonras� deneyim olarak akci�er kanamas� (baz�lar� �l�mle sonu�lanm��t�r) olu�mu� ve bu durum klinik �al��malarda da g�zlemlenmi�tir. SUTENT, akci�er kanseri olan hastalarda kullan�m i�in onayl� de�ildir.
E� zamanl� olarak antikoag�lan (�rn.; varfarin, asenokumarol) tedavisi alan hastalar tam kan say�m� (trombositler), koag�lan fakt�rler (PT/INR) ve fiziksel muayene ile periyodik olarak kontrol edilmelidir.
Gastrointestinal olaylar
Diyare, mide bulant�s�/kusma, kar�n a�r�s�, dispepsi ve stomatit en �ok rapor edilen
gastrointestinal yan etkilerdir. Ayr�ca �zofajit de rapor edilmi�tir (Bkz. B�l�m 4.8).
Tedavi gerektiren gastrointestinal advers olaylar i�in destekleyici bak�m, antiemetik, antasit veya antidiyareik ila� tedavisi ile sa�lanabilir.
SUTENT ile tedavi edilen, intra-abdominal maligniteleri olan hastalarda gastrointestinal perforasyonu kapsayabilen, ciddi bazen �l�mc�l olabilen gastrointestinal komplikasyonlar olu�mu�tur.
Hipertansiyon
Ciddi hipertansiyon (>200 mmHg sistolik veya 110 mmHg diyastolik) dahil sunitinib ile ili�kili olarak hipertansiyon bildirilmi�tir. Hastalar hipertansiyon i�in taranmal� ve uygun olduk�a kontrol edilmelidir. �la� m�dahalesiyle kontrol edilemeyen a��r hipertansiyonlu hastalarda ge�ici olarak ila� tedavisinin durdurulmas� �nerilir. Hipertansiyon uygun olarak kontrol alt�na al�nd���nda tedaviye yeniden ba�lanabilir (Bkz. B�l�m 4.8).
Hematolojik bozukluklar
Sunitinib ile ili�kili olarak azalm�� mutlak n�trofil say�lar� ve azalm�� trombosit say�s� bildirilmi�tir (Bkz. B�l�m 4.8). Yukar�da ad� ge�en olaylar k�m�latif (birikimli) de�il; tipik olarak reversibl olup genellikle tedaviye son vermeyi gerektirmemi�tir. Faz III �al��malardaki bu olaylar�n hi�biri �l�mc�l olmamakla birlikte pazarlama sonras� deneyimlerde trombositopeni ve n�tropenik enfeksiyonlar�n e�lik etti�i hemorajili durumlarda seyrek olarak �l�m olgular� rapor edilmi�tir.
Sunitinib ile tedavi esnas�nda erken ve ge� d�nemde anemi g�zlenmi�tir.
SUTENT ile tedavi g�ren hastalarda, her tedavi k�r�n�n ba�lang�c�nda tam kan say�m� yap�lmal�d�r (Bkz. B�l�m 4.8).
Kardiyak bozukluklar
SUTENT kullanan hastalarda baz�lar� �l�mle sonu�lanan, kalp yetmezli�i, kardiyomiyopati, sol ventrik�l ejeksiyon fraksiyonunun normalin alt s�n�r�n�n alt�na d��mesi, miyokardit ve miyokardiyal iskemi ve miyokard enfarkt�s�n�n de dahil oldu�u kardiyovask�ler olaylar rapor edilmi�tir. Bu veriler sunitinibin kardiyomiyopati riskini artt�rd���n� g�stermektedir.�laca�zg�etkid���nda sunitinib ile ind�klenen
kardiyomiyopati i�in tedavi g�ren hastalarda ilave risk fakt�rleri tespit edilmemi�tir. Bu olaylar a��s�ndan risk ta��yan veya kardiyovask�ler hikayesi olan hastalarda dikkatli kullan�lmal�d�r (Bkz. B�l�m 4.8).
SUTENT uygulamas�ndan �nce 12 ay i�inde miyokard enfarkt�s� (ciddi/stabil olmayan anjinay� kapsayan), koroner/periferik arter by-pass grafti, semptomatik KKY, serebrovask�ler olay veya ge�ici iskemik atak veya akci�er embolisi gibi kardiyak bulgular� olan hastalar, SUTENT klinik �al��malar�na dahil edilmemi�tir. Bunun gibi e� zamanl� rahats�zl�klar� olan hastalar�n sunitinible ili�kili sol ventrik�l i�lev bozuklu�u geli�imi i�in daha y�ksek bir riskte olup olmad�klar� bilinmemektedir.
Doktorlara SUTENT'in risk/yarar oran�n� g�z �n�nde bulundurmalar� tavsiye edilir. Kardiyak risk fakt�r� olan ve/veya koroner arter hastal��� hikayesi olan hastalar SUTENT al�rken, KKY'nin klinik belirtileri ve semptomlar� i�in dikkatli olarak g�zlenmelidirler. Hasta sunitinib al�rken, sol ventrik�l ejeksiyon fraksiyonunda (SVEF) i�in ba�lang�� ve periyodik de�erlendirmeleri de d���n�lmelidir. Kardiyak risk fakt�rleri olmayan hastalarda, ejeksiyon fraksiyonunun ba�lang�� de�eri �l��lmelidir.
KKY'nin klinik belirtilerinin varl���nda, sunitinibin kesilmesi �nerilir. Sunitinib dozu, klinik KKY bulgusu olmayan ancak, SVEF<%50 ve ba�lang�ca g�re >%20 d���� g�stermi� hastalarda kesilmeli ve /veya azalt�lmal�d�r.
QT aral��� uzamas�
Sunitinib'e maruz kalan hastalarda QT aral��� uzamas� ve Torsade de pointes g�zlemlenmi�tir. QT aral��� uzamas� Torsade de pointes de dahil olmak �zere ventrik�ler aritmilerde artm�� bir riske yol a�abilir.
Sunitinib; bilinen QT aral��� uzama �yk�s�, antiaritmik ya da QT aral���n� uzatabilen bir ila� alan veya daha �nceden bilinen �nemli kardiyak hastal���, bradikardi veya elektrolit bozuklu�u olan hastalarda dikkatle kullan�lmal�d�r. G��l� CYP3A4 inhibit�rleri ile e� zamanl� tedavi, sunitinibin plazma konsantrasyonunu y�kseltebilece�i i�in, dikkatli kullan�lmal�d�r (Bkz. B�l�m 4.2, 4.5 ve 4.8).
Ven�z tromboembolik olaylar
Derin ven trombozu ve pulmoner emboli de dahil olmak �zere sunitinib alan hastalarda tedaviye ba�l� ven�z tromboembolik olaylar bildirilmi�tir (Bkz. B�l�m 4.8). Pazarlama sonras� deneyimlerde �l�mc�l sonu� veren pulmoner emboli vakalar� g�zlenmi�tir.
Arteriyel tromboembolik olaylar
Sunitinib ile tedavi edilen hastalarda baz�lar� �l�mc�l olan arteriyel tromboembolik (ATE) olaylar raporlanm��t�r. En s�k g�zlenen olaylar serebrovask�ler olaylar, ge�ici iskemik atak ve serebral enfarkt�s �eklindeydi. Altta yatan malign hastal�k ve ya��n 65 veya �zerinde olmas�na ek olarak, hipertansiyon, diyabet ve daha �nceki tromboembolik hastal�k ATE ile ili�kili risk fakt�rleri aras�nda yer al�r.
Anevrizmalar ve arter diseksiyonlar�
Vask�ler endotelyal b�y�me fakt�r (VEGF) yolak inhibit�rlerinin, hipertansiyonu olan veya olmayan hastalarda kullan�lmas�, anevrizmalar ve/veya arter diseksiyonlar� olu�umunu kolayla�t�rabilir. SUTENT'e ba�lamadan �nce hipertansiyon veya anevrizma �yk�s� gibi risk fakt�rleri olan hastalarda bu risk dikkatle de�erlendirilmelidir.
Trombotik Mikroanjiyopati (TMA)
Trombotik trombositopenik purpura (TTP) ve hemolitik uremik sendorumu (HUS) dahil olmak �zere, bazen b�brek yetmezli�ine veya �l�mc�l bir sonuca yol a�an, hemolitik anemi, trombositopeni, yorgunluk, n�rolojik belirtilerde dalgalanma, b�brek yetmezli�i ve ate� g�r�lmesi durumunda TMA tan�s� d���n�lmelidir. TMA geli�en hastalarda sunitinib tedavisi durdurulmal� ve acilen tedaviye ba�lanmal�d�r. Sunitinib tedavisinin b�rak�lmas�ndan sonra TMA etkilerinin d�zeldi�i g�zlemlenmi�tir (Bkz. B�l�m 4.8).
Tiroid disfonksiyonu
Tiroid fonksiyonlar�n�n ba�lang��ta laboratuvar �l��mleri t�m hastalara �nerilir ve hipotiroidizmli veya hipertiroidizmli hastalar, sunitinib tedavisi ba�lat�lmadan �nce standart t�bbi uygulama ile tedavi edilmelidirler. Sunitinib tedavisi s�ras�nda her 3 ayda bir tiroid fonksiyonlar� rutin olarak izlenmelidir. Ek olarak, sunitinib tedavisindeki b�t�n hastalar, tiroid disfonksiyonun belirtileri ve semptomlar� i�in yak�ndan g�zlenmelidirler. Tiroid disfonksiyonuna dair belirtileri ve/veya semptomlar� olan hastalarda tiroid fonksiyonunun laboratuvar takibi yap�lmal� ve standart t�bbi tedavi uygulanmal�d�r.
Sunitinib ile tedavi edilen hastalarda erken veya ge� olarak hipotirodizm g�zlenmi�tir. (Bkz. B�l�m 4.8)
Pankreatit
SUTENT kullanan �e�itli solid t�m�rl� hastalarda serum lipaz� ve amilaz�nda art��lar g�r�lm��t�r. �e�itli solid t�m�rl� hastalarda lipaz seviyelerindeki art��lar ge�ici olmu� ve genellikle pankreatite ait semptomlar veya belirtilere e�lik etmemi�lerdir (Bkz. B�l�m 4.8).
Baz�lar� �l�mc�l olabilen a��r pankreatit olgular� bildirilmi�tir. E�er pankreatit bulgular� varsa sunitinib kesilmeli ve uygun destekleyici tedavi yap�lmal�d�r.
Hepatotoksisite
Sunitinib tedavisi g�ren hastalarda hepatotoksisite g�zlemlenmi�tir. Sunitinib tedavisi g�ren solid t�m�rl� hastalar�n %1'inden az�nda, baz�lar� �l�mle sonu�lanan, karaci�er yetmezli�i olgular� g�r�lm��t�r. Tedaviye ba�lanmadan, her tedavi siklusunda ve klinik olarak endike oldu�u durumlarda karaci�er fonksiyon testleri (alanin transaminaz [ALT], aspartat transaminaz [AST], bilirubin seviyeleri) takip edilmelidir. E�er karaci�er yetmezli�i semptomlar� veya belirtileri mevcutsa, sunitinib tedavisi durdurulmal� ve durum d�zelmediyse tedaviye devam edilmemeli ve uygun destekleyici tedavi yap�lmal�d�r (Bkz. B�l�m 4.8).
B�brek fonksiyonu
B�brek bozukluklar�, b�brek yetmezli�i ve/veya akut b�brek yetmezli�i olan hastalarda, baz�lar� �l�mc�l olan olgular bildirilmi�tir (Bkz. B�l�m 4.8).
Sunitinib alan hastalardaki b�brek bozuklu�u/yetmezli�i ile ili�kili risk fakt�rleri aras�nda, altta yatan renal h�creli karsinoma ek olarak, ilerlemi� ya�, diyabet, altta yatan b�brek i�lev bozuklu�u, kalp yetmezli�i, hipertansiyon, sepsis, dehidratasyon/hipovolemi ve rabdomiyoliz yer al�r.
Orta ve ileri derecede protein�risi olan hastalarda devam eden sunitinib tedavisinin g�venlili�i sistematik olarak de�erlendirilmemi�tir.
Protein�ri ve nadiren nefrotik sendrom olgular� raporlanm��t�r. Ba�lang��ta idrar tahlili yap�lmas� �nerilir ve hastalarda protein�rinin geli�imi ya da k�t�le�mesi takip edilmelidir. Nefrotik sendromlu hastalarda sunitinib kesilmelidir.
Fist�l
E�er fist�l olu�umu g�zlenirse, sunitinib tedavisi hemen kesilmelidir. Fist�le sahip hastalarda sunitinib kullan�m�na devam edilmesi ile ilgili s�n�rl� bilgi mevcuttur (Bkz. B�l�m 4.8).
Yara iyile�mesinde gecikme
SUTENT tedavisi s�resince yara iyile�mesinde gecikme olgular� rapor edilmi�tir.
Sunitinibin yara iyile�mesi �zerindeki etkisine dair resmi klinik �al��ma yap�lmam��t�r. Maj�r cerrahi giri�im ge�irecek olan hastalarda sunitinib tedavisine ge�ici olarak ara verilmesi �nerilir. Maj�r cerrahi m�dahaleden ne kadar sonra sunitinib tedavisine yeniden ba�lanaca�� konusunda s�n�rl� klinik deneyim mevcuttur. Bu nedenle, maj�r bir cerrahi m�dahaleyi takiben sunitinib tedavisine yeniden devam etme, operasyon sonras� iyile�meye ba�l� klinik de�erlendirmeyle kararla�t�r�l�r.
