SUTENT 50 mg 14 kapsül Kısa Ürün Bilgisi
{ Sunitinib Maleat }
1. BEŞERİ TIBBİ ÜRÜNÜN ADI
SUTENT® 50 mg Kapsül Sitotoksik
2. KALİTATİF VE KANTİTATİF BİLEŞİM
Etkin madde
Her bir kapsül 50 mg sunitinibe eşdeğer 66,8 mg sunitinib malat içerir.
Yardımcı maddeler
Yardımcı maddeler için 6.1'e bakınız.
3. FARMASÖTİK FORMU
Sert jelatin kapsül
İki parçalı, opak baskılı, karamel renkli, sert jelatin boyut 2 kapsüller, sarı ile turuncu arası renkte granül içerir.
4. KLİNİK ÖZELLİKLER
4.1. Terapötik endikasyonlar
Gastrointestinal Stromal Tümör (GİST)
SUTENT imatinib mesilat tedavisine dirençli veya intoleran anrezektabl ve/veya
metastatik gastrointestinal stromal tümörlerin (GİST) tedavisinde endikedir.
Metastatik Renal Hücreli Karsinom (mRHK)
SUTENT ilerlemiş ve/veya metastatik renal hücreli karsinom (mRHK) tedavisinde endikedir.
Pankreatik Nöroendokrin Tümör (pNET)
SUTENT, metastatik veya lokal ileri evrede olup cerrahi tedavisi mümkün olmayan, somatostatin analogları tedavisi sonu progresyon gelişen iyi differansiye pankreatik nöroendokrin tümörlerin (pNET) tedavisinde endikedir.
4.2. Pozoloji ve uygulama şekli
Pozoloji/uygulama sıklığı ve süresi:
Tedavi kanser ilaçlarını uygulama konusunda tecrübeli bir hekim tarafından başlatılmalıdır.
GİST ve mRHK için; önerilen SUTENT dozu 4 hafta kesintisiz günde 1 defa 50 mg ağızdan alınarak ve daha sonra 2 hafta ara vermek suretiyle 6 haftalık kürü tamamlayacak şekildedir.
pNET için SUTENT'in önerilen dozu; planlı bir ara verme dönemi olmaksızın günde bir
kez oral yolla 37,5 mg'dır.
GİST ve mRHK için, 12,5 mg'lık artış veya azaltmalarla doz modifikasyonları bireysel güvenlilik ve tolerabiliteye bağlı olarak uygulanabilir. Günlük dozlar 25 mg'ın altına düşmemeli ve 75 mg'ı geçmemelidir.
Bireysel güvenlilik ve tolere edilebilirlik göz önüne alınarak, pNET için 12,5 mg'lık adımlarla doz ayarlaması yapılabilir. Faz III pNET çalışmasında uygulanan maksimum doz günde 50 mg olmuştur.
Bireysel güvenlilik ve tolerabiliteye bağlı olarak doza ara vermek gerekebilir.
Rifampisin gibi kuvvetli CYP3A4 indükleyici ile birlikte SUTENT kullanımından sakınılmalıdır. Eğer kullanılması gerekiyorsa, kullanan hastalarda dozun 12,5 mg'lık miktarlarla (GİST ve mRHK için günde 87,5 mg'a kadar veya pNET için günde 62,5 mg'a kadar) artırılması gerekebilir. Klinik yanıt ve tolerabilite dikkatle izlenmelidir. Ketokonazol gibi CYP3A4 inhibitörü ile birlikte SUTENT kullanımından sakınılmalıdır. Eğer birlikte kullanılması mutlaka lazımsa kullanan hastalarda tolerabilite ve/veya klinik yanıta bağlı olarak SUTENT dozu 12,5 mg'lık kademelerle GİST ve mRHK için günlük minimum 37,5 mg'a veya pNET için günde 25 mg'a düşürülebilir. Eşzamanlı ilaç seçiminde CYP3A4'ü indükleyici veya inhibe edici potansiyeli çok düşük olan veya hiç olmayan alternatif ilaçlar düşünülmelidir. (Bkz. Bölüm 4.4)
Uygulama şekli:
SUTENT, aç karnına veya yemekle beraber, oral olarak alınabilir. Bir doz atlandıysa hastaya ilave doz verilmemelidir. Hasta bir sonraki gün önerilen normal doz ile devam etmelidir.
Özel popülasyonlara ilişkin ek bilgiler:
Böbrek/Karaciğer yetmezliği:
Hafif (Child-Pugh Sınıf A) ve orta (Child-Pugh Sınıf B) karaciğer yetmezliği olan hastalarda başlangıç dozunun ayarlamasına gerek yoktur. Ciddi (Child-Pugh Sınıf C) karaciğer yetersizliği olan hastalarda çalışma yapılmamıştır. Bu nedenle şiddetli karaciğer yetmezliği olan hastalarda sunitinib kullanımı ile ilgili bir öneride bulunulamamaktadır. (Bkz. Bölüm 5.2)
Orta-ciddi böbrek yetmezliği olan hastalarda veya hemodiyalize giren son dönem böbrek yetmezliği hastalarına sunitinib verilirken başlangıç doz ayarlaması gerekli değildir. Takip eden dozlar güvenlilik ve tolerabiliteye göre ayarlanmalıdır. (Bkz. Bölüm 5.2)
Pediyatrik popülasyon:
SUTENT'in 18 yaşın altındaki hastalardaki güvenlilik ve etkililiği değerlendirilmemiştir. Şu an var olan veriler Bölüm 4.8, 5.1 ve 5.2'de anlatılmıştır, fakat bu popülasyonda SUTENT kullanımı önerilmemektedir.
Geriyatrik popülasyon:
SUTENT'e ait klinik çalışmalardaki hastaların yaklaşık %34'ünün yaşı 65 veya üzeridir. Genç ve yaşlı hastalar arasında güvenlilik ve etkililik açısından herhangi bir anlamlı fark gözlenmemiştir.
4.3. Kontrendikasyonlar
Sunitinib malat veya SUTENT kapsülleri bileşenlerinden herhangi birine aşırı duyarlılığı olan hastalarda kontrendikedir (Bkz. Bölüm 6.1).
4.4. Özel kullanım uyarıları ve önlemleri
Potent CYP3A4 indükleyicileri sunitinibin plazma konsantrasyonunu azaltabileceğinden
birlikte kullanımından kaçınılmalıdır (Bkz. Bölüm 4.2 ve 4.5).
Cilt ve doku bozuklukları
Hastalar aynı zamanda SUTENT ile tedavi boyunca saç veya ciltte depigmentasyon olabileceği konusunda uyarılmalıdır. Ciltte kuruluk, kalınlık veya çatlama, ayak tabanları veya avuç içlerinde nadiren kızarıklık veya kabarcıklar diğer olası dermatolojik etkiler arasında sayılabilir.
Yukarıda adı geçen olaylar kümülatif (birikimli) değildir; tipik olarak reversibl olup genellikle tedaviye son vermeyi gerektirmemiştir. Piyoderma gangrenosum (genellikle sunitinib kullanımı bırakıldıktan sonra geri dönüşlü olan ağrılı deri ülseri) vakaları bildirilmiştir. Bazıları ölümle sonuçlanmış eritema multiforme (EM), Steven-Johnson sendromu (SJS) ve toksik epidermal nekroliz (TEN) dahil ciddi kutanöz reaksiyonlar bildirilmiştir. SJS, TEN veya EM belirtileri veya semptomları (örn. genellikle su toplaması ve mukozal lezyonlar ile seyreden ilerlemiş deri döküntüleri) mevcutsa, sunitinib tedavisi sonlandırılmalıdır. SJS veya TEN teşhisi doğrulanmışsa, tedaviye tekrar başlanmamalıdır. EM'den şüphelenilen bazı olgularda, reaksiyonun sona ermesinin ardından daha düşük dozda sunitinib tedavisine yeniden başlanması hastalar tarafından tolere edilmiştir. Bu hastalardan bazıları eş zamanlı olarak kortikosteroid ve antihistaminik tedavisi almıştır (Bkz. Bölüm 4.8).
Hemoraji ve tümörlerin kanaması
SUTENT ile yapılan klinik çalışmalarda ve pazarlama sonrası deneyimlerde, gastrointestinal sistem, solunum, üriner sistem ve beyin hemorajisi gibi bazıları ölümcül olabilecek hemorajik olaylar bildirilmiştir (Bkz. Bölüm 4.8).
Kanama olaylarının rutin değerlendirmesi, tam kan sayımı ve fizik muayene ile
yapılmalıdır.
Hemorajik olaylar yaşamış solid tümörlü hastaların, yaklaşık yarısında en çok görülen hemorajik advers olay burun kanamasıdır. Burun kanaması olaylarından bazıları ciddi olup, çok seyrek olarak ölüme yol açmıştır.
Tümörlerin kanama olayları bazen tümör nekrozuna eşlik eder, bu kanamalar fatal olabilir.
Tümör hemorajisi aniden oluşabilir ve akciğer tümörleri olgularında, ciddi ve hayatı tehdit edici hemoptizi veya akciğer kanaması olarak görülebilir. mRHK, akciğer kanseri ve GİST tedavisi için SUTENT kullanan hastalarda pazarlama sonrası deneyim olarak akciğer kanaması (bazıları ölümle sonuçlanmıştır) oluşmuş ve bu durum klinik çalışmalarda da gözlemlenmiştir. SUTENT, akciğer kanseri olan hastalarda kullanım için onaylı değildir.
Eş zamanlı olarak antikoagülan (örn.; varfarin, asenokumarol) tedavisi alan hastalar tam kan sayımı (trombositler), koagülan faktörler (PT/INR) ve fiziksel muayene ile periyodik olarak kontrol edilmelidir.