�enede osteonekroz
SUTENT ile tedavi edilen kanser hastalar�nda �ene osteonekrozu olgular� bildirilmi�tir. Olgular�n �o�u daha �nceden veya e� zamanl� olarak i.v. bifosfonat tedavisi alan hastalarda bildirilmi� olup bu durum �ene osteonekrozu i�in belirlenmi� bir risk fakt�r�d�r. SUTENT ve i.v. bifosfonatlar ayn� anda veya ard� ard�na kullan�ld���nda dikkatli olunmal�d�r.
�nvaziv dental giri�imler de tan�mlanm�� risk fakt�r�d�r. SUTENT ile tedaviye ba�lamadan �nce dental muayene ve preventif dental i�lemler �zerinde d���n�lmelidir. �nceden veya hali haz�rda i.v. bifosfonat alan hastalarda m�mk�nse invaziv dental prosed�rlerden ka��n�lmal�d�r (Bkz. B�l�m 4.8).
Hipersensitivite/Anjiyo�dem
E�er hipersensitivite nedeniyle anjiyo�dem olu�ursa, sunitinib tedavisi kesilmeli ve standart t�bbi bir bak�m yap�lmal�d�r (Bkz. B�l�m 4.8).
N�betler
Sunitinib klinik �al��malar�nda ve pazarlama sonras� deneyimlerde n�betler g�zlenmi�tir. Hipertansiyon, ba� a�r�s�, uyar�labilirlikte azalma, mental fonksiyonlarda de�i�iklik ve kortikal k�rl��� kapsayan g�rme kayb� gibi geri d�n��l� l�koensefalopati sendromunu (RPLS) ile tutarl� n�betleri ve bulgular�/semptomlar� olan hastalar, hipertansiyonun kontrol alt�na al�nmas� dahil t�bbi m�dahale ile kontrol edilmelidirler. Sunitinibin ge�ici olarak durdurulmas� �nerilir; d�zelmeyi takiben, tedavi eden doktorun karar� ile tedavi devam ettirilebilir (Bkz. B�l�m 4.8).
T�m�r Lizis Sendromu (TLS)
Baz�lar� �l�mc�l olan TLS olgular� klinik �al��malarda nadir olarak g�zlenmi�tir ve sunitinib kullanan hastalarda pazarlama sonras� deneyimlerde raporlanm��t�r. TLS i�in risk fakt�rleri aras�nda y�ksek t�m�r y�k�, �nceden var olan kronik b�brek yetmezli�i, olig�ri, dehidratasyon, hipotansiyon ve asidik idrar bulunur. Bu hastalar yak�ndan takip edilmeli ve klinik olarak belirtildi�i �ekilde tedavi edilmeli ve profilaktik hidrasyon g�z �n�nde tutulmal�d�r.
Enfeksiyonlar
N�tropeni ile birlikte veya n�tropeni olmaks�z�n baz�lar� �l�mle sonu�lanan ciddi enfeksiyonlar bildirilmi�tir. Perineum'un da dahil oldu�u bazen �l�mc�l olabilen nekrotizan fasiitis olgular� seyrek olarak rapor edilmi�tir (Bkz. B�l�m 4.8).
Sunitinib tedavisi nekrotizan fasiitis geli�mi� hastalarda kesilmeli ve uygun tedaviye
hemen ba�lanmal�d�r.
Hipoglisemi
SUTENT tedavisi s�ras�nda baz�lar� klinik olarak semptomatik olan ve bilin� kayb�ndan dolay� hastaneye kald�r�lmay� gerektiren kan �ekerinde d���� bildirilmi�tir. Semptomatik hipoglisemi durumunda SUTENT tedavisine ge�ici olarak ara verilmelidir. Diyabet hastalar�nda hipoglisemi riskini en aza indirmek i�in kullan�lan anti-diyabetik ila�lar�n dozunun ayarlanmas� gereklili�ini de�erlendirmek i�in kan glukoz seviyesi d�zenli olarak kontrol edilmelidir (Bkz. B�l�m 4.8).
Sodyum
Bu t�bbi �r�n her “doz”unda (kaps�lde) 1 mmol (23 mg)'dan daha az sodyum ihtiva eder; yani asl�nda “sodyum i�ermez”.
4.5. Di�er t�bbi �r�nler ile etkile�imler ve di�er etkile�im �ekilleri
Etkile�im �al��malar� sadece yeti�kinlerde yap�lm��t�r.
Sunitinib plazma konsantrasyonunu art�rabilen ila�lar:
CYP3A4 inhibit�rlerinin etkisi
Sa�l�kl� g�n�ll�lerde tek doz sunitinib ile potent CYP3A4 inhibit�r�, ketokonazol�n e� zamanl� uygulanmas� sunitinib+primer metabolit kompleksinin C ve EAA de�erlerini s�ras�yla %49 ve %51 art�r�r.
SUTENT'in potent CYP3A4 s�n�f�ndan di�er inhibit�rler (�rn. itrakonazol, ritonavir, greyfurt suyu, eritromisin, klaritromisin) ile birlikte uygulanmas� sunitinib konsantrasyonlar�n� art�rabilir. Bu nedenle, inhibit�rlerle birlikte uygulamadan ka��n�lmal�d�r veya CYP3A4 inhibe edici potansiyeli olmayan veya en az potansiyeli olan alternatif bir e� zamanl� ila� se�imi d���n�lmelidir. Bunun m�mk�n olmad��� durumlarda, tolere edilebilirlik dikkatli �ekilde izlenerek, SUTENT dozunun G�ST ve mRHK i�in g�nde minimum 37,5 mg'a ve pNET i�in 25 mg'a indirilmesi gerekebilir (Bkz. B�l�m 4.2).
Meme kanseri diren� proteini (BCRP) inhibit�rlerinin etkisi
Sunitinib ile BRCP inhibit�rleri aras�nda etkile�im ile ilgili s�n�rl� klinik veri bulunmaktad�r ve sunitinib ile di�er BCRP inhibit�rleri aras�nda etkile�im olas�l��� g�z ard� edilemez (Bkz. B�l�m 5.2).
Sunitinib plazma konsantrasyonunu azaltabilen ila�lar:
CYP3A4 ind�kleyicilerinin etkisi
Sa�l�kl� g�n�ll�lerde tek doz SUTENT'in CYP3A4 ind�kleyicisi, rifampisin ile e� zamanl� uygulanmas� sunitinib+primer metabolit kompleksinin C ve EAA de�erlerini s�ras�yla %23 ve %46 azaltm��t�r.
SUTENT'in potent CYP3A4 ind�kleyicileri (�rn. deksametazon, fenitoin, karbamazepin, rifampisin, fenobarbital veya sar� kantaron olarak da bilinen Hypericum perforatum/St. John's Wort) ile birlikte uygulanmas� sunitinib konsantrasyonlar�n� azaltabilir. Bu nedenle, ind�kleyicilerle birlikte uygulamas�ndan ka��n�lmal�d�r veya CYP3A4 ind�kleyici potansiyeli olmayan veya en az potansiyeli olan alternatif bir e� zamanl� ila� se�imi d���n�lmelidir. Bunun m�mk�n olmad��� durumlarda, tolere edilebilirlik dikkatli �ekilde izlenerek, SUTENT dozunun 12,5 mg'l�k art��larla artt�r�lmas� gerekebilir (G�ST ve mRHK i�in g�nde 87,5 mg'a ya da pNET i�in g�nde 62,5 mg'a kadar) (Bkz. B�l�m 4.2).
�zel pop�lasyonlara ili�kin ek bilgiler:
Pediyatrik pop�lasyon:
Bu pop�lasyonla ilgili herhangi bir etkile�im �al��mas� yap�lmam��t�r.
4.6. Gebelik ve laktasyon
Genel tavsiye
Gebelik kategorisi: D
�ocuk do�urma potansiyeli bulunan kad�nlar/Do�um kontrol� (Kontrasepsiyon) �ocuk do�urma potansiyeli bulunan kad�nlar SUTENT tedavisi s�ras�nda gebe kalmama konusunda uyar�lmal�d�r. Tedavi s�resince uygun do�um kontrol y�ntemi uygulanmal� ve gebe kalmaktan ka��n�lmal�d�r.
Gebelik d�nemi
Gebe kad�nlar �zerinde yap�lan bir �al��ma yoktur. Hayvanlarda yap�lan �al��malar, fetal malformasyonlar dahil olmak �zere �reme toksisitesi g�stermi�tir (Bkz. B�l�m 5.3). Gebelik d�neminde SUTENT kullan�l�rsa veya hasta SUTENT tedavisi s�ras�nda gebe kal�rsa; hasta, ilac�n fet�s �zerindeki potansiyel zararl� etkisi konusunda uyar�lmal�d�r.
Laktasyon d�nemi
Sunitinib ve/veya metabolitleri s��an s�t�ne ge�mektedir. Sunitinib veya onun primer aktif metabolitinin, insan s�t�ne ge�ip ge�medi�i bilinmemektedir. �la�lar�n genelde insan s�t�ne ge�mesi ve emzirilen bebekler �zerinde ciddi advers etki potansiyeli nedeniyle SUTENT tedavisi s�ras�nda emzirme durdurulmal�d�r.
�reme yetene�i/Fertilite
Klinik d��� bulgulara dayal� olarak, erkek ve di�i fertilitesi SUTENT tedavisinden etkilenebilir. (Bkz. B�l�m 5.3)
4.7. Ara� ve makine kullan�m� �zerindeki etkiler
SUTENT ara� ve makine kullan�m� yetene�i �zerinde minor etkilere sahiptir. Hastalar SUTENT ile tedavi s�ras�nda ba� d�nmesi olabilece�i konusunda uyar�lmal�d�r.
4.8. �stenmeyen etkiler
Sunitinib ile ili�kilendirilen ve baz�lar� �l�mc�l olan en �nemli ciddi advers etkiler b�brek yetmezli�i, kalp yetmezli�i, pulmoner emboli, gastrointestinal perforasyon ve hemorajidir (�rn. solunum yollar�, gastointestinal, t�m�r, idrar yollar� ve beyin hemorajileri). Herhangi bir derecedeki en yayg�n advers etkiler (RHK, G�ST ve pNET �al��malar�ndaki hastalarda g�r�len) i�tahta azalma, tat alma bozukluklar�, hipertansiyon, bitkinlik, gastrointestinal bozukluklar (�rn. diyare, bulant�, stomatit, dispepsi ve kusma), ciltte renk farkl�la�mas� ve palmar-plantar eritrodizestezi sendromudur. Bu semptomlar tedavi devam ederken hafifleyebilir. Hipotirodizm tedavi esnas�nda ortaya ��kabilir. Hematolojik rahats�zl�klar (�rn. n�tropeni, trombositopeni ve anemi) �ok yayg�n advers etkilerdendir.
B�l�m 4.4 ve 4.8'de belirtilen advers etkiler d���nda sunitinib tedavisi ile ilgili olmas� olas� olan �l�mc�l olaylar �oklu organ yetmezli�i, disemine intravask�ler koag�lasyon, peritoneal hemoraji, adrenal yetmezlik, pn�motoraks, �ok ve ani �l�md�r.
7115 ki�ilik bir veri setinden G�ST, mRHK ve pNET hastalar�nda bildirilen advers reaksiyonlar, sistem, organ, s�n�f ve s�kl�k derecesine g�re a�a��da listelenmi�tir. Klinik �al��malarda belirlenen pazarlama sonras� yan etkiler de yer almaktad�r.
�ok yayg�n (≥1/10), yayg�n (≥1/100 ila <1/10), yayg�n olmayan (≥1/1.000 ila <1/100), seyrek (≥1/10.000 ila <1/1.000), �ok seyrek (<1/10.000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor) olarak tan�mlanmaktad�r.
Enfeksiyonlar ve enfestasyonlar
Yayg�n: Viral enfeksiyonlara, solunum enfeksiyonlar�, abseler, mantar
enfeksiyonlar�, idrar yolu enfeksiyonlar�, deri enfeksiyonlar�, sepsis
Yayg�n olmayan: Nekrotizan fasiitis, bakteriyel enfeksiyonlar
Kan ve lenf sistemi hastal�klar�
�ok Yayg�n: Anemi, trombositopeni, n�tropeni, l�kopeni
Yayg�n: Lenfopeni
Yayg�n olmayan: Pansitopeni
Seyrek: Trombotik mikroanjiyopati
Ba����kl�k sistemi hastal�klar� Yayg�n olmayan: Hipersensitivite Seyrek: Anjiyo�dem
Endokrin hastal�klar�
�ok Yayg�n: Hipotiroidizm Yayg�n olmayan: Hipertiroidizm Seyrek: Tiroidit
Metabolizma ve beslenme hastal�klar�
�ok Yayg�n: ��tahta azalma
Yayg�n: Dehidratasyon, hipoglisemi
Seyrek: T�m�r lizis sendromu
Psikiyatrik hastal�klar �ok Yayg�n: Uykusuzluk Yayg�n: Depresyon
Sinir sistemi hastal�klar�
�ok Yayg�n: Sersemlik, ba� a�r�s�, tat alma bozukluklar� Yayg�n: Parestezi, periferal n�ropati, hipoestezi, hiperestezi
Yayg�n olmayan: Serebral hemoraji,serebrovask�ler olay, ge�ici iskemik atak
Seyrek: Geri d�n��l� posterior ensefalopati sendromu
G�z hastal�klar�
Yayg�n: Periorbital �dem, lakrimasyon art���, g�z kapa��nda �dem
Kardiyak hastal�klar
Yayg�n: Miyokardiyal iskemiejeksiyon fraksiyonunda azalma
Yayg�n olmayan: Miyokardiyal enfarkt�s, kalp yetmezli�i, konjestif kalp yetmezli�i, kardiyomiyopati, perikard ef�zyonu, elektrokardiyogramda QT uzamas�
Seyrek: Sol ventrik�ler yetmezlik, Torsades de pointes
Vask�ler hastal�klar
�ok Yayg�n: Hipertansiyon
Yayg�n: Derin ven trombozu, s�cak basma, y�z�n k�zarmas� Yayg�n olmayan: T�m�r hemoraji
Bilinmiyor: Anevrizmalar ve arter diseksiyonlar�
Solunum, g���s hastal�klar� ve mediastinal hastal�klar
�ok Yayg�n: Burun kanamas�, dispne, �ks�r�k
Yayg�n: Pulmoner emboli, plevral ef�zyon, hemoptizi, orofarengeal a�r�, nazal konjesyon, burunda kuruluk
Yayg�n olmayan: Pulmoner hemoraji, solunum yetmezli�i
Gastrointestinal hastal�klar
�ok Yayg�n: Stomatit, abdominal a�r�, kusma, diyare, dispepsi, mide bulant�s�, kab�zl�k
Yayg�n: Gastro-�zofageal refl� hastal���, disfaji, gastrointestinal hemoraji �zofajit,
abdominal distansiyon, abdominal rahats�zl�k, rektal hemoraji, di� eti kanamas�, a��z �lseri, proktalji, keilitis, hemoroid, glossodini, oral a�r�, gaz, a��z kurulu�u, a��zda rahats�zl�k hissi, ge�irme
Yayg�n olmayan: Gastrointestinal perforasyonpankreatit, anal fist�l, kolit
Hepato-biliyer hastal�klar
Yayg�n olmayan: Karaci�er yetmezli�i, kolesistitkaraci�er fonksiyon anormallikleri
Seyrek: Hepatit
Deri ve derialt� doku hastal�klar�
�ok Yayg�n: Ciltte renk de�i�ikli�i, palmar-plantar eritrodizestezi sendromu, d�k�nt�, sa� renginde de�i�im, cilt kurulu�u
Yayg�n: Ciltte soyulma, deri reaksiyonu, egzema, su toplamas�, eritem, sa� d�k�lmesi, akne, prurit, cilt hiperpigmentasyonu, deride lezyon, hiperkeratoz, dermatit, t�rnak bozukluklar�
Seyrek: Eritema multiforme, Stevens-Johnson sendromu, gangrenli piyodermi, toksik
epidermal nekroz
Kas-iskelet hastal�klar�, ba� dokusu ve kemik hastal�klar� �ok Yayg�n: Ekstremitelerde a�r�, eklemlerde a�r�, s�rt a�r�s� Yayg�n: Kas iskelet a�r�s�, kas g��s�zl���, miyalji, kas spazm� Yayg�n olmayan: �enede osteonekroz, fist�l
Seyrek: Rabdomiyoliz, miyopati
B�brek ve idrar yolu hastal�klar�
Yayg�n: Kromat�ri, b�brek yetmezli�i, akut b�brek yetmezli�protein�ri
Yayg�n olmayan: �drar yolunda hemoraji
Seyrek: Nefrotik sendrom
Genel bozukluklar ve uygulama b�lgesine ili�kin hastal�klar �ok Yayg�n: Mukozal inflamasyon, bitkinlik, �dem, ate� Yayg�n: G���s a�r�s�, a�r�, grip benzeri belirtiler, �rperme Yayg�n olmayan: Yara iyile�mesinde gecikme
Ara�t�rmalar
Yayg�n: Kilo kayb�, beyaz kan h�cresi say�s�nda azalma, lipaz art���, trombosit say�s�nda azalma, hemoglobin azalmas�, amilaz art���, aspartat amino transferazda art��, alanin amino transferaz art���, kanda kreatinin art���, kan bas�nc� art���, kanda �rik asit art��� Yayg�n olmayan: Kanda kreatin fosfokinaz art���, kanda tiroid stim�lan hormon (TSH) art���
*�l�mc�l olaylar� i�ermektedir
A�a��daki terimler birle�tirilmi�tir:
sendrom
uPs�riyaziform dermatit, eksfoliyatif d�k�nt�, d�k�nt�, eritremat�z d�k�nt�, folik�ler d�k�nt�, generalize d�k�nt�, mak�ler d�k�nt�, mak�lo pap�ler d�k�nt�, pap�ler d�k�nt�, pruritik d�k�nt�
Se�ilmi� advers etkilerin tan�mlar�:
Enfeksiyonlar ve enfestasyonlar
Baz� olgularda, n�tropeninin e�lik etti�i veya etmedi�i kimileri �l�mc�l olan ciddi enfeksiyonlar rapor edilmi�tir. Bazen fatal olabilen, perineumunki de dahil olmak �zere nekrotizan fasiit g�zlenmi�tir (Bkz. B�l�m 4.4).
Kan ve lenf sistemi bozukluklar�
Grade 3 ve 4 �iddetindeki olgularda azalm�� mutlak n�trofil say�s�, Faz III G�ST �al��mas�ndaki hastalar�n s�ras�yla %10 ve %1,7'sinde, Faz III mRHK �al��mas�ndaki hastalar�n %16 ve %1,6's�nda ve Faz III pNET �al��mas�ndaki hastalar�n %13 ve
%2,4'�nde bildirilmi�tir. Grade 3 ve 4 �iddetindeki olgularda azalm�� trombosit say�s�, Faz III G�ST �al��mas�ndaki hastalar�n s�ras�yla %3,7 ve %0,4'�nde, Faz III mRHK �al��mas�ndaki hastalar�n %8,2 ve %1,1'inde ve Faz III pNET �al��mas�ndaki hastalar�n
%3,7 ve %1,2'sinde bildirilmi�tir (Bkz. B�l�m 4.4).
Bir Faz III G�ST �al��mas�nda, plasebo alan hastalar�n %17'sine kar��l�k, SUTENT alan hastalar�n %18'inde kanama bulgular� olu�mu�tur. Daha �nceden tedavi almam�� mRHK hastalar�nda, interferon-α (IFN-α) alan hastalar�n %11'ine kar��l�k, SUTENT alan hastalar�n %39'u kanama bulgular� g�stermi�tir. IFN-α grubundaki hastalar�n 5'ine (%1,7) kar��, SUTENT kullananlardan 17 (%4,5) hasta Grade 3 kanama bulgusu g�stermi�tir. Sitokin-refrakter mRHK i�in SUTENT alan hastalar�n %26's�nda kanama g�zlenmi�tir. Faz III pNET �al��mas�nda plasebo kullanan hastalar�n %9,85'ine k�yasla sunitinib kullanan hastalar�n %21,7'sinde burun kanamas� d���nda kanama olaylar� g�zlenmi�tir (Bkz. B�l�m 4.4).
Klinik ara�t�rmalarda G�ST'i olan hastalar�n yakla��k %2'sinde t�m�r hemorajisi meydana gelmi�tir.
Ba����kl�k sistemi bozukluklar�
Anjiyo�demi de i�eren a��r� duyarl�l�k reaksiyonlar� raporlanm��t�r (Bkz. B�l�m 4.4).
Endokrin hastal�klar�
Daha �nceden tedavi almam�� hastalarla yap�lan bir mRHK �al��mas�nda, sunitinib kullanan hastalar�n 61'inde (%16) ve IFN-α kolunda 3 hastada (<%1) ve sitokine- refrakter hastalarla yap�lan iki mRHK �al��mas�nda hastalar�n 7'sinde (%4) advers etki olarak hipotiroidizm rapor edilmi�tir.
Ayr�ca, sitokin-refrakter mRHK hastalar�n 4 (%2) tanesinde TSH y�kselmeleri rapor edilmi�tir. Sonu� olarak, mRHK pop�lasyonunun %7'sinde, tedaviyle ortaya ��kan hipotiroidizmin klinik ve laboratuvar kan�t� vard�r. Geli�en hipotiroidizm sunitinib kullanan G�ST'i olan hastalar�n %6,2'sinde g�r�l�rken bu oran plasebo kullanan hastalarda %1'dir. Faz III pNET �al��mas�nda sunitinib alan 6 hastada (%7,2) ve plasebo alan bir hastada (%1,2) hipotiroidizm bildirilmi�tir.
Meme kanseri olan hastalardaki prospektif olarak y�r�t�len iki �al��mada tiroid fonksiyonu izlendi; SUTENT'in meme kanserinde kullan�m� onaylanmam��t�r. Bir �al��mada, sunitinib alan 15 (%13,6) hastada ve standard bak�m g�ren 3 (%2,9) hastada hipotiroidizm raporlanm��t�r. Kan TSH'sinde art��, sunitinib alan 1 (%0,9) hastada bildirilmi� ve standart bak�m g�ren kimsede rastlanmam��t�r. Hipertiroidizm, sunitinib ile tedavi edilen herhangi bir hastada raporlanmam��t�r ve standard bak�m g�ren 1 hastada (%1,0) raporlanm��t�r. Bir di�er �al��mada; hipotiroidizm, sunitinib alan toplam 31 (%13) hastada ve kapesitabin alan 2 (%0,8) hastada bildirilmi�tr. Kan TSH art���, sunitinib alan 12 (%5,0) hastada bildirilmi�tir ve kapesitabin alan hi�bir hastada g�r�lmemi�tir. Hipertiroidizm, sunitinib alan 4 (%1,7) hastada bildirildi ve kapesitabin alan hi�bir hastada bildirilmemi�tir.KanTSHd�����,sunitinib alan 3 (%1,3) hastada
bildirilmi�tir ve kapesitabin alan hi�bir hastada bildirilmemi�tir. T4 art���, sunitinib alan 2 (%0,8) hastada bildirilmi�tir ve kapesitabin alan 1 (%0,4) hastada bildirilmi�tir. T3 art���, sunitinib alan 1 (%0,8) hastada ve kapesitabin alan hi�bir hastada bildirilmemi�tir. Bildirilen t�m tiroid-ili�kili olaylar Grade 1 ve 2 ‘de olmu�tur (Bkz. B�l�m 4.4).
Metabolizma ve beslenme bozukluklar�
mRHK ve G�ST hastalar� ile kar��la�t�r�ld���nda pNET hastalar�nda daha fazla s�kl�kta hipoglisemi olaylar� rapor edilmi�tir. Yine de klinik �al��malarda g�zlemlenen bu yan etkilerin �o�unun �al��ma tedavisi ile ilgili oldu�u d���n�lmemi�tir (Bkz. B�l�m 4.4).
Sinir sistemi bozukluklar�
Sunitinib'in klinik �al��malar�nda ve pazarlama sonras� deneyimlerinde, geri d�n���ml� posterior l�koensefalopati'nin n�bet ve radyolojik kan�t� bulunan hastalarda, kimisi �l�mc�l olan az say�da (%<1) bildirim olmu�tur. Beyin metastaz�na ait radyolojik kan�t� olan veya olmayan hastalarda n�bet g�zlemlenmi�tir (Bkz. B�l�m 4.4).
Kardiyak bozukluklar
SUTENT ile tedavi g�ren G�ST'i olan hastalar�n yakla��k %2'sinde, sitokine refrakter mRHK hastalar�n�n %4'�nde ve plasebo ile tedavi g�ren hastalar�n %2'sinde, SVEF normalin en d���k s�n�r�n�n alt�nda ve %20'den daha fazla azalmalar olmu�tur. SVEF'deki bu d����ler d�zenli bir progresyon g�stermeyip, s�kl�kla tedavinin devam�nda iyile�meyle sonu�lanm��t�r. Daha �nceden tedavi almam�� mRHK �al��mas�nda, SUTENT ve IFN-α'daki hastalar�n s�ras�yla, %27'si ve %15'i, normalin alt s�n�r�n�n alt�nda bir SVEF de�eri g�stermi�tir. Sunitinib alan iki hastada (<%1) konjestif kalp yetmezli�i (KKY) te�his edilmi�tir.
G�ST hastalar�n�n %1,2'sinde ve plasebo ile tedavi g�ren hastalar�n %1'inde 'kalp yetmezli�i', 'konjestif kalp yetmezli�i veya 'sol ventrik�l yetmezli�i' gibi advers olaylar bildirilmi�tir. Pivotal Faz III G�ST �al��mas�nda (n=312) tedaviye ba�l� �l�mc�l kardiyak reaksiyonlar �al��man�n her iki kolunda (sunitinib ve plasebo) %1 oran�nda g�r�lm��t�r. Sitokine refrakter mRHK hastalar�nda yap�lan Faz II �al��mada hastalardan %0,9'unda tedaviye ba�l� �l�mc�l miyokard enfarkt�s� g�r�l�rken daha �nceden tedavi almam�� mRHK hastalar�nda yap�lan Faz III �al��mada IFN-α alan hastalar�n %0,6's�nda �l�mc�l kardiyak olaylar g�r�lm�� olup sunitinib alan hastalarda bu oran %0'd�r. Faz III pNET �al��mas�nda, sunitinib alan bir (%1) hastada tedavi ile ili�kili fatal kalp yetmezli�i meydana gelmi�tir.