Gastrointestinal olaylar
Diyare, mide bulantısı/kusma, karın ağrısı, dispepsi ve stomatit en çok rapor edilen
gastrointestinal yan etkilerdir. Ayrıca özofajit de rapor edilmiştir (Bkz. Bölüm 4.8).
Tedavi gerektiren gastrointestinal advers olaylar için destekleyici bakım, antiemetik,
antasit veya antidiyareik ilaç tedavisi ile sağlanabilir.
SUTENT ile tedavi edilen, intra-abdominal maligniteleri olan hastalarda gastrointestinal perforasyonu kapsayabilen, ciddi bazen ölümcül olabilen gastrointestinal komplikasyonlar oluşmuştur.
Hipertansiyon
Ciddi hipertansiyon (>200 mmHg sistolik veya 110 mmHg diyastolik) dahil sunitinib ile ilişkili olarak hipertansiyon bildirilmiştir. Hastalar hipertansiyon için taranmalı ve uygun oldukça kontrol edilmelidir. İlaç müdahalesiyle kontrol edilemeyen ağır hipertansiyonlu hastalarda geçici olarak ilaç tedavisinin durdurulması önerilir. Hipertansiyon uygun olarak kontrol altına alındığında tedaviye yeniden başlanabilir (Bkz. Bölüm 4.8).
Hematolojik bozukluklar
Sunitinib ile ilişkili olarak azalmış mutlak nötrofil sayıları ve azalmış trombosit sayısı bildirilmiştir (Bkz. Bölüm 4.8). Yukarıda adı geçen olaylar kümülatif (birikimli) değil; tipik olarak reversibl olup genellikle tedaviye son vermeyi gerektirmemiştir. Faz III çalışmalardaki bu olayların hiçbiri ölümcül olmamakla birlikte pazarlama sonrası deneyimlerde trombositopeni ve nötropenik enfeksiyonların eşlik ettiği hemorajili durumlarda seyrek olarak ölüm olguları rapor edilmiştir.
Sunitinib ile tedavi esnasında erken ve geç dönemde anemi gözlenmiştir.
SUTENT ile tedavi gören hastalarda, her tedavi kürünün başlangıcında tam kan sayımı yapılmalıdır (Bkz. Bölüm 4.8).
Kardiyak bozukluklar
SUTENT kullanan hastalarda bazıları ölümle sonuçlanan, kalp yetmezliği, kardiyomiyopati, sol ventrikül ejeksiyon fraksiyonunun normalin alt sınırının altına düşmesi, miyokardit ve miyokardiyal iskemi ve miyokard enfarktüsünün de dahil olduğu kardiyovasküler olaylar rapor edilmiştir. Bu veriler sunitinibin kardiyomiyopati riskini arttırdığını göstermektedir.İlacaözgüetkidışında sunitinib ile indüklenen
kardiyomiyopati için tedavi gören hastalarda ilave risk faktörleri tespit edilmemiştir. Bu olaylar açısından risk taşıyan veya kardiyovasküler hikayesi olan hastalarda dikkatli kullanılmalıdır (Bkz. Bölüm 4.8).
SUTENT uygulamasından önce 12 ay içinde miyokard enfarktüsü (ciddi/stabil olmayan anjinayı kapsayan), koroner/periferik arter by-pass grafti, semptomatik KKY, serebrovasküler olay veya geçici iskemik atak veya akciğer embolisi gibi kardiyak bulguları olan hastalar, SUTENT klinik çalışmalarına dahil edilmemiştir. Bunun gibi eş zamanlı rahatsızlıkları olan hastaların sunitinible ilişkili sol ventrikül işlev bozukluğu gelişimi için daha yüksek bir riskte olup olmadıkları bilinmemektedir.
Doktorlara SUTENT'in risk/yarar oranını göz önünde bulundurmaları tavsiye edilir. Kardiyak risk faktörü olan ve/veya koroner arter hastalığı hikayesi olan hastalar SUTENT alırken, KKY'nin klinik belirtileri ve semptomları için dikkatli olarak gözlenmelidirler. Hasta sunitinib alırken, sol ventrikül ejeksiyon fraksiyonunda (SVEF) için başlangıç ve periyodik değerlendirmeleri de düşünülmelidir. Kardiyak risk faktörleri olmayan hastalarda, ejeksiyon fraksiyonunun başlangıç değeri ölçülmelidir.
KKY'nin klinik belirtilerinin varlığında, sunitinibin kesilmesi önerilir. Sunitinib dozu, klinik KKY bulgusu olmayan ancak, SVEF<%50 ve başlangıca göre >%20 düşüş göstermiş hastalarda kesilmeli ve /veya azaltılmalıdır.
QT aralığı uzaması
Sunitinib'e maruz kalan hastalarda QT aralığı uzaması ve Torsade de pointes gözlemlenmiştir. QT aralığı uzaması Torsade de pointes de dahil olmak üzere ventriküler aritmilerde artmış bir riske yol açabilir.
Sunitinib; bilinen QT aralığı uzama öyküsü, antiaritmik ya da QT aralığını uzatabilen bir ilaç alan veya daha önceden bilinen önemli kardiyak hastalığı, bradikardi veya elektrolit bozukluğu olan hastalarda dikkatle kullanılmalıdır. Güçlü CYP3A4 inhibitörleri ile eş zamanlı tedavi, sunitinibin plazma konsantrasyonunu yükseltebileceği için, dikkatli kullanılmalıdır (Bkz. Bölüm 4.2, 4.5 ve 4.8).
Venöz tromboembolik olaylar
Derin ven trombozu ve pulmoner emboli de dahil olmak üzere sunitinib alan hastalarda tedaviye bağlı venöz tromboembolik olaylar bildirilmiştir (Bkz. Bölüm 4.8). Pazarlama sonrası deneyimlerde ölümcül sonuç veren pulmoner emboli vakaları gözlenmiştir.
Arteriyel tromboembolik olaylar
Sunitinib ile tedavi edilen hastalarda bazıları ölümcül olan arteriyel tromboembolik (ATE) olaylar raporlanmıştır. En sık gözlenen olaylar serebrovasküler olaylar, geçici iskemik atak ve serebral enfarktüs şeklindeydi. Altta yatan malign hastalık ve yaşın 65 veya üzerinde olmasına ek olarak, hipertansiyon, diyabet ve daha önceki tromboembolik hastalık ATE ile ilişkili risk faktörleri arasında yer alır.
Anevrizmalar ve arter diseksiyonları
Vasküler endotelyal büyüme faktör (VEGF) yolak inhibitörlerinin, hipertansiyonu olan veya olmayan hastalarda kullanılması, anevrizmalar ve/veya arter diseksiyonları oluşumunu kolaylaştırabilir. SUTENT'e başlamadan önce hipertansiyon veya anevrizma öyküsü gibi risk faktörleri olan hastalarda bu risk dikkatle değerlendirilmelidir.
Trombotik Mikroanjiyopati (TMA)
Trombotik trombositopenik purpura (TTP) ve hemolitik uremik sendorumu (HUS) dahil olmak üzere, bazen böbrek yetmezliğine veya ölümcül bir sonuca yol açan, hemolitik anemi, trombositopeni, yorgunluk, nörolojik belirtilerde dalgalanma, böbrek yetmezliği ve ateş görülmesi durumunda TMA tanısı düşünülmelidir. TMA gelişen hastalarda sunitinib tedavisi durdurulmalı ve acilen tedaviye başlanmalıdır. Sunitinib tedavisinin bırakılmasından sonra TMA etkilerinin düzeldiği gözlemlenmiştir (Bkz. Bölüm 4.8).
Tiroid disfonksiyonu
Tiroid fonksiyonlarının başlangıçta laboratuvar ölçümleri tüm hastalara önerilir ve hipotiroidizmli veya hipertiroidizmli hastalar, sunitinib tedavisi başlatılmadan önce standart tıbbi uygulama ile tedavi edilmelidirler. Sunitinib tedavisi sırasında her 3 ayda bir tiroid fonksiyonları rutin olarak izlenmelidir. Ek olarak, sunitinib tedavisindeki bütün hastalar, tiroid disfonksiyonun belirtileri ve semptomları için yakından gözlenmelidirler. Tiroid disfonksiyonuna dair belirtileri ve/veya semptomları olan hastalarda tiroid fonksiyonunun laboratuvar takibi yapılmalı ve standart tıbbi tedavi uygulanmalıdır.
Sunitinib ile tedavi edilen hastalarda erken veya geç olarak hipotirodizm gözlenmiştir. (Bkz. Bölüm 4.8)
Pankreatit
SUTENT kullanan çeşitli solid tümörlü hastalarda serum lipazı ve amilazında artışlar görülmüştür. Çeşitli solid tümörlü hastalarda lipaz seviyelerindeki artışlar geçici olmuş ve genellikle pankreatite ait semptomlar veya belirtilere eşlik etmemişlerdir (Bkz. Bölüm 4.8).
Bazıları ölümcül olabilen ağır pankreatit olguları bildirilmiştir. Eğer pankreatit bulguları varsa sunitinib kesilmeli ve uygun destekleyici tedavi yapılmalıdır.
Hepatotoksisite
Sunitinib tedavisi gören hastalarda hepatotoksisite gözlemlenmiştir. Sunitinib tedavisi gören solid tümörlü hastaların %1'inden azında, bazıları ölümle sonuçlanan, karaciğer yetmezliği olguları görülmüştür. Tedaviye başlanmadan, her tedavi siklusunda ve klinik olarak endike olduğu durumlarda karaciğer fonksiyon testleri (alanin transaminaz [ALT], aspartat transaminaz [AST], bilirubin seviyeleri) takip edilmelidir. Eğer karaciğer yetmezliği semptomları veya belirtileri mevcutsa, sunitinib tedavisi durdurulmalı ve durum düzelmediyse tedaviye devam edilmemeli ve uygun destekleyici tedavi yapılmalıdır (Bkz. Bölüm 4.8).