Vask�ler hastal�klar
Hipertansiyon: Hipertansiyon klinik �al��malarda �ok yayg�n olarak bildirilmi�tir. Bu hasta pop�lasyonunun yakla��k %2,7'sinde SUTENT dozu azalt�lm�� veya ge�ici olarak ertelenmi�tir. Bu hastalar�n hi�birinde SUTENT ile tedaviye son verilmemi�tir. Bu hasta pop�lasyonunun yakla��k %4,7'sinde ciddi hipertansiyon (>200 mmHg sistolik veya 110 mmHg diyastolik) meydana gelmi�tir. Hipertansiyon, daha �nceden tedavi g�rmemi� mRHK i�in IFN-α alan hastalar�n %3,6's�na kar��l�k, SUTENT alan hastalar�n yakla��k
%33,9'unda rapor edilmi�tir. Ciddi hipertansiyon, �nceden tedavi edilmemi� SUTENT hastalar�n�n %12'sinde ve IFN-α alan hastalar�n %1'inden az�nda olu�mu�tur. Faz III pNET �al��mas�nda hipertansiyon plasebo alan hastalar�n %4,9'unda raporlanm��ken
sunitinib alanlar hastalardabuoran%26,5'dir.pNETolan hastalarda �iddetli
hipertansiyon, sunitinib alanlar�n %10'unda ve plasebo alanlar�n %3'�nde meydana gelmi�tir.
Ven�z tromboembolik olaylar: G�ST ve mRHK dahil olmak �zere yap�lan klinik �al��malarda sunitinib alan solid t�m�rl� hastalar�n yakla��k %1'inde tedaviye ba�l� ven�z tromboembolik olaylar raporlanm��t�r.
Bir Faz III G�ST �al��mas�nda plasebo alan herhangi bir hastada ven�z tromboembolik olay tespit edilmemesine kar��n SUTENT alan yedi hastada (%3) ven�z tromboembolik olay tespit edilmi�tir: bu yedi hastan�n be�inde, Grade 3 derin ven trombozu (DVT) ve ikisinde Grade 1 ya da 2 derin ven trombozu (DVT) geli�mi�tir. Bu yedi G�ST hastas�n�n d�rd�nde, ilk DVT incelemesini takiben tedavi kesilmi�tir.
Tedavi g�rmemi� mRHK hastalar�nda yap�lan Faz III �al��mada sunitinib alan on �� hastada (%3) ve iki sitokine refrakter mRHK �al��mas�ndaki d�rt hastada (%2) ven�z trombolik olay raporlanm��t�r. Bu hastalar�n dokuzunda pulmoner emboli mevcut olup, birinde Grade 2, di�er sekiz tanesinde ise Grade 4 derecesindedir. Bu hastalardan 8'inde birinde Grade 1, ikisinde Grade 2, d�rd�nde Grade 3 ve birinde Grade 4 derecesinde olmak �zere DVT bulunmaktad�r. Sitokine refrakter mRHK �al��mas�ndaki pulmoner embolisi mevcut olan bir hastada doz kesilmesine gerek duyulmu�tur.
IFN-α alan tedavi g�rmemi� mRHK hastalar�n�n alt�s�nda (%2), ven�z tromboembolik olaylar g�zlenmi�tir; bir hastada (<%1) Grade 3 DVT ve be� hastan�n (%1) hepsinde Grade 4 olan pulmoner emboli g�r�lm��t�r.
Faz III pNET �al��mas�nda sunitinib alan 1 hastada (%1,2) ve plasebo alan 5 hastada (%6,1) ven�z tromboembolik olaylar bildirilmi�tir. Plasebo kullanan 2 hastada biri Grade 2 biri Grade 3 olmak �zere DVT bulunmaktad�r.
G�ST, mRHK ve pNET �al��malar�n�n hi� birinde �l�mc�l bir olgu bildirilmemi�tir. �l�m ile sonu�lanan olgular pazarlama sonras� g�r�lm��t�r.
Faz III �al��malar�nda sunitinib alan G�ST hastalar�n�n yakla��k %3,1'inde ve mRHK hastalar�n�n yakla��k %1,2'sinde pulmoner emboli olgular� g�zlenmi�tir. Faz III �al��malar�nda sunitinib alan pNET hastalar�nda pulmoner emboli g�r�lmemi�tir. Pazarlama sonras� �al��malarda �l�mle sonu�lanan seyrek olgular g�zlenmi�tir.
SUTENT uygulamas�ndan �nce 12 ay i�inde pulmoner emboli ya�ayan hastalar sunitinib klinik �al��malar�na dahil edilmemi�tir.
Faz III �al��malar�nda sunitinib alan hastalardan, G�ST hastalar�n�n yakla��k
%17,8'inde, mRHK hastalar�n�n yakla��k %26,7'sinde ve pNET hastalar�n�n %12'sinde pulmoner olaylar (dispne, plevral ef�zyon, pulmoner emboli veya pulmoner �dem gibi) bildirilmi�tir.
Klinik �al��malarda sunitinib alan G�ST ve mRHK hastalar� dahil solid t�m�rl� hastalar�n yakla��k %22,2'sinde pulmoner olaylar g�zlemlenmi�tir.
Gastrointestinal bozukluklar
G�ST veya mRHK tedavisii�insunitinibalanhastalardanadiren (%1'inden az) pankreatit
g�r�lm��t�r. Faz III pNET �al��mas�nda tedavi ile ili�kili pankreatit bildirilmemi�tir (Bkz. B�l�m 4.4).
Bir Faz III G�ST �al��mas�nda plasebo alan hastalar�n %0,98'inde �l�mc�l gastrointestinal kanama g�r�lm��t�r.
Hepato-biliyer bozukluklar
Karaci�er fonksiyon testi anormalliklerini, hepatit veya karaci�er yetmezli�ini i�erebilen hepatik disfonksiyon bildirilmi�tir (Bkz. B�l�m 4.4).
Deri ve deri alt� doku hastal�klar�
�lac�n b�rak�lmas�yla geri d�n��l� olan piyoderma gangrenosum (a�r�l� deri �lseri) bildirilmi�tir (Bkz. B�l�m 4.4).
Kas-iskelet sistemi ve ba� dokusu bozukluklar�
Baz�lar� akut b�brek yetmezli�i ile birlikte olan miyopati ve/veya rabdomiyoliz olgular� rapor edilmi�tir. Kas toksisitesinin belirtileri veya semptomlar� g�r�len hastalar�n bak�mlar� standart t�bbi uygulamalar do�rultusunda yap�lmal�d�r (Bkz. B�l�m 4.4).
Baz� olgularda �l�mle sonu�lanan, bazen t�m�r nekrozu ve regresyonu ile bazen ili�kili
olan fist�l olu�umu rapor edilmi�tir (Bkz. B�l�m 4.4).
SUTENT ile tedavi edilen kanser hastalar�nda �ene osteonekrozu olgular� bildirilmi�tir; olgular�n �o�u daha �nceden veya e� zamanl� olarak i.v. bisfosfonat tedavisi alm��lard�r ve/veya invazif dental i�lemler gerektiren dental hastal�k hikayesine sahiptirler ve bu durumlar �ene osteonekrozu i�in belirlenmi� risk fakt�rleridir (Bkz. B�l�m 4.4).
Ara�t�rmalar
Klinik olmayan �al��malardan (in vitro ve in vivo ) elde edilen veriler, �nerilen insan dozunun �zerindeki dozlarda sunitinibin kalp aksiyon potansiyelinin repolarizasyon s�recini inhibe etti�ini yani QT uzamas�na neden oldu�unu ortaya koymaktad�r.
Solid t�m�rl� 450 hastan�n %1,1'inde 60 msn'den fazla s�rede ba�lang�ca k�yasla de�i�imler ve %0,5'inde ise QTc aral���nda 500 msn'yi a�an art��lar meydana gelmi�tir; bu parametrelerin her ikisi de potansiyel anlaml� de�i�iklikler olarak kabul edilmi�tir. Yakla��k olarak terap�tik konsantrasyonun iki kat�nda sunitinibin QTkF (Frederika konsantrasyonu) aral���n� uzatt��� g�sterilmi�tir.
QT aral��� uzamas�, ilerlemi� maligniteli 20-87 ya�lar� aras�nda 24 hastan�n kat�ld��� bir deney ile ara�t�r�lm��t�r. Bu �al��man�n terap�tik konsantrasyonlarda (3. g�n) baseline correction metodu kullan�larak ve terap�tik dozdan daha fazla (9. g�n) dozda her iki ba�lang�� d�zeltme metodu kullan�larak elde edilen sonu�lar� sunitinibin QT aral��� (%90 GA, �st limit >15 msn ile ortalama plasebo ayarl� de�i�iklik >10 msn olarak tan�mlanm��t�r) �zerinde etkisi oldu�unu g�stermi�tir. Hi�bir hastan�n QT aral��� >500 msn de�ildir. 3. g�nde dozdan 24 saat sonra (yani 50 mg'lik �nerilen ba�lang�� dozundan sonra beklenen terap�tik plazma konsantrasyonunda) baseline correction metodu kullan�larak QTcF aral��� �zerinde bir etki g�zlenmi� olmas�na ra�men bu bulgunun klinik olarak �nemi belirsizdir.
Terap�tik doza ya da terap�tik maruz kalmadan daha b�y�k doza kar��l�k gelen zamanlarda kapsaml� bir seri EKG de�erlendirmesi yap�ld���nda de�erlendirilebilir veya tedavisi ama�lanan hasta (ITT) grubundan hi�birinde g�r�len QT aral���nda uzama “ciddi” (yani yan etkiler i�in ortak terminoloji kriteri (CTCAE) versiyon 3.0'a g�re Grade 3'e e�it veya b�y�k) de�ildir.
Tedavi edici plazma konsantrasyonlar�nda, ba�lang�ca g�re maksimum ortalama QTcF de�i�ikli�i 9 msn (%90 GA: 15,1 msn) olmu�tur. Tedavi edici konsantrasyonlar�n yakla��k olarak iki kat�nda, ba�lang�ca g�re maksimum QTcF ortalama de�i�ikli�i 15,4 msn (%90 GA: 22,4 msn) olmu�tur. Bir pozitif kontrol olarak kullan�lan moksifloksasin (400 mg), ba�lang�ca g�re 5,6 msn'lik bir maksimum ortalama QTcF de�i�ikli�i g�stermi�tir. Herhangi bir hastada Grade 2'den (CTCAE v.3.0) daha b�y�k QTc aral��� g�zlenmemi�tir (Bkz. B�l�m 4.4).
mRHK'da uzun d�nem g�venlilik
mRHK hastalar�nda sunitinibin uzun d�nem g�venlili�i; ilk basamak; bevasizumab- refrakter ve sitokin-refrakter tedavi rejimlerinde 5739 hastaya ait verilere dayanarak (bu hastalar�n 807'si (%14) ≥2 y�ldan 6 y�la kadar tedavi g�rm��t�r) tamamlanm�� 9 klinik �al��maya dahil olan hastalar analiz edilmi�tir. Uzun d�nem sunitinib tedavisi alan 807 hastada; 6 y�ll�k periyod boyunca yeni vaka olarak ortaya ��kabilen ve zaman i�inde artan hipotiroidi hari� olmak �zere; tedavi ile ili�kili advers olaylar�n (T�AO) �o�u �ncelikle ilk 6 ay-1 y�lda olu�mu� ve sonras�nda stabil seyretmi� ya da zamanla s�kl�kta azalma g�stermi�tir. Sunitinib ile uzat�lm�� tedavi yeni T�AO tipleri ile ili�kili g�r�nmemektedir.
�zel pop�lasyonlara ili�kin ek bilgiler:
Pediyatrik pop�lasyon:
Sunitinib'in g�venlilik profili, a�a��da a��kland��� gibi bir Faz I doz artt�rma �al��mas�ndan, bir Faz II a��k etiketli �al��madan, bir Faz 1/2 tek kollu �al��madan ve yay�nlardan elde edilmi�tir.
Refrakter solid t�m�rleri olup, b�y�k b�l�m� primer beyin t�m�r� tan�s�yla kat�lan 35 hastada; 30'u pediyatrik hastalardan (3-17 ya�) ve 5 'i gen� eri�kin hastalardan (18-21 ya�) olu�an, oral sunitinibe ili�kin bir Faz I doz art�� �al��mas� ger�ekle�tirilmi�tir. �al��maya kat�lan herkeste yan etkiler g�zlemlenmi�tir; kardiyak toksisite de dahil bu yan etkilerin �o�u ciddidir (toksisite grade ≥3). En yayg�n g�r�len yan etkiler, gastrointestinal (GI) toksisite, n�tropeni, halsizlik ve ALT y�kselmesidir. Kardiyak yan etki riski daha �nceden antrasiklinlere veya kardiyak irradyasyona maruz kalm�� pediyatrik hastalarda kalmam��lara oranla daha y�ksek olmu�tur. Daha �nceden antrasiklinlere veya kardiyak irradyasyona maruz kalmas�ndan ba��ms�z olarak bu pediyatrik hastalarda maksimum tolere edilen doz (MTD) tan�mlanm��t�r (Bkz. B�l�m 5.1).
Rek�ran/progresif/refrakter ileri dereceli gliomu (HGG) veya epandimomu bulunan, 27'si pediyatrik (3-16 ya�) ve 2'si gen� eri�kin hastadan (18-19 ya�) olu�an 29 hastada, Faz II a��k etiketli bir �al��ma ger�ekle�tirilmi�tir. Her iki grupta da Grade 5 advers reaksiyon g�r�lmemi�tir. En yayg�n (≥% 10) tedaviye ba�l� advers reaksiyonlar n�trofil say�s�n�n azalmas� (6 [%20,7] hasta) ve intrakraniyal hemoraji (3 [%10,3] hasta) olmu�tur.
�leri anrezektabl G�ST'i olan 6 pediyatrik hastada (13-16 ya�) Faz 1/2 tek kollu bir �al��ma yap�lm��t�r. En s�k g�r�len advers ila� reaksiyonlar�, diyare, mide bulant�s�, beyaz kan h�cresi say�s�nda azalma, n�tropeni ve ba� a�r�s� olmu�tur ve her biri 3 (%50) hastada primer olarak Grade 1 veya 2 seviyesinde g�r�lm��t�r. 6 hastadan 4'�nde (%66,7) Grade 3-4 tedaviye ba�l� advers reaksiyon (Grade 3 hipofosfatemi, n�tropeni ve trombositopenin her biri 1 hastada ve 1 hastada Grade 4 n�tropeni) g�r�lm��t�r. Bu �al��mada ciddi advers reaksiyonlar veya Grade 5 advers reaksiyonlar bildirilmemi�tir. Hem klinik �al��mada hem de yay�nlarda, g�venlilik profili yeti�kinlerde bilinen g�venlilik profili ile uyumlu olmu�tur.
��pheli advers reaksiyonlar�n raporlanmas�
Ruhsatland�rma sonras� ��pheli ila� advers reaksiyonlar�n�n raporlanmas� b�y�k �nem ta��maktad�r. Raporlama yap�lmas�, ilac�n yarar/risk dengesinin s�rekli olarak izlenmesine olanak sa�lar. Sa�l�k mesle�i mensuplar�n�n herhangi bir ��pheli advers reaksiyonu T�rkiye Farmakovijilans Merkezi (TUFAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e- posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks: 0 312 218
35 99)
4.9. Doz a��m� ve tedavisi
SUTENT kullan�m�nda doz a��m�n�n tedavisi i�in spesifik bir antidot yoktur ve doz a��m� tedavisi i�in genel destekleyici �l��mler gerekmektedir. Endike ise, emilmemi� ilac�n eliminasyonu emesis ve gastrik lavaj ile yap�labilir. Doz a��m� olgular� bildirilmi�tir; baz� olgular SUTENT'in bilinen g�venlilik profiliyle uyumlu olan advers olaylarla ili�kilidir.
5. FARMAKOLOJ�K �ZELL�KLER
5.1. Farmakodinamik �zellikler
Farmakoterap�tik grup: Antinoeplastik ve imm�nomod�lat�r ila�lar, antineoplastik ajanlar, protein kinaz inhibit�rleri, di�er protein kinaz inhibit�rleri
ATC kodu: L01EX01
Sunitinib; t�m�r geli�iminde, neoanjiyogenezde ve kanserin metastatik progresyonunda rol oynayan bir �ok tirozin kinaz resept�r�n� (TKR) inhibe eder. Sunitinib trombosit kaynakl� b�y�me fakt�r� resept�rleri (PDGFRα ve PDGFRβ), VEGF resept�rleri (VEGFR1, VEGFR2 ve VEGFR3), k�k h�cre fakt�r resept�r� (KIT), Fms-tipi tirozin kinaz-3 (FLT3), koloni uyar�c� fakt�r resept�r� (CSF- 1R) ve glial h�cre kaynakl� n�rotrofik fakt�r resept�r�n�n (RET) inhibit�r� olarak tan�mlanm��t�r. Ba�l�ca metaboliti olan desetil sunitinibin, biyokimyasal ve h�cresel testlerde sunitinibe benzer etkide oldu�u g�sterilmi�tir.
Klinik �al��malar
SUTENT'in klinik g�venlilik ve etkilili�i imatinibe tolerans� olmayan veya imatinib rezistan olan (imatinib tedavisi s�resince veya sonras�nda hastal�k progresyonu g�r�len) malign gastrointestinal stromal t�m�rl� (G�ST) hastalar�n tedavisinde ve metastatik renal h�cre karsinomlu (mRHK) hastalar�n tedavisinde ve anrezektabl pNET olan hastalar�n tedavisinde ara�t�r�lm��t�r.
Etkililik; G�ST'de t�m�r progresyonuna kadar ge�en s�reyi (TTP) ve sa�kal�mdaki art���, tedavi uygulanmam�� mRHK i�in progresyonsuz sa�kal�m� ve sitokine diren�li mRHK i�in objektif yan�t oranlar�n�, pNET i�in de progresyonsuz sa�kal�m� temel alm��t�r.
Gastrointestinal Stromal T�m�rler (G�ST)
�matinibe (medyan maksimum g�nl�k doz 800 mg) diren�li ve intolerans� olmas�ndan dolay� G�ST tedavisinde ba�ar�s�z olunan hastalarda a��k etiketli, doz ayarlama �al��mas� ger�ekle�tirilmi�tir. �al��maya ilac� farkl� doz ve doz �emas� uygulanan s�relerde uygulayan 97 adet hasta dahil edilmi�tir; bunlardan 55'i ilac� �nerilen tedavi s�resi olan 4 hafta kullan�p, 2 hafta ara vermek suretiyle (''4/2 �emas�'') 50 mg olarak alm��t�r. Bu �al��mada medyan TTP 34,0 haftad�r ( %95 GA = 22,0 hafta-46,0 hafta).
�matinibi tolere edemeyen veya imatinib (medyan maksimum g�nl�k doz 800 mg) ile tedavi esnas�nda veya sonras�nda hastal�k progresyonu g�r�len G�ST'i olan hastalarda randomize, �ift-k�r ve plasebo-kontroll� bir Faz III �al��ma ger�ekle�tirilmi�tir. Bu �al��mada 312 hasta, hastal�k progresyonu veya �al��madan ba�ka bir sebeple �ekilme olmad��� s�rece 50 mg SUTENT veya plaseboyu a��zdan g�nde bir kez ve 4/2 �emas�na g�re alacak �ekilde randomize (2:1) edilmi�tir (hastalar�n 207'si SUTENT, 105'i plasebo alm��t�r). �al��man�n primer etkililik sonlan�m noktas�, randomizasyondan objektif t�m�r progresyona kadar ge�en s�re olarak tan�mlanan TTP idi.
�nceden belirlenen ara d�nem analizinde, SUTENT i�in medyan TTP (progresyona kadar ge�en s�re) ara�t�rmac� de�erlendirmesinde 28,9 hafta (%95 GA=21,3-34,1 hafta) ve ba��ms�z de�erlendirmeye g�re 27,3 hafta (%95 GA=16,0-32,1) olup, plasebo kolundaki ara�t�rmac� de�erlendirmesine g�re 5,1 haftal�k (%95 GA=4,4-10,1), ba��ms�z de�erlendirmedeki 6,4 haftal�k (%95 GA=4,4-10,0) TTP'den istatistiksel anlaml� olarak daha uzun olmu�tur. Genel sa�kal�m (OS)'deki fark istatistiksel olarak sunitinib lehinedir [HR: 0,491 (%95 GA:0,290-0,831)]. �l�m riski sunitinib koluyla kar��la�t�r�ld���nda plasebo kolunda 2 kat fazlad�r.
Etkililik ve g�venlilik interim analiz sonu�lar� sonras� �al��ma k�rlemeden ��kar�lm�� ve plasebo kolundaki hastalara a��k etiketli sunitinib tedavisi �nerilmi�tir.
Ba�ta plasebo alan 99 hastada dahil �al��man�n a��k etiketli tedavi faz�nda toplam 255
hasta sunitinib alm��t�r.
�al��man�n a��k etiket faz�ndaki primer ve sekonder sonlan�m noktas� analizleri zaman�nda elde edilen interim analiz sonu�lar�n� do�rulam��t�r (Bkz. Tablo 1).
Tablo 1 G�ST etkilik sonlan�m noktas� �zetleri (ITT pop�lasyonu)
| �ift-k�r tedavi |
| |||
| Medyan (%95 GA) | Risk Oran� | Plasebo / �apraz ge�i� grubu | ||
Sonlan�m noktas� | SUTENT | Plasebo | (%95 GA) | p | tedavi |
Primer |
| ||||
TTP (hafta) |
| ||||
Ara | 27,3 (16,0-32,1) | 6,4 (4,4-10,0) | 0,329 (0,233-0,466) | <0,001 | - |
Final | 26,6 (16,0-32,1) | 6,4 (4,4-10,0 ) | 0,339 (0,244-0,472) | <0,001 | 10,4 (4,3-2,0) |
Sekonder |
| ||||
PFS (hafta) |
| ||||
Ara | 24,1 (11,1-28,3) | 6,0 (4,4-9,9) | 0,333 (0,238-0,467) | <0,001 | - |
Final | 22,9 (10,9-28,0) | 6,0 (4,4-9,7) | 0,347 (0,253-0,475) | <0,001 | - |
ORR (%) |
| ||||
Ara | 6,8 (3,7-11,1) | 0 (-) | NA | 0,006 | - |
Final | 6,6 (3,8-10,5) | 0 (-) | NA | 0,004 | 10,1 (5,0-17,8) |
OS (hafta)e |
|
|
|
|
|
Ara | - | - | 0,491 (0,290-0,831) | 0,007 | - |
Final | 72,7 (61,3-83,0) | 64,9 (45,7-96,0) | 0,876 (0,679-1,129) | 0,306 | - |
K�saltmalar: GA = g�ven aral���; ITT = tedavisi ama�lanan hasta; NA = uygulanamaz; ORR = objektif yan�t oranlar�; OS = genel sa�kal�m; PFS = progresyonsuz sa�kal�m; TTP = t�m�r progresyonuna kadar ge�en s�re.
Ba�lang�� noktas�, �aprazlama oldu�unda s�f�rlanm�� ve etkililik analizleri ara�t�rmac�n�n de�erlendirmesi baz al�narak yap�lm��t�r.
ITT pop�lasyonundaki medyan OS sunitinib ve plasebo kollar�nda s�ras�yla 72,7 hafta ve 64,9 haftad�r (HR 0,876, %95 GA: 0,679-1,129, p = 0,306). Bu analizde plasebo kolu daha �nceden a��k etiketli sunitinib tedavisi alm�� ve plaseboya randomize edilmi� hastalar� i�ermektedir.
Tedavi edilmemi� renal h�creli karsinomu (mRHK)
Tedavi-edilmemi� mRHK'li hastalarda tek ajan olarak sunitinib ve IFN-α'y� kar��la�t�ran bir �ok merkezli, uluslararas� faz III randomize �al��ma yap�lm��t�r. Yedi y�z elli (750) hasta ya sunitinib ile tekrarlayan 6 haftal�k sikl�sler halinde 4 hafta 50 mg g�nl�k oral doz takiben 2 hafta ara ( 4/2 doz �emas�) veya i lk hafta 3 milyon �nite (MU) ikinci hafta 6 MU ve ���nc� hafta 9 MU ve bundan sonra her hafta ard���k olmayan 3 g�n subk�tan olarak IFN-α almak �zere birebir (1:1) randomize edilmi�tir .
Sunitinib tedavisinin medyan s�resi 11,1 ay (0,4-46,1 aral���nda), IFN-α tedavisinin medyan s�resi ise 4,1 ayd�r (0,1-45,6 aral���nda). Tedavi ile alakal� ciddi yan etkiler sunitinib ve IFN-α alan hastalarda s�ras�yla %23,7 ve %6,9 olarak raporlanm��t�r. Bunun yan�nda yan etkilerden dolay� tedavinin yar�m b�rak�lma oran� sunitinib i�in %20 iken IFN- α i�in %23'd�r. Doz kesilmesi sunitinib kullanan 202 hastada (%54) g�r�lm��ken IFN-α kullanan hastalarda bu say� 141'dir (%39). Doz azalt�lmas� ise sunitinib ve IFN-α kullanan hastalar�n s�ras�yla 194 (%52) ve 98 (%27)'inde g�r�lm��t�r. Hastalar progresyon
g�r�lene kadar ya da tedaviden ��kar�lana kadar tedavi edilmi�lerdir.
Primer etkililik sonlan�m noktas� progresyonsuz sa�kal�md�r (PFS). Planl� bir ara analiz, sunitinib i�in IFN-α'n�n �st�nde istatistiksel olarak anlaml� bir avantaj g�stermi�tir. Bu �al��mada medyan PFS s�ras�yla 47,3 ve 22,0; HR 0,415'tir. (%95 GA: 0,320-0,539, p<0,001). Di�er sonlan�m noktalar� objektif yan�t oran� (ORR), OS ve g�venliliktir. Primer sonlan�m noktas� elde edildikten sonra �ekirdek radyolojik de�erlendirmeye devam edilmemi�tir. Final analizde ara�t�r�c�n�n de�erlendirmesi ile ORR sunitinib kolu i�in %46 (%95 GA: 41-51) IFN-α kolu i�in %12 (%95 GA: 9-16) olarak belirlenmi�tir (p<0,001).