Böbrek fonksiyonu
Böbrek bozuklukları, böbrek yetmezliği ve/veya akut böbrek yetmezliği olan hastalarda, bazıları ölümcül olan olgular bildirilmiştir (Bkz. Bölüm 4.8).
Sunitinib alan hastalardaki böbrek bozukluğu/yetmezliği ile ilişkili risk faktörleri arasında, altta yatan renal hücreli karsinoma ek olarak, ilerlemiş yaş, diyabet, altta yatan böbrek işlev bozukluğu, kalp yetmezliği, hipertansiyon, sepsis, dehidratasyon/hipovolemi ve rabdomiyoliz yer alır.
Orta ve ileri derecede proteinürisi olan hastalarda devam eden sunitinib tedavisinin güvenliliği sistematik olarak değerlendirilmemiştir.
Proteinüri ve nadiren nefrotik sendrom olguları raporlanmıştır. Başlangıçta idrar tahlili yapılması önerilir ve hastalarda proteinürinin gelişimi ya da kötüleşmesi takip edilmelidir. Nefrotik sendromlu hastalarda sunitinib kesilmelidir.
Fistül
Eğer fistül oluşumu gözlenirse, sunitinib tedavisi hemen kesilmelidir. Fistüle sahip hastalarda sunitinib kullanımına devam edilmesi ile ilgili sınırlı bilgi mevcuttur (Bkz. Bölüm 4.8).
Yara iyileşmesinde gecikme
SUTENT tedavisi süresince yara iyileşmesinde gecikme olguları rapor edilmiştir.
Sunitinibin yara iyileşmesi üzerindeki etkisine dair resmi klinik çalışma yapılmamıştır. Majör cerrahi girişim geçirecek olan hastalarda sunitinib tedavisine geçici olarak ara verilmesi önerilir. Majör cerrahi müdahaleden ne kadar sonra sunitinib tedavisine yeniden başlanacağı konusunda sınırlı klinik deneyim mevcuttur. Bu nedenle, majör bir cerrahi müdahaleyi takiben sunitinib tedavisine yeniden devam etme, operasyon sonrası iyileşmeye bağlı klinik değerlendirmeyle kararlaştırılır.
Çenede osteonekroz
SUTENT ile tedavi edilen kanser hastalarında çene osteonekrozu olguları bildirilmiştir. Olguların çoğu daha önceden veya eş zamanlı olarak i.v. bifosfonat tedavisi alan hastalarda bildirilmiş olup bu durum çene osteonekrozu için belirlenmiş bir risk faktörüdür. SUTENT ve i.v. bifosfonatlar aynı anda veya ardı ardına kullanıldığında dikkatli olunmalıdır.
İnvaziv dental girişimler de tanımlanmış risk faktörüdür. SUTENT ile tedaviye başlamadan önce dental muayene ve preventif dental işlemler üzerinde düşünülmelidir. Önceden veya hali hazırda i.v. bifosfonat alan hastalarda mümkünse invaziv dental prosedürlerden kaçınılmalıdır (Bkz. Bölüm 4.8).
Hipersensitivite/Anjiyoödem
Eğer hipersensitivite nedeniyle anjiyoödem oluşursa, sunitinib tedavisi kesilmeli ve standart tıbbi bir bakım yapılmalıdır (Bkz. Bölüm 4.8).
Nöbetler
Sunitinib klinik çalışmalarında ve pazarlama sonrası deneyimlerde nöbetler gözlenmiştir. Hipertansiyon, baş ağrısı, uyarılabilirlikte azalma, mental fonksiyonlarda değişiklik ve kortikal körlüğü kapsayan görme kaybı gibi geri dönüşlü lökoensefalopati sendromunu (RPLS) ile tutarlı nöbetleri ve bulguları/semptomları olan hastalar, hipertansiyonun kontrol altına alınması dahil tıbbi müdahale ile kontrol edilmelidirler. Sunitinibin geçici olarak durdurulması önerilir; düzelmeyi takiben, tedavi eden doktorun kararı ile tedavi devam ettirilebilir (Bkz. Bölüm 4.8).
Tümör Lizis Sendromu (TLS)
Bazıları ölümcül olan TLS olguları klinik çalışmalarda nadir olarak gözlenmiştir ve sunitinib kullanan hastalarda pazarlama sonrası deneyimlerde raporlanmıştır. TLS için risk faktörleri arasında yüksek tümör yükü, önceden var olan kronik böbrek yetmezliği, oligüri, dehidratasyon, hipotansiyon ve asidik idrar bulunur. Bu hastalar yakından takip edilmeli ve klinik olarak belirtildiği şekilde tedavi edilmeli ve profilaktik hidrasyon göz önünde tutulmalıdır.
Enfeksiyonlar
Nötropeni ile birlikte veya nötropeni olmaksızın bazıları ölümle sonuçlanan ciddi enfeksiyonlar bildirilmiştir. Perineum'un da dahil olduğu bazen ölümcül olabilen nekrotizan fasiitis olguları seyrek olarak rapor edilmiştir (Bkz. Bölüm 4.8).
Sunitinib tedavisi nekrotizan fasiitis gelişmiş hastalarda kesilmeli ve uygun tedaviye
hemen başlanmalıdır.
Hipoglisemi
SUTENT tedavisi sırasında bazıları klinik olarak semptomatik olan ve bilinç kaybından dolayı hastaneye kaldırılmayı gerektiren kan şekerinde düşüş bildirilmiştir. Semptomatik hipoglisemi durumunda SUTENT tedavisine geçici olarak ara verilmelidir. Diyabet hastalarında hipoglisemi riskini en aza indirmek için kullanılan anti-diyabetik ilaçların dozunun ayarlanması gerekliliğini değerlendirmek için kan glukoz seviyesi düzenli olarak kontrol edilmelidir (Bkz. Bölüm 4.8).
Sodyum
Bu tıbbi ürün her “dozâ€unda (kapsülde) 1 mmol (23 mg)'dan daha az sodyum ihtiva eder; yani aslında “sodyum içermezâ€.
4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri
Etkileşim çalışmaları sadece yetişkinlerde yapılmıştır.
Sunitinib plazma konsantrasyonunu artırabilen ilaçlar:
CYP3A4 inhibitörlerinin etkisi
Sağlıklı gönüllülerde tek doz sunitinib ile potent CYP3A4 inhibitörü, ketokonazolün eş zamanlı uygulanması sunitinib+primer metabolit kompleksinin C ve EAA değerlerini sırasıyla %49 ve %51 artırır.
SUTENT'in potent CYP3A4 sınıfından diğer inhibitörler (örn. itrakonazol, ritonavir, greyfurt suyu, eritromisin, klaritromisin) ile birlikte uygulanması sunitinib konsantrasyonlarını artırabilir. Bu nedenle, inhibitörlerle birlikte uygulamadan kaçınılmalıdır veya CYP3A4 inhibe edici potansiyeli olmayan veya en az potansiyeli olan alternatif bir eş zamanlı ilaç seçimi düşünülmelidir. Bunun mümkün olmadığı durumlarda, tolere edilebilirlik dikkatli şekilde izlenerek, SUTENT dozunun GİST ve mRHK için günde minimum 37,5 mg'a ve pNET için 25 mg'a indirilmesi gerekebilir (Bkz. Bölüm 4.2).
Meme kanseri direnç proteini (BCRP) inhibitörlerinin etkisi
Sunitinib ile BRCP inhibitörleri arasında etkileşim ile ilgili sınırlı klinik veri bulunmaktadır ve sunitinib ile diğer BCRP inhibitörleri arasında etkileşim olasılığı göz ardı edilemez (Bkz. Bölüm 5.2).
Sunitinib plazma konsantrasyonunu azaltabilen ilaçlar:
CYP3A4 indükleyicilerinin etkisi
Sağlıklı gönüllülerde tek doz SUTENT'in CYP3A4 indükleyicisi, rifampisin ile eş zamanlı uygulanması sunitinib+primer metabolit kompleksinin C ve EAA değerlerini sırasıyla %23 ve %46 azaltmıştır.
SUTENT'in potent CYP3A4 indükleyicileri (örn. deksametazon, fenitoin, karbamazepin, rifampisin, fenobarbital veya sarı kantaron olarak da bilinen Hypericum perforatum/St. John's Wort) ile birlikte uygulanması sunitinib konsantrasyonlarını azaltabilir. Bu nedenle, indükleyicilerle birlikte uygulamasından kaçınılmalıdır veya CYP3A4 indükleyici potansiyeli olmayan veya en az potansiyeli olan alternatif bir eş zamanlı ilaç seçimi düşünülmelidir. Bunun mümkün olmadığı durumlarda, tolere edilebilirlik dikkatli şekilde izlenerek, SUTENT dozunun 12,5 mg'lık artışlarla arttırılması gerekebilir (GİST ve mRHK için günde 87,5 mg'a ya da pNET için günde 62,5 mg'a kadar) (Bkz. Bölüm 4.2).
Özel popülasyonlara ilişkin ek bilgiler:
Pediyatrik popülasyon:
Bu popülasyonla ilgili herhangi bir etkileşim çalışması yapılmamıştır.