IFN-α ile kar��la�t�r�ld���nda sunitinib tedavisi daha uzun sa�kal�m s�releriyle ili�kilendirilmi�tir. Medyan OS sunitinib kolu i�in 114,6 hafta (%95 GA: 100,1-142,9) ve IFN-α kolu i�in 94,9 hafta (%95 GA: 77,7-117,0) olmu�tur [HR= 0,821 (%95 GA: 0,673-1,001); log-rank testi ile p=0,0510.]
ITT pop�lasyonunda g�zlemlenen ve �ekirdek radyolojik laboratuvar de�erlendirmesine ile
belirlenen genel PFS ve OS tablo 2'de �zetlenmi�tir:
Tablo 2 – Daha �nce tedavi edilmemi� mRHK etkililik sonlan�m noktas� �zetleri (ITT
pop�lasyonu)
PFS �zeti | Sunitinib (N = 375) | IFN- (N = 375) | |
Progrese olmayan veya �len hastalar [n (%)] | 161 (42,9) | 176 (46,9) | |
Progrese olmayan veya �len hastalar [n (%)] | 214 (57,1) | 199 (53,1) | |
PFS (hafta) | |||
�eyrek (%95 GA) | |||
%25 | 22,7 (18,0-34,0) | 10,0 (7,3-10,3) | |
%50 | 48,3 (46,4-58,3) | 22,1 (17,1-24,0) | |
%75 | 84,3 (72,9-95,1) | 58,1 (45,6-82,1) | |
Tabakaland�r�lmam�� analiz | |||
Risk oran� (sunitinib vs IFN-) | 0,5268 | ||
Risk oran� i�in %95 GA | (0,4316-0,6430) | ||
p-de�eri |
| <0,0001 | |
OS �zeti | Sunitinib (N = 375) | IFN- (N = 375) | |
Progrese olmayan veya �len hastalar [n (%)] | 185 (49,3) | 175 (46,7) | |
Progrese olmayan veya �len hastalar [n (%)] | 190 (50,7) | 200 (53,3) | |
OS (hafta) | |||
�eyrek (%95 GA) | |||
%25 | 56,6 (48,7-68,4) | 41,7 (32,6-51,6) | |
%50 | 114,6 (100,1-142,9) | 94,9 (77,7-117,0) | |
%75 | NA (NA-NA) | NA (NA-NA) | |
Tabakaland�r�lmam�� analiz | |||
Risk oran� (sunitinib vs IFN-) | 0,8209 | ||
Risk oran� i�in %95 GA | (0,6730-1,0013) | ||
p-de�eri | 0,0510 | ||
K�saltmalar: GA = g�ven aral���; IFN-α = interferon-alfa; ITT = tedavisi ama�lanan hasta; N = hasta say�s�;
NA = uygulanamaz; OS = genel sa�kal�m; PFS = progresyonsuz sa�kal�m
Sitokin-refrakter metastatik renal h�creli karsinom
Sunitinib ile bir Faz II �al��ma ger�ekle�tirilmi�tir. �al��ma interleukin-2 ya da IFN- ile birlikte �nceki sitokin tedavisine refrakter hastalar� kapsamaktad�r. 63 hasta 50 mg oral sunitinib (6 haftal�k tamamlanm�� sikl�s-4 hafta boyunca g�nde bir kere ve takiben 2 hafta dinlenme periyodu (4/2 doz �emas�)) ile ba�lam��t�r. Primer sonlan�m noktas� ORR'dir. RECIST (solid t�m�rlerde yan�t de�erlendirme kriteri) kriterlerine g�re belirlenmi�tir.
Bu �al��mada objektif yan�t oran� % 36,5'd�r (%95 GA: %24,7-%49,6). Progresyona kadar ge�en s�re (TTP) 37,7 haftad�r (%95 GA: 24,0-46,4 hafta).
A��k etiketli, tek kollu, �ok merkezli, do�rulay�c� bir �al��mada sunitinibin etkililik ve g�venlili�i bir �nceki sitokin tedavisine refrakter mRHK hastalar�nda de�erlendirilmi�tir. 106 hasta 4/2 �emas�na g�re en az 1 doz 50 mg sunitinib alm��t�r.
Bu �al��man�n primer sonlan�m noktas� ORR'dir. Sekonder sonlan�m noktas� TTP, yan�t s�resi (DoR) ve OS'yi i�ermektedir. Bu �al��mada ORR %35,8 (%95 GA: %26,8-%47,5). Medyan DoR'ye ve OS'ye hen�z ula��lamam��t�r.
Pankreatik n�roendokrin t�m�rler (pNET)
Destekleyici Faz II, a��k etiketli, �ok merkezli �al��ma, rezektabl olmayan pNET'li hastalarda g�nde tek ajan olarak sunitinibin etkilili�ini ve g�venlili�ini, 4/2 �emas�nda [4 haftal�k tedavi, 2 haftal�k dinlenme periyodu, g�nde bir kere 50 mg] de�erlendirdi. 66 hastan�n pankreatik adac�k h�creli t�m�r kohortunda birincil sonlan�m noktas� yan�t oran�
%17 idi.
Unrezektabl pNET olan hastalarda tek ba��na sunitinib ile ilgili Faz III, �ok merkezli, uluslararas�, randomize, �ift k�r plasebo kontroll� bir pivot �al��ma yap�lm��t�r.
Hastalar, RECIST'e dayal� olarak, �nceki 12 ay i�inde belgelenmi� progresyona ihtiya� duydu ve planlanm�� bir istirahat d�nemi (n = 86) veya plasebo (n = 85) olmadan, g�nde bir kez 37,5 mg sunitinib almak �zere randomize edildi (1: 1).
Primer objektif, plasebo alan hastalara kar�� sunitinib alan hastalarda PFS'yi kar��la�t�rmakt�. Di�er sonlan�m noktalar� aras�nda OS, ORR, Hasta Taraf�ndan Bildirilen Sonu�lar ve g�venlik yer almaktad�r.
Demografik veriler, sunitinib ve plasebo gruplar� aras�nda kar��la�t�r�labilir d�zeydeydi. Buna ek olarak, sunitinib hastalar�n�n %49'unda plasebo hastalar�n�n %52'sinde fonksiyonel olmayan t�m�rler vard� ve her iki kolun %92'sinde karaci�er metastaz� vard�.
�al��mada somatostatin analoglar�n�n kullan�m�na izin verildi.
Sunitinib hastalar�n�n toplam %66's� buna kar��n plasebo hastalar�n�n %72'si daha �nce sistemik tedavi ald�. Buna ek olarak, sunitinib hastalar�n�n %24'� plasebo hastalar�n�n ise
%22'si somatostatin analoglar� alm��t�r.
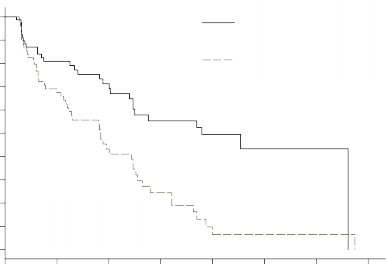
Plaseboya k�yasla sunitinib i�in ara�t�rmac�lar taraf�ndan de�erlendirilen PFS'de klinik olarak anlaml� bir avantaj g�zlenmi�tir. Medyan PFS, sunitinib kolunda 11,4 ay ve plasebo kolunda 5,5 ay olmu�tur [risk oran� (nispi risk) : 0,418 (%95 GA 0,263-0,662), p = 0,0001].
Hastal���n progresyonunu belirlemek i�in RECIST'in ara�t�rmac� t�m�r �l��mleri uygulanmas�na dayanan t�retilmi� t�m�r yan�t de�erlendirmeleri yap�ld���nda benzer sonu�lar g�zlemlenmi�tir (Tablo 3). De�erlendirilen temel �zelliklerin t�m alt gruplar�nda, �nceki al�nan sistemik tedavilerin say�s�na g�re bir analizi de i�erecek �ekilde sunitinib lehine bir risk oran� g�zlenmi�tir. Sunitinib kolunda 29 hasta ve plasebo kolunda 24 hasta �nceden sistemik tedavi almad�; bu hastalardan PFS i�in risk oran� 0,365 (%95 GA: 0,156- 0,857), p = 0,0156 idi. Benzer �ekilde, sunitinib kolundaki 57 hastada (28'i �nceden bir sistemik terapi ve 29'u �nceden 2 veya daha fazla sistemik terapi alm��) ve plasebo kolundaki 61 hastada (25'i �nceden bir sistemik terapi ve 36's� �nceden 2 veya daha fazla sistemik terapi alm��), PFS i�in risk oran� 0,456 (%95 GA: 0,264-0,787), p = 0,0036 idi.
Progresyon karar�n�n, ara�t�rmac� taraf�ndan bildirilen t�m�r �l��mlerine dayand���, PFS olay� olarak muamele edilen �al��ma sonland�rma haricindeki nedenlerle sans�rlenmi� t�m hastalarda PFS duyarl�l�k analizi yap�ld�. Bu analiz, sunitinibin tedavi etkisinin konservatif bir tahminini sa�lam�� ve 0,507 de�erinde bir risk oran� (%95 GA: 0,350-0,733), p = 0,000193 ortaya koyarak birincil analizi desteklemi�tir.
pNET ile ilgili pivot �al��ma, ba��ms�z bir “ �la� �zleme Komitesi”'nin �nerisi �zerine erken sonland�r�lm��t�r ve birincil sonlan�m noktas�, her ikisi de tedavi etkisinin tahminlerini etkilemi� olan ara�t�rmac� de�erlendirmesine dayand�r�lm��t�r.
PFS'nin ara�t�rmac� temelli de�erlendirmesinde yanl�l��� bertaraf edebilmek i�in k�r ba��ms�z merkez incelemesi yap�ld�; bu inceleme ara�t�rmac� de�erlendirmesini desteklemi�tir. Tablo 3'te g�sterilmi�tir.
Tablo 3 - Faz III �al��mas�ndan elde edilen pNET etkinli�i sonu�lar�
Etkililik parametresi | SUTENT (n = 86) | Plasebo (n = 85) | HR (%95 GA) | p-de�eri |
Ara�t�rmac� de�erlendirmesine g�re progresyonsuz sa�kal�m [medyan, aylar (%95 GA)] | 11,4 (7,4–19,8) | 5,5 (3,6–7,4) | 0,418 (0,263–0,662) |
0,0001 |
Ara�t�r�c� t�m�r de�erlendirmelerine RECIST uygulanmas�na dayanan t�retilmi� t�m�r yan�t� de�erlendirmesi ile progresyonsuz sa�kal�m [medyan, aylar (%95 GA)] |
12,6 (7,4–16,9) |
5,4 (3,5–6,0) |
0,401 (0,252–0,640) |
0,000066 |
T�m�r de�erlendirmelerinin k�r ba��ms�z merkez g�zden ge�irilmesiyle progresyonsuz sa�kal�m [medyan, aylar (%95 GA)], |
12,6 (11,1–20,6) |
5,8 (3,8–7,2) |
0,315 (0,181–0,546) |
0,000015 |
Genel sa�kal�m [5 y�l takip] [medyan, aylar (%95 GA)] | 38,6 (25,6–56,4) | 29,1 (16,4–36,8) | 0,730 (0,504–1,057) | 0,0940 |
Objektif yan�t oran� [%, (%95 GA)] | 9,3 (3,2–15,4) | 0 | NA | 0,0066 |
�ekil 1 – pNET Faz 3 �al��mas�nda, Kaplan Meier Progresyonsuz Sa�kal�m Grafi�i
Progression Free Survival Probability (%)
![]()
Progresyonsuz Sa�kal�m Olas�l��� (%)
Time (Months)
![]()
S�re (Ay)
![]()
OS verileri, �al��ma sonunda olgunla�mam��t�r. Sunitinib kolu i�in [20,6 ay (%95 GA 20,6, NR) verileri, plasebo kolu i�in NR (%95 GA 15,5, NR) verileri ile kar��la�t�r�ld���nda risk oran�: 0,409 (%95 GA: 0,187-0,894), p-de�eri = 0,0204]'d�r. Sunitinib kolunda 9 ve plasebo kolunda 21 �l�m meydana gelmi�tir.
Hastal�k progresyonu s�ras�nda hastalar k�rle�tirilmemi�tir ve plasebo alan hastalara, ayr� bir uzant� �al��mas�nda a��k etiketli SUTENT'e eri�im sunulmu�tur. �al��man�n erken d�nemde sonlanmas� nedeniyle, kalan hastalar k�rle�tirilmemi�tir ve bu hastalara, ayr� bir uzant� �al��mas�nda a��k etiketli SUTENT'e eri�im sa�lanm��t�r. Plasebo kolundaki 85 hastadan (%69,4) 59'u, hastal���n ilerlemesi veya �al��ma sonlanmas�ndaki k�rleme kalkt�ktan sonra a��k etiketli sunitinibe ge�ti. Uzatma �al��mas�nda 5 y�ll�k izlem sonras�nda g�zlemlenen OS, 0,730 (%95 GA 0,504-1,057) risk oran�n� g�sterdi.
Avrupa Kanser Ara�t�rma ve Tedavisi Organizasyonu Ya�am Kalitesi Anketinden (EORTC QLQC-30) al�nan sonu�lar; toplamda genel sa�l�k ile ili�kili ya�am kalitesinin ve be� fonksiyon alan�n�n (fiziksel, rol, bili�sel, duygusal ve sosyal), s�n�rl� advers semptomatik etkilerle, plaseboya kar�� sunitinib tedavisi alan hastalarda korundu�unu g�stermi�tir.