4.6. Gebelik ve laktasyon
Genel tavsiye
Gebelik kategorisi: D
Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon) Çocuk doğurma potansiyeli bulunan kadınlar SUTENT tedavisi sırasında gebe kalmama konusunda uyarılmalıdır. Tedavi süresince uygun doğum kontrol yöntemi uygulanmalı ve gebe kalmaktan kaçınılmalıdır.
Gebelik dönemi
Gebe kadınlar üzerinde yapılan bir çalışma yoktur. Hayvanlarda yapılan çalışmalar, fetal malformasyonlar dahil olmak üzere üreme toksisitesi göstermiştir (Bkz. Bölüm 5.3). Gebelik döneminde SUTENT kullanılırsa veya hasta SUTENT tedavisi sırasında gebe kalırsa; hasta, ilacın fetüs üzerindeki potansiyel zararlı etkisi konusunda uyarılmalıdır.
Laktasyon dönemi
Sunitinib ve/veya metabolitleri sıçan sütüne geçmektedir. Sunitinib veya onun primer aktif metabolitinin, insan sütüne geçip geçmediği bilinmemektedir. İlaçların genelde insan sütüne geçmesi ve emzirilen bebekler üzerinde ciddi advers etki potansiyeli nedeniyle SUTENT tedavisi sırasında emzirme durdurulmalıdır.
Üreme yeteneği/Fertilite
Klinik dışı bulgulara dayalı olarak, erkek ve dişi fertilitesi SUTENT tedavisinden etkilenebilir. (Bkz. Bölüm 5.3)
4.7. Araç ve makine kullanımı üzerindeki etkiler
SUTENT araç ve makine kullanımı yeteneği üzerinde minor etkilere sahiptir. Hastalar SUTENT ile tedavi sırasında baş dönmesi olabileceği konusunda uyarılmalıdır.
4.8. İstenmeyen etkiler
Sunitinib ile ilişkilendirilen ve bazıları ölümcül olan en önemli ciddi advers etkiler böbrek yetmezliği, kalp yetmezliği, pulmoner emboli, gastrointestinal perforasyon ve hemorajidir (örn. solunum yolları, gastointestinal, tümör, idrar yolları ve beyin hemorajileri). Herhangi bir derecedeki en yaygın advers etkiler (RHK, GİST ve pNET çalışmalarındaki hastalarda görülen) iştahta azalma, tat alma bozuklukları, hipertansiyon, bitkinlik, gastrointestinal bozukluklar (örn. diyare, bulantı, stomatit, dispepsi ve kusma), ciltte renk farklılaşması ve palmar-plantar eritrodizestezi sendromudur. Bu semptomlar tedavi devam ederken hafifleyebilir. Hipotirodizm tedavi esnasında ortaya çıkabilir. Hematolojik rahatsızlıklar (örn. nötropeni, trombositopeni ve anemi) çok yaygın advers etkilerdendir.
Bölüm 4.4 ve 4.8'de belirtilen advers etkiler dışında sunitinib tedavisi ile ilgili olması olası olan ölümcül olaylar çoklu organ yetmezliği, disemine intravasküler koagülasyon, peritoneal hemoraji, adrenal yetmezlik, pnömotoraks, şok ve ani ölümdür.
7115 kişilik bir veri setinden GİST, mRHK ve pNET hastalarında bildirilen advers reaksiyonlar, sistem, organ, sınıf ve sıklık derecesine göre aşağıda listelenmiştir. Klinik çalışmalarda belirlenen pazarlama sonrası yan etkiler de yer almaktadır.
Çok yaygın (≥1/10), yaygın (≥1/100 ila <1/10), yaygın olmayan (≥1/1.000 ila <1/100), seyrek (≥1/10.000 ila <1/1.000), çok seyrek (<1/10.000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor) olarak tanımlanmaktadır.
Enfeksiyonlar ve enfestasyonlar
Yaygın: Viral enfeksiyonlar, solunum enfeksiyonları, abseler, mantar
enfeksiyonları, idrar yolu enfeksiyonları, deri enfeksiyonları, sepsis
Yaygın olmayan: Nekrotizan fasiitis, bakteriyel enfeksiyonlar
Kan ve lenf sistemi hastalıkları
Çok Yaygın: Anemi, trombositopeni, nötropeni, lökopeni
Yaygın: Lenfopeni
Yaygın olmayan: Pansitopeni
Seyrek: Trombotik mikroanjiyopati
Bağışıklık sistemi hastalıkları Yaygın olmayan: Hipersensitivite Seyrek: Anjiyoödem
Endokrin hastalıkları
Çok Yaygın: Hipotiroidizm Yaygın olmayan: Hipertiroidizm Seyrek: Tiroidit
Metabolizma ve beslenme hastalıkları
Çok Yaygın: İştahta azalma
Yaygın: Dehidratasyon, hipoglisemi
Seyrek: Tümör lizis sendromu
Psikiyatrik hastalıklar Çok Yaygın: Uykusuzluk Yaygın: Depresyon
Sinir sistemi hastalıkları
Çok Yaygın: Sersemlik, baş ağrısı, tat alma bozuklukları Yaygın: Parestezi, periferal nöropati, hipoestezi, hiperestezi
Yaygın olmayan: Serebral hemoraji,serebrovasküler olay, geçici iskemik atak
Seyrek: Geri dönüşlü posterior ensefalopati sendromu
Göz hastalıkları
Yaygın: Periorbital ödem, lakrimasyon artışı, göz kapağında ödem
Kardiyak hastalıklar
Yaygın: Miyokardiyal iskemiejeksiyon fraksiyonunda azalma
Yaygın olmayan: Miyokardiyal enfarktüs*, kalp yetmezliği, konjestif kalp yetmezliği,
kardiyomiyopati, perikard efüzyonu, elektrokardiyogramda QT uzaması Seyrek: Sol ventriküler yetmezlik, Torsades de pointes
Vasküler hastalıklar
Çok Yaygın: Hipertansiyon
Yaygın: Derin ven trombozu, sıcak basma, yüzün kızarması Yaygın olmayan: Tümör hemoraji
Bilinmiyor: Anevrizmalar ve arter diseksiyonları
Solunum, göğüs hastalıkları ve mediastinal hastalıklar
Çok Yaygın: Burun kanaması, dispne, öksürük
Yaygın: Pulmoner emboli, plevral efüzyon, hemoptizi, orofarengeal ağrı, nazal konjesyon, burunda kuruluk
Yaygın olmayan: Pulmoner hemoraji, solunum yetmezliği
Gastrointestinal hastalıklar
Çok Yaygın: Stomatit, abdominal ağrı, kusma, diyare, dispepsi, mide bulantısı, kabızlık
Yaygın: Gastro-özofageal reflü hastalığı, disfaji, gastrointestinal hemoraji özofajit,
abdominal distansiyon, abdominal rahatsızlık, rektal hemoraji, diş eti kanaması, ağız ülseri, proktalji, keilitis, hemoroid, glossodini, oral ağrı, gaz, ağız kuruluğu, ağızda rahatsızlık hissi, geğirme
Yaygın olmayan: Gastrointestinal perforasyonpankreatit, anal fistül, kolit
Hepato-biliyer hastalıklar
Yaygın olmayan: Karaciğer yetmezliği, kolesistitkaraciğer fonksiyon anormallikleri
Seyrek: Hepatit
Deri ve derialtı doku hastalıkları
Çok Yaygın: Ciltte renk değişikliği, palmar-plantar eritrodizestezi sendromu, döküntü, saç renginde değişim, cilt kuruluğu
Yaygın: Ciltte soyulma, deri reaksiyonu, egzema, su toplaması, eritem, saç dökülmesi, akne, prurit, cilt hiperpigmentasyonu, deride lezyon, hiperkeratoz, dermatit, tırnak bozuklukları
Seyrek: Eritema multiforme, Stevens-Johnson sendromu, gangrenli piyodermi, toksik
epidermal nekroz
Kas-iskelet hastalıkları, bağ dokusu ve kemik hastalıkları Çok Yaygın: Ekstremitelerde ağrı, eklemlerde ağrı, sırt ağrısı Yaygın: Kas iskelet ağrısı, kas güçsüzlüğü, miyalji, kas spazmı Yaygın olmayan: Çenede osteonekroz, fistül
Seyrek: Rabdomiyoliz, miyopati
Böbrek ve idrar yolu hastalıkları
Yaygın: Kromatüri, böbrek yetmezliği, akut böbrek yetmezliği
proteinüri
Yaygın olmayan: İdrar yolunda hemoraji
Seyrek: Nefrotik sendrom
Genel bozukluklar ve uygulama bölgesine ilişkin hastalıklar Çok Yaygın: Mukozal inflamasyon, bitkinlik, ödem, ateş Yaygın: Göğüs ağrısı, ağrı, grip benzeri belirtiler, ürperme Yaygın olmayan: Yara iyileşmesinde gecikme
Araştırmalar
Yaygın: Kilo kaybı, beyaz kan hücresi sayısında azalma, lipaz artışı, trombosit sayısında azalma, hemoglobin azalması, amilaz artışı, aspartat amino transferazda artış, alanin amino transferaz artışı, kanda kreatinin artışı, kan basıncı artışı, kanda ürik asit artışı Yaygın olmayan: Kanda kreatin fosfokinaz artışı, kanda tiroid stimülan hormon (TSH) artışı
*Ölümcül olayları içermektedir
Aşağıdaki terimler birleştirilmiştir:
sendrom
uPsöriyaziform dermatit, eksfoliyatif döküntü, döküntü, eritrematöz döküntü, foliküler döküntü, generalize döküntü, maküler döküntü, makülo papüler döküntü, papüler döküntü, pruritik döküntü
Seçilmiş advers etkilerin tanımları:
Enfeksiyonlar ve enfestasyonlar
Bazı olgularda, nötropeninin eşlik ettiği veya etmediği kimileri ölümcül olan ciddi enfeksiyonlar rapor edilmiştir. Bazen fatal olabilen, perineumunki de dahil olmak üzere nekrotizan fasiit gözlenmiştir (Bkz. Bölüm 4.4).