Progresif, ileri/metastatik, iyi diferansiye edilmi�, rezeke edilemeyen pNET'li hastalarda sunitinibin etkilili�ini ve g�venilirli�ini de�erlendiren Faz IV �ok uluslu, �ok merkezli, tek kollu, a��k etiketli bir �al��ma ger�ekle�tirildi.
Y�z alt� hasta (hi�bir tedavi almam�� kohortunda 61 hasta ve sonraki basamak kohortunda 45 hasta) g�nde bir kez 37,5 mg oral yoldan sunitinib ile kesintisiz g�nl�k dozlama program� ile tedavi g�rd�.
Ara�t�rmac� taraf�ndan de�erlendirilen progresyonsuz sa�kal�m hem genel pop�lasyonda (%95 GA: 10,9-16,7) hem de hi�bir tedavi almam�� kohortta (%95 GA: 7,4-16,8) 13,2 ay idi.
�zel pop�lasyonlara ili�kin ek bilgiler:
Pediyatrik pop�lasyon:
Pediyatrik hastalarda sunitinib kullan�m�na ili�kin deneyimler k�s�tl�d�r (Bkz. B�l�m
4.2).
Refrakter solid t�m�rleri olup, b�y�k b�l�m� primer beyin t�m�r� tan�s� olan 35 hastada; 30'u pediyatrik hastalardan (3-17 ya�) ve 5'i gen� eri�kin hastalardan (18-21 ya�) olu�an, oral sunitinibe ili�kin, bir Faz I doz art�� �al��mas� ger�ekle�tirilmi�tir. �al��man�n birinci k�sm�nda doz k�s�tlay�c� kardiyotoksisite g�zlenmi� ve bu nedenle �nceden potansiyel kardiyotoksik tedaviler (antrasiklinler dahil) veya kardiyak radyasyon uygulanan hastalar d��lanacak �ekilde d�zeltme yap�lm��t�r. Daha �nce kanser tedavisi alan fakat kardiyak toksisite a��s�ndan risk fakt�rleri bulunmayan hastalar�n yer ald��� �al��man�n ikinci k�sm�nda, 4/2 �emas�nda g�nl�k 15 mg/m dozunda (MTD) sunitinib genellikle tolere edilebilir ve klinik a��dan kontrol alt�na al�nabilir olmu�tur. Olgular�n hi�birinde tam yan�t veya k�smi yan�t elde edilmemi�tir. 6 hastada (%17) stabil hastal�k g�zlenmi�tir. G�ST'i olan bir hasta, 15 mg/m doz d�zeyinde dahil edilmi� ve yarara ili�kin kan�t g�zlenmemi�tir. G�zlenen advers ila� reaksiyonlar� genel olarak eri�kinlerde g�r�lenlere benzer bulunmu�tur (Bkz. B�l�m 4.8).
HGG veya epandimomu bulunan, 27'si pediyatrik hastadan (3-16 ya�) ve 2'si gen� eri�kin hastadan (18-19 ya�) olu�an 29 hastada, Faz II a��k etiketli bir �al��ma ger�ekle�tirilmi�tir. �al��ma, hastal�k kontrol�n�n olmamas� nedeniyle planl� bir ara d�nem analizi s�ras�nda kapat�lm��t�r. Medyan PFS, HGG grubunda 2,3 ay ve epandimoma grubunda 2,7 ay olmu�tur. Medyan genelOS,HGGgrubunda5,1ayveepandimoma grubunda 12,3 ay
olmu�tur. En yayg�n (≥% 10) tedaviye ba�l� advers olaylar�n, her iki grupta kombine olarak n�trofil say�s�n�n azalmas� (6 hasta [%20,7]) ve intrakraniyal hemoraji (3 hasta [%10,3]) oldu�u bildirilmi�tir (Bkz. B�l�m 4.8).
G�nde 15 mg/m ila 30 mg/m aras�nda de�i�en dozlarda 4/2 �emas�na g�re sunitinib alan,
ya�lar� 13-16 aras�nda de�i�en G�ST'i olan 6 pediyatrik hastada oral sunitinibin bir Faz
½ �al��mas�ndan elde edilen kan�tlar ve mevcut yay�nlanm�� veriler (G�ST'i olan 20 pediyatrik veya gen� eri�kin hasta) sunitinib tedavisinin, 26 hastadan 18'inde (%69,2) hem imatinib yetersizli�inden veya intolerans�ndan sonra (21 hastadan 16's�nda stabil hastal�k) hem de novo/cerrahi ameliyat sonras�nda (5 hastadan 2'sinde stabil hastal�k) hastal�k stabilizasyonu sa�lad��� g�zlenmi�tir. Faz ½ �al��mas�nda, 6 hastan�n 3'�nde stabil hastal�k ve 3'�nde hastal�k ilerlemesi g�zlenmi�tir (s�ras�yla 1 hastaya neo adjuvan ve 1 hastaya adjuvan imatinib verilmi�tir). Ayn� �al��mada, 6 hastan�n 4'�nde (%66,7) Grade 3-4 tedaviye ba�l� advers olaylar (Grade 3 hipofosfatemi, n�tropeni ve trombositopeninin her biri 1 hastada ve 1 hastada Grade 4 n�tropeni) g�r�lm��t�r. Ek olarak, yay�nlar, 5 hastada g�zlenen �u Grade 3 advers ila� reaksiyonlar� bildirmi�tir: Yorgunluk (2), gastrointestinal advers ila� reaksiyonlar� (diyare dahil) (2), hematolojik advers ila� reaksiyonlar� (anemi dahil) (2), kolesistit (1), hipertiroidizm (1) ve mukozit (1).
G�ST'i olan pediyatrik hastalarda (6-17 ya� grubu) sunitinibin farmakokinetik (PK) ve kilit g�venlilik ile etkililik sonlan�m noktalar�n�n ekstrapolasyonu amac�yla, bir pop�lasyon PK ve farmakokinetik/farmakodinamik (PK/PD) analizi ger�ekle�tirilmi�tir. Bu analizde G�ST veya solid t�m�rleri olan eri�kinlerden ve solid t�m�rleri olan pediyatrik hastalardan toplanan veriler temel al�nm��t�r. Modelleme analizleri do�rultusunda, k���k ya� ve d���k v�cut �l��mlerinin plazma ila� maruziyetine g�venlilik ve etkililik yan�tlar�n� olumsuz etkilemedi�i belirlenmi�tir. Sunitinib yarar/riskinin, k���k ya� veya d���k v�cut �l��mlerinden olumsuz etkilenmedi�i ve ba�l�ca plazma ila� maruziyetine ba�l� oldu�u g�r�lm��t�r.
Avrupa �la� Ajans�, b�brek veya renal pelvis karsinomunun (nefroblastom, nefroblastomatoz, berrak h�creli sarkom, mezoblastik nefrom, renal med�ller karsinom ve b�brek rabdoid t�m�r� hari� olmak �zere) tedavisinde �ocuk n�fusun t�m alt gruplar�nda SUTENT ile yap�lan �al��malar�n sonu�lar�n� sunma y�k�ml�l���nden feragat etti (�ocuklarda kullan�m hakk�nda bilgi i�in b�l�m 4.2'ye bak�n).
Avrupa �la� Ajans�, gastroenteropankreatik n�roendokrin t�m�rlerin (n�roblastom, n�roganglioblastoma, feokromasitom hari�) tedavisinde �ocuk n�fusunun t�m alt gruplar�nda SUTENT ile yap�lan �al��malar�n sonu�lar�n� sunma y�k�ml�l���nden feragat etti (bkz. �ocuklarda kullan�m hakk�nda bilgi i�in b�l�m 4.2).
5.2. Farmakokinetik �zellikler
Genel �zellikler
Sunitinibin farmakokineti�i 135 sa�l�kl� g�n�ll�de ve solid t�m�rl� 266 hastada de�erlendirilmi�tir. Her iki grupta da farmakokinetik benzerdir.
Emilim:
Sunitinib a��zdan uygulanmas�n� takiben 6-12 saat (T) i�inde maksimum konsantrasyona (C) ula��r. Yiyeceklerin sunitinibin biyoyararlan�m�na herhangi bir etkisi yoktur. Sunitinib ve primer aktif metabolitin kararl� durum konsantrasyonlar�na 10-14 g�n i�inde ula��l�r. 14. g�n itibariyle sunitinib ve aktif metabolitinin kombine plazma konsantrasyonlar� 62,9-101 ng/ml olup, bu konsantrasyonlar klinik verilerde �ng�r�len in vitro olarak resept�r fosforilasyonunu inhibe edecek ve in vivo olarak t�m�r staz�/b�y�mesini azaltacak hedef konsantrasyonlard�r. Primer aktif metabolit toplam maruziyetin %23-%37'sini olu�turmaktad�r. Tekrarlayan g�nl�k uygulamalarda veya test edilen doz rejimlerinin tekrarlayan k�rlerinde sunitinib veya primer aktif metabolitinin farmakokineti�inde anlaml� de�i�iklikler olmam��t�r.
Da��l�m:
Sunitinib ve primer aktif metabolitinin in vitro �al��malarda konsantrasyondan ba��ms�z olarak plazma proteinine ba�lanma derecesi s�ras�yla %95 ve %90 olmu�tur. Sunitinib i�in da��l�m hacmi (V), dokulara da��l�m� g�sterecek �ekilde, b�y�kt�r (2230 litre).
Metabolik etkile�imler
Test edilen t�m sitokrom P450 izoformlar� (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5, ve CYP4A9/11) i�in hesaplanan in vitro Ki de�erleri; sunitinib ve primer aktif metabolitinin, bu enzimlerle metabolize olan di�er etkin maddelerin metabolizmas�n� ind�kleme ihtimalinin az olaca��n� g�stermektedir.
Biyotransformasyon:
Sunitinib as�l olarak, bir sitokrom P450 enzimi olan CYP3A4 taraf�ndan metabolize edilir. Primer aktif metaboliti desetil sunitinibtir ve bu metabolit tekrardan CYP3A4 taraf�ndan metabolize edilir.
Sunitinibin potent CYP3A4 ind�kleyicileri veya inhibit�rleri ile birlikte kullan�m� ile sunitinibin plazma d�zeyi de�i�ebilece�inden, birlikte kullan�m�ndan ka��n�lmal�d�r (Bkz. B�l�m 4.4 ve 4.5).
Eliminasyon:
At�l�m primer olarak fe�es yoluyla (%61) ger�ekle�ir. Renal eliminasyon metabolitler ile birlikte uygulanan dozun %16's�d�r. Sunitinib ve primer aktif metaboliti plazmada, idrarda ve fe�este g�r�len ila� ba�lant�l� esas bile�iklerdir ve s�ras�yla %91,5, %86,4 ve
%73,8 oran�nda g�r�lmektedir. Min�r metabolitler idrar ve fe�este g�r�lm��, ancak genellikle plazmada g�r�lmemi�lerdir. Total oral klerens (CL/F) 34-62 litre/saat olmu�tur. Sa�l�kl� g�n�ll�lere tek doz oral uygulaman�n ard�ndan sunitinibin terminal yar�lanma �mr� yakla��k olarak 40-60 saat iken, primer aktif desetil metabolitininki 80- 110 saat olmu�tur.
Do�rusall�k / Do�rusal olmayan durum:
25-100 mg'l�k doz aral���nda plazma konsantrasyon-zaman e�risi alt�ndaki alan (EAA) ve C dozla orant�l� olarak artar. G�nl�k tekrarlayan uygulamalarda sunitinib miktar� 3-4 kat�na ��karken, primer metabolitinin miktar� 7-10 kat�na ��kar.
BCRP inhibit�r� olan ila�larla birlikte kullan�m
In vitro ortamda sunitinib, efluks ta��y�c� BCRP'n�n substrat�d�r. A6181038 �al��mas�nda bir BCRP inhibit�r� olan gefitinibin, sunitinib veya toplam ilac�n (sunitinib + metabolit) Cve EAA de�erleri �zerinde klinik olarak anlaml� bir etkisi olmam��t�r (Bkz. B�l�m 4.5). Bu �al��ma mRHK'l� hastalarda sunitinibin gefitinib ile birlikte kullan�ld���nda g�venlilik/tolere edilebilirli�i, maksimum tolere edilen dozu ve antit�m�r aktivitesinin ara�t�r�ld��� �ok merkezli, a��k etiketli, Faz 1/2 bir �al��mad�r. Gefitinib (g�nl�k 250 mg) ve sunitinib (4 hafta kullan�m sonras� 2 hafta ara verecek �ekilde g�nl�k 37,5 mg [Kohort 1, n=4] veya 50 mg [Kohort 2, n=7]) birlikte uyguland���ndaki famakokinetikleri sekonder �al��ma objektifi olarak de�erlendirilmi�tir. Sunitinib farmakokinetik parametrelerindeki de�i�iklikler klinik olarak anlaml� bulunmam�� ve bir ila�-ila� etkile�imi oldu�unu g�stermemi�tir; ancak g�receli d���k hasta say�s� (N=7+4) ve farmakokinetik parametrelerdeki hastalar aras� varyasyonun orta-y�ksek seviyede olmas� g�z�n�ne al�nd���nda, bu �al��man�n farmakokinetik ila�-ila� etkile�imi sonu�lar�n� de�erlendirirken dikkatli olunmas� gerekmektedir.