Kan ve lenf sistemi bozuklukları
Grade 3 ve 4 şiddetindeki olgularda azalmış mutlak nötrofil sayısı, Faz III GİST çalışmasındaki hastaların sırasıyla %10 ve %1,7'sinde, Faz III mRHK çalışmasındaki hastaların %16 ve %1,6'sında ve Faz III pNET çalışmasındaki hastaların %13 ve
%2,4'ünde bildirilmiştir. Grade 3 ve 4 şiddetindeki olgularda azalmış trombosit sayısı,
Faz III GİST çalışmasındakihastalarınsırasıyla%3,7ve %0,4'ünde, Faz III mRHK
çalışmasındaki hastaların %8,2 ve %1,1'inde ve Faz III pNET çalışmasındaki hastaların
%3,7 ve %1,2'sinde bildirilmiştir (Bkz. Bölüm 4.4).
Bir Faz III GİST çalışmasında, plasebo alan hastaların %17'sine karşılık, SUTENT alan hastaların %18'inde kanama bulguları oluşmuştur. Daha önceden tedavi almamış mRHK hastalarında, interferon-α (IFN-α) alan hastaların %11'ine karşılık, SUTENT alan hastaların %39'u kanama bulguları göstermiştir. IFN-α grubundaki hastaların 5'ine (%1,7) karşı, SUTENT kullananlardan 17 (%4,5) hasta Grade 3 kanama bulgusu göstermiştir. Sitokin-refrakter mRHK için SUTENT alan hastaların %26'sında kanama gözlenmiştir. Faz III pNET çalışmasında plasebo kullanan hastaların %9,85'ine kıyasla sunitinib kullanan hastaların %21,7'sinde burun kanaması dışında kanama olayları gözlenmiştir (Bkz. Bölüm 4.4).
Klinik araştırmalarda GİST'i olan hastaların yaklaşık %2'sinde tümör hemorajisi meydana gelmiştir.
Bağışıklık sistemi bozuklukları
Anjiyoödemi de içeren aşırı duyarlılık reaksiyonları raporlanmıştır (Bkz. Bölüm 4.4).
Endokrin hastalıkları
Daha önceden tedavi almamış hastalarla yapılan bir mRHK çalışmasında, sunitinib kullanan hastaların 61'inde (%16) ve IFN-α kolunda 3 hastada (<%1) ve sitokine- refrakter hastalarla yapılan iki mRHK çalışmasında hastaların 7'sinde (%4) advers etki olarak hipotiroidizm rapor edilmiştir.
Ayrıca, sitokin-refrakter mRHK hastaların 4 (%2) tanesinde TSH yükselmeleri rapor edilmiştir. Sonuç olarak, mRHK popülasyonunun %7'sinde, tedaviyle ortaya çıkan hipotiroidizmin klinik ve laboratuvar kanıtı vardır. Gelişen hipotiroidizm sunitinib kullanan GİST'i olan hastaların %6,2'sinde görülürken bu oran plasebo kullanan hastalarda %1'dir. Faz III pNET çalışmasında sunitinib alan 6 hastada (%7,2) ve plasebo alan bir hastada (%1,2) hipotiroidizm bildirilmiştir.
Meme kanseri olan hastalardaki prospektif olarak yürütülen iki çalışmada tiroid fonksiyonu izlendi; SUTENT'in meme kanserinde kullanımı onaylanmamıştır. Bir çalışmada, sunitinib alan 15 (%13,6) hastada ve standard bakım gören 3 (%2,9) hastada hipotiroidizm raporlanmıştır. Kan TSH'sinde artış, sunitinib alan 1 (%0,9) hastada bildirilmiş ve standart bakım gören kimsede rastlanmamıştır. Hipertiroidizm, sunitinib ile tedavi edilen herhangi bir hastada raporlanmamıştır ve standard bakım gören 1 hastada (%1,0) raporlanmıştır. Bir diğer çalışmada; hipotiroidizm, sunitinib alan toplam 31 (%13) hastada ve kapesitabin alan 2 (%0,8) hastada bildirilmiştr. Kan TSH artışı, sunitinib alan 12 (%5,0) hastada bildirilmiştir ve kapesitabin alan hiçbir hastada görülmemiştir. Hipertiroidizm, sunitinib alan 4 (%1,7) hastada bildirildi ve kapesitabin alan hiçbir hastada bildirilmemiştir. Kan TSH düşüşü, sunitinib alan 3 (%1,3) hastada bildirilmiştir ve kapesitabin alan hiçbir hastada bildirilmemiştir. T4 artışı, sunitinib alan 2 (%0,8) hastada bildirilmiştir ve kapesitabin alan 1 (%0,4) hastada bildirilmiştir. T3 artışı, sunitinib alan 1 (%0,8) hastada ve kapesitabin alan hiçbir hastada bildirilmemiştir. Bildirilen tüm tiroid-ilişkili olaylar Grade 1 ve 2 ‘de olmuştur (Bkz. Bölüm 4.4).
Metabolizma ve beslenme bozuklukları
mRHK ve GİST hastaları ile karşılaştırıldığında pNET hastalarında daha fazla sıklıkta hipoglisemi olayları rapor edilmiştir. Yine de klinik çalışmalarda gözlemlenen bu yan etkilerin çoğunun çalışma tedavisi ile ilgili olduğu düşünülmemiştir (Bkz. Bölüm 4.4).
Sinir sistemi bozuklukları
Sunitinib'in klinik çalışmalarında ve pazarlama sonrası deneyimlerinde, geri dönüşümlü posterior lökoensefalopati'nin nöbet ve radyolojik kanıtı bulunan hastalarda, kimisi ölümcül olan az sayıda (%<1) bildirim olmuştur. Beyin metastazına ait radyolojik kanıtı olan veya olmayan hastalarda nöbet gözlemlenmiştir (Bkz. Bölüm 4.4).
Kardiyak bozukluklar
SUTENT ile tedavi gören GİST'i olan hastaların yaklaşık %2'sinde, sitokine refrakter mRHK hastalarının %4'ünde ve plasebo ile tedavi gören hastaların %2'sinde, SVEF normalin en düşük sınırının altında ve %20'den daha fazla azalmalar olmuştur. SVEF'deki bu düşüşler düzenli bir progresyon göstermeyip, sıklıkla tedavinin devamında iyileşmeyle sonuçlanmıştır. Daha önceden tedavi almamış mRHK çalışmasında, SUTENT ve IFN-α'daki hastaların sırasıyla, %27'si ve %15'i, normalin alt sınırının altında bir SVEF değeri göstermiştir. Sunitinib alan iki hastada (<%1) konjestif kalp yetmezliği (KKY) teşhis edilmiştir.
GİST hastalarının %1,2'sinde ve plasebo ile tedavi gören hastaların %1'inde 'kalp yetmezliği', 'konjestif kalp yetmezliği veya 'sol ventrikül yetmezliği' gibi advers olaylar bildirilmiştir. Pivotal Faz III GİST çalışmasında (n=312) tedaviye bağlı ölümcül kardiyak reaksiyonlar çalışmanın her iki kolunda (sunitinib ve plasebo) %1 oranında görülmüştür. Sitokine refrakter mRHK hastalarında yapılan Faz II çalışmada hastalardan %0,9'unda tedaviye bağlı ölümcül miyokard enfarktüsü görülürken daha önceden tedavi almamış mRHK hastalarında yapılan Faz III çalışmada IFN-α alan hastaların %0,6'sında ölümcül kardiyak olaylar görülmüş olup sunitinib alan hastalarda bu oran %0'dır. Faz III pNET çalışmasında, sunitinib alan bir (%1) hastada tedavi ile ilişkili fatal kalp yetmezliği meydana gelmiştir.
Vasküler hastalıklar
Hipertansiyon: Hipertansiyon klinik çalışmalarda çok yaygın olarak bildirilmiştir. Bu hasta popülasyonunun yaklaşık %2,7'sinde SUTENT dozu azaltılmış veya geçici olarak ertelenmiştir. Bu hastaların hiçbirinde SUTENT ile tedaviye son verilmemiştir. Bu hasta popülasyonunun yaklaşık %4,7'sinde ciddi hipertansiyon (>200 mmHg sistolik veya 110 mmHg diyastolik) meydana gelmiştir. Hipertansiyon, daha önceden tedavi görmemiş mRHK için IFN-α alan hastaların %3,6'sına karşılık, SUTENT alan hastaların yaklaşık
%33,9'unda rapor edilmiştir. Ciddi hipertansiyon, önceden tedavi edilmemiş SUTENT hastalarının %12'sinde ve IFN-α alan hastaların %1'inden azında oluşmuştur. Faz III pNET çalışmasında hipertansiyon plasebo alan hastaların %4,9'unda raporlanmışken sunitinib alanlar hastalarda bu oran %26,5'dir. pNET olan hastalarda şiddetli hipertansiyon, sunitinib alanların %10'unda ve plasebo alanların %3'ünde meydana gelmiştir.
Venöz tromboembolik olaylar: GİST ve mRHK dahil olmak üzere yapılan klinik çalışmalarda sunitinib alan solid tümörlü hastaların yaklaşık %1'inde tedaviye bağlı venöz tromboembolik olaylar raporlanmıştır.
Bir Faz III GİST çalışmasında plasebo alan herhangi bir hastada venöz tromboembolik olay tespit edilmemesine karşın SUTENT alan yedi hastada (%3) venöz tromboembolik olay tespit edilmiştir: bu yedi hastanın beşinde, Grade 3 derin ven trombozu (DVT) ve ikisinde Grade 1 ya da 2 derin ven trombozu (DVT) gelişmiştir. Bu yedi GİST hastasının dördünde, ilk DVT incelemesini takiben tedavi kesilmiştir.
Tedavi görmemiş mRHK hastalarında yapılan Faz III çalışmada sunitinib alan on üç hastada (%3) ve iki sitokine refrakter mRHK çalışmasındaki dört hastada (%2) venöz trombolik olay raporlanmıştır. Bu hastaların dokuzunda pulmoner emboli mevcut olup, birinde Grade 2, diğer sekiz tanesinde ise Grade 4 derecesindedir. Bu hastalardan 8'inde birinde Grade 1, ikisinde Grade 2, dördünde Grade 3 ve birinde Grade 4 derecesinde olmak üzere DVT bulunmaktadır. Sitokine refrakter mRHK çalışmasındaki pulmoner embolisi mevcut olan bir hastada doz kesilmesine gerek duyulmuştur.
IFN-α alan tedavi görmemiş mRHK hastalarının altısında (%2), venöz tromboembolik olaylar gözlenmiştir; bir hastada (<%1) Grade 3 DVT ve beş hastanın (%1) hepsinde Grade 4 olan pulmoner emboli görülmüştür.
Faz III pNET çalışmasında sunitinib alan 1 hastada (%1,2) ve plasebo alan 5 hastada (%6,1) venöz tromboembolik olaylar bildirilmiştir. Plasebo kullanan 2 hastada biri Grade 2 biri Grade 3 olmak üzere DVT bulunmaktadır.
GİST, mRHK ve pNET çalışmalarının hiç birinde ölümcül bir olgu bildirilmemiştir. Ölüm ile sonuçlanan olgular pazarlama sonrası görülmüştür.
Faz III çalışmalarında sunitinib alan GİST hastalarının yaklaşık %3,1'inde ve mRHK hastalarının yaklaşık %1,2'sinde pulmoner emboli olguları gözlenmiştir. Faz III çalışmalarında sunitinib alan pNET hastalarında pulmoner emboli görülmemiştir. Pazarlama sonrası çalışmalarda ölümle sonuçlanan seyrek olgular gözlenmiştir.
SUTENT uygulamasından önce 12 ay içinde pulmoner emboli yaşayan hastalar sunitinib klinik çalışmalarına dahil edilmemiştir.
Faz III çalışmalarında sunitinib alan hastalardan, GİST hastalarının yaklaşık
%17,8'inde, mRHK hastalarının yaklaşık %26,7'sinde ve pNET hastalarının %12'sinde pulmoner olaylar (dispne, plevral efüzyon, pulmoner emboli veya pulmoner ödem gibi) bildirilmiştir.
Klinik çalışmalarda sunitinib alan GİST ve mRHK hastaları dahil solid tümörlü hastaların yaklaşık %22,2'sinde pulmoner olaylar gözlemlenmiştir.
Gastrointestinal bozukluklar
GİST veya mRHK tedavisi için sunitinib alan hastalarda nadiren (%1'inden az) pankreatit görülmüştür. Faz III pNET çalışmasında tedavi ile ilişkili pankreatit bildirilmemiştir (Bkz. Bölüm 4.4).
Bir Faz III GİST çalışmasında plasebo alan hastaların %0,98'inde ölümcül gastrointestinal kanama görülmüştür.
Hepato-biliyer bozukluklar
Karaciğer fonksiyon testi anormalliklerini, hepatit veya karaciğer yetmezliğini içerebilen hepatik disfonksiyon bildirilmiştir (Bkz. Bölüm 4.4).
Deri ve deri altı doku hastalıkları
İlacın bırakılmasıyla geri dönüşlü olan piyoderma gangrenosum (ağrılı deri ülseri) bildirilmiştir (Bkz. Bölüm 4.4).
Kas-iskelet sistemi ve bağ dokusu bozuklukları
Bazıları akut böbrek yetmezliği ile birlikte olan miyopati ve/veya rabdomiyoliz olguları rapor edilmiştir. Kas toksisitesinin belirtileri veya semptomları görülen hastaların bakımları standart tıbbi uygulamalar doğrultusunda yapılmalıdır (Bkz. Bölüm 4.4).
Bazı olgularda ölümle sonuçlanan, bazen tümör nekrozu ve regresyonu ile bazen ilişkili
olan fistül oluşumu rapor edilmiştir (Bkz. Bölüm 4.4).
SUTENT ile tedavi edilen kanser hastalarında çene osteonekrozu olguları bildirilmiştir; olguların çoğu daha önceden veya eş zamanlı olarak i.v. bisfosfonat tedavisi almışlardır ve/veya invazif dental işlemler gerektiren dental hastalık hikayesine sahiptirler ve bu durumlar çene osteonekrozu için belirlenmiş risk faktörleridir (Bkz. Bölüm 4.4).
Araştırmalar
Klinik olmayan çalışmalardan (in vitro ve in vivo ) elde edilen veriler, önerilen insan dozunun üzerindeki dozlarda sunitinibin kalp aksiyon potansiyelinin repolarizasyon sürecini inhibe ettiğini yani QT uzamasına neden olduğunu ortaya koymaktadır.
Solid tümörlü 450 hastanın %1,1'inde 60 msn'den fazla sürede başlangıca kıyasla değişimler ve %0,5'inde ise QTc aralığında 500 msn'yi aşan artışlar meydana gelmiştir; bu parametrelerin her ikisi de potansiyel anlamlı değişiklikler olarak kabul edilmiştir. Yaklaşık olarak terapötik konsantrasyonun iki katında sunitinibin QTkF (Frederika konsantrasyonu) aralığını uzattığı gösterilmiştir.
QT aralığı uzaması, ilerlemiş maligniteli 20-87 yaşları arasında 24 hastanın katıldığı bir deney ile araştırılmıştır. Bu çalışmanın terapötik konsantrasyonlarda (3. gün) baseline correction metodu kullanılarak ve terapötik dozdan daha fazla (9. gün) dozda her iki başlangıç düzeltme metodu kullanılarak elde edilen sonuçları sunitinibin QT aralığı (%90 GA, üst limit >15 msn ile ortalama plasebo ayarlı değişiklik >10 msn olarak tanımlanmıştır) üzerinde etkisi olduğunu göstermiştir. Hiçbir hastanın QT aralığı >500 msn değildir. 3. günde dozdan 24 saat sonra (yani 50 mg'lik önerilen başlangıç dozundan sonra beklenen terapötik plazma konsantrasyonunda) baseline correction metodu kullanılarak QTcF aralığı üzerinde bir etki gözlenmiş olmasına rağmen bu bulgunun klinik olarak önemi belirsizdir.
Terapötik doza ya da terapötik maruz kalmadan daha büyük doza karşılık gelen zamanlarda kapsamlı birseriEKGdeğerlendirmesi yapıldığında değerlendirilebilir veya
tedavisi amaçlanan hasta (ITT) grubundan hiçbirinde görülen QT aralığında uzama “ciddi†(yani yan etkiler için ortak terminoloji kriteri (CTCAE) versiyon 3.0'a göre Grade 3'e eşit veya büyük) değildir.
Tedavi edici plazma konsantrasyonlarında, başlangıca göre maksimum ortalama QTcF değişikliği 9 msn (%90 GA: 15,1 msn) olmuştur. Tedavi edici konsantrasyonların yaklaşık olarak iki katında, başlangıca göre maksimum QTcF ortalama değişikliği 15,4 msn (%90 GA: 22,4 msn) olmuştur. Bir pozitif kontrol olarak kullanılan moksifloksasin (400 mg), başlangıca göre 5,6 msn'lik bir maksimum ortalama QTcF değişikliği göstermiştir. Herhangi bir hastada Grade 2'den (CTCAE v.3.0) daha büyük QTc aralığı gözlenmemiştir (Bkz. Bölüm 4.4).
mRHK'da uzun dönem güvenlilik
mRHK hastalarında sunitinibin uzun dönem güvenliliği; ilk basamak; bevasizumab- refrakter ve sitokin-refrakter tedavi rejimlerinde 5739 hastaya ait verilere dayanarak (bu hastaların 807'si (%14) ≥2 yıldan 6 yıla kadar tedavi görmüştür) tamamlanmış 9 klinik çalışmaya dahil olan hastalar analiz edilmiştir. Uzun dönem sunitinib tedavisi alan 807 hastada; 6 yıllık periyod boyunca yeni vaka olarak ortaya çıkabilen ve zaman içinde artan hipotiroidi hariç olmak üzere; tedavi ile ilişkili advers olayların (TİAO) çoğu öncelikle ilk 6 ay-1 yılda oluşmuş ve sonrasında stabil seyretmiş ya da zamanla sıklıkta azalma göstermiştir. Sunitinib ile uzatılmış tedavi yeni TİAO tipleri ile ilişkili görünmemektedir.
Özel popülasyonlara ilişkin ek bilgiler:
Pediyatrik popülasyon:
Sunitinib'in güvenlilik profili, aşağıda açıklandığı gibi bir Faz I doz arttırma çalışmasından, bir Faz II açık etiketli çalışmadan, bir Faz 1/2 tek kollu çalışmadan ve yayınlardan elde edilmiştir.