Hastalardaki karakteristik �zellikler
Karaci�er yetmezli�i:
Sunitinib ve aktif metaboliti esas olarak karaci�er taraf�ndan metabolize edilir. Hafif (Child-Pugh S�n�f A) veya orta (Child-Pugh S�n�f B) karaci�er yetmezli�i olan hastalarda sunitinibin tek bir dozuna sistemik maruz kal�m, normal karaci�er fonksiyonu olan deneklerle kar��la�t�r�ld���nda benzer olmu�tur. Sunitinib, ciddi (Child- Pugh S�n�f C) karaci�er yetersizli�i olan hastalarda �al���lmam��t�r.
�al��malara ALT veya AST de�erleri >2,5xULN (normalin en �st s�n�r�) veya karaci�er metastaz� > 5,0 x ULN olan hastalar dahil edilmemi�tir.
B�brek yetmezli�i:
Pop�lasyon farmakokinetik analizleri, kreatinin klerensi 42-347 ml/dak olan hastalarda, sunitinibin klerensinin de�i�medi�ini g�stermektedir. Tek doz SUTENT uygulamas�ndan sonra, ciddi b�brek yetmezli�i (kreatinin klerensi <30ml/dak) olan ki�ilerle normal renal fonksiyona (kreatinin klerensi >80ml/dak) sahip ki�ilerde sistemik maruziyet ayn� olmu�tur. Son evre b�brek yetmezli�i hastalar�nda sunitinib ve primer metabolitinin hemodiyalizle eliminasyonu yap�lamasa da, normal b�brek fonksiyonuna sahip ki�ilerle kar��la�t�r�ld���nda sistemik maruziyet sunitinib i�in %47, primer metaboliti i�in %31 daha az olmu�tur.
Kilo ve performans durumu:
Demografik verinin pop�lasyon farmakokinetik analizi, v�cut a��rl��� veya performans durumunda, ba�lang�� doz ayarlamalar�n�n gerekli olmad���n� g�sterir.
Cinsiyet:
Eldeki veriler kad�nlarda g�r�n�r sunitinib klerensinin (CL/F) erkeklere oranla %30 daha az oldu�unu g�stermektedir; ancak bu fark ba�lang�� dozunun de�i�tirilmesini gerektirmemektedir.
Pediyatrik pop�lasyon:
Pediyatrik hastalarda sunitinib kullan�m�na ili�kin deneyimler k�s�tl�d�r (Bkz. b�l�m 4.2). G�ST ve solid t�m�rleri olan eri�kin hastalar� ve solid t�m�rleri olan pediyatrik hastalar� i�eren toplu veritaban�na ili�kin pop�lasyon PK analizleri tamamlanm��t�r. Ya� ve v�cut �l��mlerinin (toplam v�cut a��rl��� veya v�cut y�zey alan�) yan� s�ra di�er e� de�i�kenlerin sunitinib ve metabolitlerinin �nemli PK parametreleri �zerindeki etkisini de�erlendirmek �zere ad�msal e� de�i�ken modelleme analizleri yap�lm��t�r. Test edilen ya� ve v�cut �l��m� ile ili�kili e� de�i�kenlerden ya�, sunitinibin g�r�nen klerensi �zerinde anlaml� bir e� de�i�ken olarak belirlenmi�tir (pediyatrik hasta ne kadar k���kse, g�r�nen klerens o kadar d���kt�r). Benzer �ekilde, v�cut y�zey alan�, aktif metabolitin g�r�nen klerensi �zerinde anlaml� bir e� de�i�ken olarak belirlenmi�tir (v�cut y�zey alan� ne kadar d���kse, g�r�nen klerens o kadar d���kt�r).
Ayr�ca, 3 pediyatrik �al��madan toplu veri setine ili�kin birle�tirilmi� pop�lasyon PK analizine g�re (2 pediyatrik solid t�m�r �al��mas� ve 1 pediyatrik G�ST �al��mas�: 6-11 ya� ve 12-17 ya�) ba�lang�� v�cut y�zey alan� (BSA), sunitinibin ve aktif metabolitinin g�r�nen klirensi �zerinde anlaml� bir e� de�i�ken olarak belirlenmi�tir. Bu analize dayanarak, BSA de�erleri 1,10 ve 1,87 m aras�nda olan pediyatrik hastalarda g�nl�k yakla��k 20 mg/m'lik bir doz, sunitinib ve aktif metabolitine plazma maruziyetinin (EAA'n�n %75 ila %125'i aras�nda), G�ST'i olan yeti�kinlere, 4/2 �emas�na g�re (EAA 1233 ng.hr/mL) g�nl�k 50 mg sunitinib uygulamas� ile g�zlenen plazma maruziyetine benzer olmas� beklenir. Pediyatrik �al��malarda, 15 mg/m olan ba�lang�� sunitinib dozu (Faz I doz-art�� �al��mas�nda tan�mlanan MTD'ye g�re, Bkz. B�l�m 5.1), G�ST'i olan pediyatrik hastalarda 22,5 mg/m'ye ve ard�ndan bireysel hasta g�venlili�i/tolere edilebilirli�ine ba�l� olarak 30 mg/m'ye (toplam g�nl�k 50 mg dozu a�mayacak �ekilde) y�kselmi�tir. Ayr�ca, G�ST'i olan pediyatrik hastalarda yay�nlanm�� literat�rlere g�re, 16,6 mg/m ila 36 mg/m aras�nda de�i�mekte olan hesaplanm�� ba�lang�� dozu, toplam g�nl�k 50 mg dozu a�mayacak �ekilde 40,4 mg/m‘ye kadar y�kselmi�tir.
5.3. Klinik �ncesi g�venlilik verileri
Maymun ve s��anlarda 9 aya kadar devam eden tekrarlayan doz toksisite �al��malar�nda primer hedef organ etkileri gastrointestinal sistem (maymunlarda emezis ve diyare), adrenal bez (s��anlarda fibrozis g�r�len nekrozu takiben ve maymunlarda kortikal konjesyon ve/veya hemoraji), hemolenfopoetik sistem (kemik ili�i hiposel�laritesi ve timusta lenfoid dokunun azalmas�, dalak ve lenf nod�l�), ekzokrin pankreas (tek h�cre nekrozuyla asinar h�cre degran�lasyonu), t�kr�k bezi (asinar hipertrofi), eklem (b�y�me pla�� kal�nla�mas�), uterus (atrofi), overler (azalm�� folik�ler geli�im) g�r�lm��t�r. T�m bu bulgular klinik olarak anlaml� bir sunitinibin plazma maruziyeti seviyesinde g�r�lm��t�r. QT aral���nda uzama, b�brekte mezangiyal matriks, gastrointestinal sistemde ve oral mukozada hemoraji ve testislerde (t�b�ler atrofi) ve anterior pit�iter h�creleri hipertrofisi di�er �al��malarda g�r�len ilave etkiler aras�ndad�r. Uterustaki (endometriyal atrofi) ve kemik b�y�me pla��ndaki (fizeal kal�nla�ma veya k�k�rdak displazisi) de�i�imler sunitinibin farmakolojik etkisiyle ili�kilendirilmi�tir. Bu bulgular�n �o�u tedavi kesildi�inde 2-6 hafta i�inde geri d�n���ml� olmu�tur.
Genotoksisite
Sunitinibin genotoksik potansiyeli in vitro ve in vivo olarak de�erlendirilmi�tir. Sunitinib, s��an karaci�eri ile sa�lanan metabolik aktivasyonu kullanan bakterilerde mutajenik de�ildi. Sunitinib in vitro olarak insan periferik kan lenfosit h�crelerinde yap�sal kromozom aberasyonuna neden olmam��t�r. �nsan periferik kan lenfositlerinde in vitro olarak metabolik aktivasyon varl���nda ve yoklu�unda poliploidi (say�sal kromozom aberasyonu) g�zlenmi�tir. Sunitinib, s��an kemik ili�inde in vivo olarak klastojenik de�ildi. Esas aktif metabolit genotoksisite a��s�ndan de�erlendirilmemi�tir.
Karsinojenite
ayl�k oral gavaj doz-aral��� belirleme �al��mas�nda (0, 10, 25, 75 veya 200 mg/kg/g�n dozlar�nda) devaml� g�nl�k dozlama yap�lan rasH2 transgenik farelerde test edilen en y�ksek dozda (200 mg/kg/g�n) dudenumun Brunner bezinin karsinomas� ve hiperplazisi g�zlenmi�tir.
RasH2 transgenik farelerde g�nl�k dozlama ile 6 ayl�k bir oral gavaj karsinojenite �al��mas� (0, 8, 25, 75 (50'ye azalt�lan) mg/kg/g�n) yap�lm��t�r. G�nl�k 25 mg/kg'l�k ve daha fazla dozlarda 1-veya 6- ayl�k s�releri (g�nl�k �nerilen dozu kullanan hastalar�n EAA's�n�n 7,3 kat� veya daha fazlas�) takiben gastroduodenal karsinomalar, arka plan hemajiyosarkom insidans�nda art��, ve/veya gastrik mukozal hiperplazi g�zlenmi�tir.
6. FARMAS�T�K �ZELL�KLER
6.1. Yard�mc� maddelerin listesi
Kaps�l i�eri�i:
Mannitol (E421) Kroskarmeloz Sodyum (E468) Povidon
Saf Su
Magnezyum Stearat (E572)
Kaps�l k�l�f� (Bask�l�, Opak, Standart Sar�, Boyut #3, Sert Jelatin Kaps�l): Titanyum Dioksit (E171)
Sar� Demir Oksit (E172iii) Jelatin (E441)
Bask� M�rekkebi:
�ellak (E904)
Propilen Glikol (E1520) G��l� amonyak ��zeltisi Potasyum Hidroksit Siyah Demir Oksit Dehidraft Alkol �zopropil Alkol
B�til Alkol
�r�n, s���r kaynakl� jelatin hammaddesi i�ermektedir.
6.2. Ge�imsizlikler
Ge�erli de�ildir.
6.3. Raf �mr�
36 ay
6.4. Saklamaya y�nelik �zel tedbirler
25°C'nin alt�nda oda s�cakl���nda saklanmal�d�r.
6.5. Ambalaj�n niteli�i ve i�eri�i
Her biri 7 kaps�l i�eren Aluminyum/PVC/Aclar® blisterlerde ambalajlanm��t�r. Paket
b�y�kl��� 28 (7×4) kaps�l halindedir.
6.6. Be�eri t�bbi �r�nden arta kalan maddelerin imhas� ve di�er �zel �nlemler
Kullan�lmam�� olan �r�nler ya da at�k materyaller “T�bbi At�klar�n Kontrol� Y�netmeli�i” ve “Ambalaj At�klar�n�n Kontrol� Y�netmeli�i”ne uygun olarak imha edilmelidir.
 Rahim Boyu ( Serviks ) Kanseri
Rahim boynu (serviks) kanseri 35 ya� alt� kad�nlarda g�r�len vakalarda meme kanserinden
sonra ikinci s�ray� al�r.Serviks kanserinin geli�mesi y�llarca s�rebilir.
Rahim Boyu ( Serviks ) Kanseri
Rahim boynu (serviks) kanseri 35 ya� alt� kad�nlarda g�r�len vakalarda meme kanserinden
sonra ikinci s�ray� al�r.Serviks kanserinin geli�mesi y�llarca s�rebilir. |
 L�semi Kan Kanseri
L�semi, kan kanseridir ve v�cudunun kan olu�turan dokular�n�n hastalanmas� anlam�na gelir. Bir�ok l�semi t�r� vard�r; baz� l�semi t�rleri �ocuklarda baz�lar� da yeti�kinlerde s�k g�r�l�r.
L�semi Kan Kanseri
L�semi, kan kanseridir ve v�cudunun kan olu�turan dokular�n�n hastalanmas� anlam�na gelir. Bir�ok l�semi t�r� vard�r; baz� l�semi t�rleri �ocuklarda baz�lar� da yeti�kinlerde s�k g�r�l�r. |
�LA� E�DE�ERLER�
| E�de�er �la� Ad� | Barkodu | �la� Fiyat� |
|---|---|---|
| SUNIKSA | 8699717150089 | 23,255.08TL |
| SUNO | 8699541153706 | 11,682.81TL |
| SUNUBIS | 8699540023260 | 11,682.81TL |
| SUTENT | 8699532158710 | |
| SUTESGO | 8699525159809 | 11,682.81TL |
| Di�er E�de�er �la�lar |
 |
Kalp Krizi Kalbe giden kan ak��� durdu�unda kalp krizi meydana gelir. |
 |
Diyabet Hastal��� Diyabet, ins�lin hormonu ile ilgili problemlerden kaynaklanan bir hastal�kt�r. |
 |
S�rt A�r�s� S�rt a�r�s� birden bire ortaya ��k�p �iddetli (akut) olabilir veya zamanla geli�ip daha uzun s�reli sorunlara (kronik) neden olabilir. |
�LA� GENEL B�LG�LER�
Pfizer �la�lar� Ltd.�ti.
| Sat�� Fiyat� | 34827.89 TL [ 1 Dec 2025 ] |
| �nceki Sat�� Fiyat� | 34827.89 TL [ 24 Nov 2025 ] |
| Original / Jenerik | Original �la� |
| Re�ete Durumu | K�rm�z� Re�eteli bir ila�d�r. |
| Barkodu | 8681308159571 |
| Etkin Madde | Sunitinib Maleat |
| �thal ( ref. �lke : Fransa ) ve Be�eri bir ila�d�r. |