Refrakter solid tümörleri olup, büyük bölümü primer beyin tümörü tanısıyla katılan 35 hastada; 30'u pediyatrik hastalardan (3-17 yaş) ve 5 'i genç erişkin hastalardan (18-21 yaş) oluşan, oral sunitinibe ilişkin bir Faz I doz artış çalışması gerçekleştirilmiştir. Çalışmaya katılan herkeste yan etkiler gözlemlenmiştir; kardiyak toksisite de dahil bu yan etkilerin çoğu ciddidir (toksisite grade ≥3). En yaygın görülen yan etkiler, gastrointestinal (GI) toksisite, nötropeni, halsizlik ve ALT yükselmesidir. Kardiyak yan etki riski daha önceden antrasiklinlere veya kardiyak irradyasyona maruz kalmış pediyatrik hastalarda kalmamışlara oranla daha yüksek olmuştur. Daha önceden antrasiklinlere veya kardiyak irradyasyona maruz kalmasından bağımsız olarak bu pediyatrik hastalarda maksimum tolere edilen doz (MTD) tanımlanmıştır (Bkz. Bölüm 5.1).
Reküran/progresif/refrakter ileri dereceli gliomu (HGG) veya epandimomu bulunan, 27'si pediyatrik (3-16 yaş) ve 2'si genç erişkin hastadan (18-19 yaş) oluşan 29 hastada, Faz II açık etiketli bir çalışma gerçekleştirilmiştir. Her iki grupta da Grade 5 advers reaksiyon görülmemiştir. En yaygın (≥% 10) tedaviye bağlı advers reaksiyonlar nötrofil sayısının azalması (6 [%20,7] hasta) ve intrakraniyal hemoraji (3 [%10,3] hasta) olmuştur.
İleri anrezektabl GİST'i olan 6 pediyatrik hastada (13-16 yaş) Faz 1/2 tek kollu bir çalışma yapılmıştır. En sık görülen advers ilaç reaksiyonları, diyare, mide bulantısı, beyaz kan hücresi sayısında azalma, nötropeni ve baş ağrısı olmuştur ve her biri 3 (%50) hastada primer olarak Grade 1 veya 2 seviyesinde görülmüştür. 6 hastadan 4'ünde (%66,7) Grade 3-4 tedaviye bağlı advers reaksiyon (Grade 3 hipofosfatemi, nötropeni ve trombositopenin her biri 1 hastada ve 1 hastada Grade 4 nötropeni) görülmüştür. Bu çalışmada ciddi advers reaksiyonlar veya Grade 5 advers reaksiyonlar bildirilmemiştir. Hem klinik çalışmada hem de yayınlarda, güvenlilik profili yetişkinlerde bilinen güvenlilik profili ile uyumlu olmuştur.
Şüpheli advers reaksiyonların raporlanması
Ruhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar/risk dengesinin sürekli olarak izlenmesine olanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu Türkiye Farmakovijilans Merkezi (TUFAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e- posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks: 0 312 218
35 99)
4.9. Doz aşımı ve tedavisi
SUTENT kullanımında doz aşımının tedavisi için spesifik bir antidot yoktur ve doz aşımı tedavisi için genel destekleyici ölçümler gerekmektedir. Endike ise, emilmemiş ilacın eliminasyonu emesis ve gastrik lavaj ile yapılabilir. Doz aşımı olguları bildirilmiştir; bazı olgular SUTENT'in bilinen güvenlilik profiliyle uyumlu olan advers olaylarla ilişkilidir.
5. FARMAKOLOJİK ÖZELLİKLER
5.1. Farmakodinamik özellikler
Farmakoterapötik grup: Antinoeplastik ve immünomodülatör ilaçlar, antineoplastik ajanlar,
protein kinaz inhibitörleri, diğer protein kinaz inhibitörleri ATC kodu: L01EX01
Sunitinib; tümör gelişiminde, neoanjiyogenezde ve kanserin metastatik progresyonunda rol oynayan bir çok tirozin kinaz reseptörünü (TKR) inhibe eder. Sunitinib trombosit kaynaklı büyüme faktörü reseptörleri (PDGFRα ve PDGFRβ), VEGF reseptörleri (VEGFR1, VEGFR2 ve VEGFR3), kök hücre faktör reseptörü (KIT), Fms-tipi tirozin kinaz-3 (FLT3), koloni uyarıcı faktör reseptörü (CSF- 1R) ve glial hücre kaynaklı nörotrofik faktör reseptörünün (RET) inhibitörü olarak tanımlanmıştır. Başlıca metaboliti olan desetil sunitinibin, biyokimyasal ve hücresel testlerde sunitinibe benzer etkide olduğu gösterilmiştir.
Klinik Çalışmalar
SUTENT'in klinik güvenlilik ve etkililiği imatinibe toleransı olmayan veya imatinib rezistan olan (imatinib tedavisi süresince veya sonrasında hastalık progresyonu görülen) malign gastrointestinal stromal tümörlü (GİST) hastaların tedavisinde ve metastatik renal hücre karsinomlu (mRHK) hastaların tedavisinde ve anrezektabl pNET olan hastaların tedavisinde araştırılmıştır.
Etkililik; GİST'de tümör progresyonuna kadar geçen süreyi (TTP) ve sağkalımdaki artışı, tedavi uygulanmamış mRHK için progresyonsuz sağkalımı ve sitokine dirençli mRHK için objektif yanıt oranlarını, pNET için de progresyonsuz sağkalımı temel almıştır.
Gastrointestinal Stromal Tümörler (GİST)
İmatinibe (medyan maksimum günlük doz 800 mg) dirençli ve intoleransı olmasından dolayı GİST tedavisinde başarısız olunan hastalarda açık etiketli, doz ayarlama çalışması gerçekleştirilmiştir. Çalışmaya ilacı farklı doz ve doz şeması uygulanan sürelerde uygulayan 97 adet hasta dahil edilmiştir; bunlardan 55'i ilacı önerilen tedavi süresi olan 4 hafta kullanıp, 2 hafta ara vermek suretiyle (''4/2 şeması'') 50 mg olarak almıştır. Bu çalışmada medyan TTP 34,0 haftadır ( %95 GA = 22,0 hafta-46,0 hafta).
İmatinibi tolere edemeyen veya imatinib (medyan maksimum günlük doz 800 mg) ile tedavi esnasında veya sonrasında hastalık progresyonu görülen GİST'i olan hastalarda randomize, çift-kör ve plasebo-kontrollü bir Faz III çalışma gerçekleştirilmiştir. Bu çalışmada 312 hasta, hastalık progresyonu veya çalışmadan başka bir sebeple çekilme olmadığı sürece 50 mg SUTENT veya plaseboyu ağızdan günde bir kez ve 4/2 şemasına göre alacak şekilde randomize (2:1) edilmiştir (hastaların 207'si SUTENT, 105'i plasebo almıştır). Çalışmanın primer etkililik sonlanım noktası, randomizasyondan objektif tümör progresyona kadar geçen süre olarak tanımlanan TTP idi.
Önceden belirlenen ara dönem analizinde, SUTENT için medyan TTP (progresyona kadar geçen süre) araştırmacı değerlendirmesinde 28,9 hafta (%95 GA=21,3-34,1 hafta) ve bağımsız değerlendirmeye göre 27,3 hafta (%95 GA=16,0-32,1) olup, plasebo kolundaki araştırmacı değerlendirmesine göre 5,1 haftalık (%95 GA=4,4-10,1), bağımsız değerlendirmedeki 6,4 haftalık (%95 GA=4,4-10,0) TTP'den istatistiksel anlamlı olarak daha uzun olmuştur. Genel sağkalım (OS)'deki fark istatistiksel olarak sunitinib lehinedir [HR: 0,491 (%95 GA:0,290-0,831)]. Ölüm riski sunitinib koluyla karşılaştırıldığında plasebo kolunda 2 kat fazladır.
Etkililik ve güvenlilik interim analiz sonuçları sonrası çalışma körlemeden çıkarılmış ve plasebo kolundaki hastalara açık etiketli sunitinib tedavisi önerilmiştir.
Başta plasebo alan 99 hastada dahil çalışmanın açık etiketli tedavi fazında toplam 255
hasta sunitinib almıştır.
Çalışmanın açık etiket fazındaki primer ve sekonder sonlanım noktası analizleri zamanında elde edilen interim analiz sonuçlarını doğrulamıştır (Bkz. Tablo 1).
Tablo 1 GİST etkilik sonlanım noktası özetleri (ITT popülasyonu)
| Çift-kör tedavi |
| |||
| Medyan (%95 GA) | Risk Oranı | Plasebo / Çapraz geçiş grubu | ||
Sonlanım noktası | SUTENT | Plasebo | (%95 GA) | p | tedavi |
Primer |
| ||||
TTP (hafta) |
| ||||
Ara | 27,3 (16,0-32,1) | 6,4 (4,4-10,0) | 0,329 (0,233-0,466) | <0,001 | - |
Final | 26,6 (16,0-32,1) | 6,4 (4,4-10,0 ) | 0,339 (0,244-0,472) | <0,001 | 10,4 (4,3-2,0) |
Sekonder |
| ||||
PFS (hafta) |
| ||||
Ara | 24,1 (11,1-28,3) | 6,0 (4,4-9,9) | 0,333 (0,238-0,467) | <0,001 | - |
Final | 22,9 (10,9-28,0) | 6,0 (4,4-9,7) | 0,347 (0,253-0,475) | <0,001 | - |
ORR (%) |
| ||||
Ara | 6,8 (3,7-11,1) | 0 (-) | NA | 0,006 | - |
Final | 6,6 (3,8-10,5) | 0 (-) | NA | 0,004 | 10,1 (5,0-17,8) |
OS (hafta)e |
|
|
|
|
|
Ara | - | - | 0,491 (0,290-0,831) | 0,007 | - |
Final | 72,7 (61,3-83,0) | 64,9 (45,7-96,0) | 0,876 (0,679-1,129) | 0,306 | - |
Kısaltmalar: GA = güven aralığı; ITT = tedavisi amaçlanan hasta; NA = uygulanamaz; ORR = objektif yanıt oranları; OS = genel sağkalım; PFS = progresyonsuz sağkalım; TTP = tümör progresyonuna kadar geçen süre.
Başlangıç noktası, çaprazlama olduğunda sıfırlanmış ve etkililik analizleri araştırmacının değerlendirmesi baz alınarak yapılmıştır.
ITT popülasyonundaki medyan OS sunitinib ve plasebo kollarında sırasıyla 72,7 hafta ve 64,9 haftadır (HR 0,876, %95 GA: 0,679-1,129, p = 0,306). Bu analizde plasebo kolu daha önceden açık etiketli sunitinib tedavisi almış ve plaseboya randomize edilmiş hastaları içermektedir.
Tedavi edilmemiş renal hücreli karsinomu (mRHK)
Tedavi-edilmemiş mRHK'li hastalarda tek ajan olarak sunitinib ve IFN-α'yı karşılaştıran bir çok merkezli, uluslararası Faz III randomize çalışma yapılmıştır. Yedi yüz elli (750) hasta ya sunitinib ile tekrarlayan 6 haftalık siklüsler halinde 4 hafta 50 mg günlük oral doz takiben 2 hafta ara ( 4/2 doz şeması) veya i lk hafta 3 milyon ünite (MU) ikinci hafta 6 MU ve üçüncü hafta 9 MU ve bundan sonra her hafta ardışık olmayan 3 gün subkütan olarak IFN-α almak üzere birebir (1:1) randomize edilmiştir.
Sunitinib tedavisinin medyan süresi 11,1 ay (0,4-46,1 aralığında), IFN-α tedavisinin medyan süresi ise 4,1 aydır (0,1-45,6 aralığında). Tedavi ile alakalı ciddi yan etkiler sunitinib ve IFN-α alan hastalarda sırasıyla %23,7 ve %6,9 olarak raporlanmıştır. Bunun yanında yan etkilerden dolayı tedavinin yarım bırakılma oranı sunitinib için %20 iken IFN- α için %23'dür. Doz kesilmesi sunitinib kullanan 202 hastada (%54) görülmüşken IFN-α kullanan hastalarda bu sayı 141'dir (%39). Doz azaltılması ise sunitinib ve IFN-α kullanan hastaların sırasıyla 194 (%52) ve 98 (%27)'inde görülmüştür. Hastalar progresyon
görülene kadar ya da tedaviden çıkarılana kadar tedavi edilmişlerdir.
Primer etkililik sonlanım noktası progresyonsuz sağkalımdır (PFS). Planlı bir ara analiz, sunitinib için IFN-α'nın üstünde istatistiksel olarak anlamlı bir avantaj göstermiştir. Bu çalışmada medyan PFS sırasıyla 47,3 ve 22,0; HR 0,415'tir. (%95 GA: 0,320-0,539, p<0,001). Diğer sonlanım noktaları objektif yanıt oranı (ORR), OS ve güvenliliktir. Primer sonlanım noktası elde edildikten sonra çekirdek radyolojik değerlendirmeye devam edilmemiştir. Final analizde araştırıcının değerlendirmesi ile ORR sunitinib kolu için %46 (%95 GA: 41-51) IFN-α kolu için %12 (%95 GA: 9-16) olarak belirlenmiştir (p<0,001).
IFN-α ile karşılaştırıldığında sunitinib tedavisi daha uzun sağkalım süreleriyle ilişkilendirilmiştir. Medyan OS sunitinib kolu için 114,6 hafta (%95 GA: 100,1-142,9) ve IFN-α kolu için 94,9 hafta (%95 GA: 77,7-117,0) olmuştur [HR= 0,821 (%95 GA: 0,673-1,001); log-rank testi ile p=0,0510.]
ITT popülasyonunda gözlemlenen ve çekirdek radyolojik laboratuvar değerlendirmesine ile
belirlenen genel PFS ve OS tablo 2'de özetlenmiştir:
Tablo 2 – Daha önce tedavi edilmemiş mRHK etkililik sonlanım noktası özetleri (ITT
popülasyonu)
PFS özeti | Sunitinib (N = 375) | IFN-ï¡ (N = 375) | |
Progrese olmayan veya ölen hastalar [n (%)] | 161 (42,9) | 176 (46,9) | |
Progrese olmayan veya ölen hastalar [n (%)] | 214 (57,1) | 199 (53,1) | |
PFS (hafta) | |||
Çeyrek (%95 GA) | |||
%25 | 22,7 (18,0-34,0) | 10,0 (7,3-10,3) | |
%50 | 48,3 (46,4-58,3) | 22,1 (17,1-24,0) | |
%75 | 84,3 (72,9-95,1) | 58,1 (45,6-82,1) | |
Tabakalandırılmamış analiz | |||
Risk oranı (sunitinib vs IFN-ï¡) | 0,5268 | ||
Risk oranı için %95 GA | (0,4316-0,6430) | ||
p-değeri |
| <0,0001 | |
OS özeti | Sunitinib (N = 375) | IFN-ï¡ (N = 375) | |
Progrese olmayan veya ölen hastalar [n (%)] | 185 (49,3) | 175 (46,7) | |
Progrese olmayan veya ölen hastalar [n (%)] | 190 (50,7) | 200 (53,3) | |
OS (hafta) | |||
Çeyrek (%95 GA) | |||
%25 | 56,6 (48,7-68,4) | 41,7 (32,6-51,6) | |
%50 | 114,6 (100,1-142,9) | 94,9 (77,7-117,0) | |
%75 | NA (NA-NA) | NA (NA-NA) | |
Tabakalandırılmamış analiz | |||
Risk oranı (sunitinib vs IFN-ï¡) | 0,8209 | ||
Risk oranı için %95 GA | (0,6730-1,0013) | ||
p-değeri | 0,0510 | ||
Kısaltmalar: GA = güven aralığı; IFN-α = interferon-alfa; ITT = tedavisi amaçlanan hasta; N = hasta sayısı;
NA = uygulanamaz; OS = genel sağkalım; PFS = progresyonsuz sağkalım
Sitokin-refrakter metastatik renal hücreli karsinom
Sunitinib ile bir Faz II çalışma gerçekleştirilmiştir. Çalışma interleukin-2 ya da IFN- ile birlikte önceki sitokin tedavisine refrakter hastaları kapsamaktadır. 63 hasta 50 mg oral sunitinib (6 haftalık tamamlanmış siklüs-4 hafta boyunca günde bir kere ve takiben 2 hafta dinlenme periyodu (4/2 doz şeması)) ile başlamıştır. Primer sonlanım noktası ORR'dir. RECIST (solid tümörlerde yanıt değerlendirme kriteri) kriterlerine göre belirlenmiştir.
Bu çalışmada objektif yanıt oranı % 36,5'dır (%95 GA: %24,7-%49,6). Progresyona kadar geçen süre (TTP) 37,7 haftadır (%95 GA: 24,0-46,4 hafta).
Açık etiketli, tek kollu, çok merkezli, doğrulayıcı bir çalışmada sunitinibin etkililik ve güvenliliği bir önceki sitokin tedavisine refrakter mRHK hastalarında değerlendirilmiştir. 106 hasta 4/2 şemasına göre en az 1 doz 50 mg sunitinib almıştır.
Bu çalışmanın primer sonlanım noktası ORR'dir. Sekonder sonlanım noktası TTP, yanıt süresi (DoR) ve OS'yi içermektedir. Bu çalışmada ORR %35,8 (%95 GA: %26,8-%47,5). Medyan DoR'ye ve OS'ye henüz ulaşılamamıştır.
Pankreatik nöroendokrin tümörler (pNET)
Destekleyici Faz II, açık etiketli, çok merkezli çalışma, rezektabl olmayan pNET'li hastalarda günde tek ajan olarak sunitinibin etkililiğini ve güvenliliğini, 4/2 şemasında [4 haftalık tedavi, 2 haftalık dinlenme periyodu, günde bir kere 50 mg] değerlendirdi. 66 hastanın pankreatik adacık hücreli tümör kohortunda birincil sonlanım noktası yanıt oranı
%17 idi.
Unrezektabl pNET olan hastalarda tek başına sunitinib ile ilgili Faz III, çok merkezli, uluslararası, randomize, çift kör plasebo kontrollü bir pivot çalışma yapılmıştır.
Hastalar, RECIST'e dayalı olarak, önceki 12 ay içinde belgelenmiş progresyona ihtiyaç duydu ve planlanmış bir istirahat dönemi (n = 86) veya plasebo (n = 85) olmadan, günde bir kez 37,5 mg sunitinib almak üzere randomize edildi (1: 1).
Primer objektif, plasebo alan hastalara karşı sunitinib alan hastalarda PFS'yi karşılaştırmaktı. Diğer sonlanım noktaları arasında OS, ORR, Hasta Tarafından Bildirilen Sonuçlar ve güvenlik yer almaktadır.
Demografik veriler, sunitinib ve plasebo grupları arasında karşılaştırılabilir düzeydeydi. Buna ek olarak, sunitinib hastalarının %49'unda plasebo hastalarının %52'sinde fonksiyonel olmayan tümörler vardı ve her iki kolun %92'sinde karaciğer metastazı vardı.
Çalışmada somatostatin analoglarının kullanımına izin verildi.
Sunitinib hastalarının toplam %66'sı buna karşın plasebo hastalarının %72'si daha önce sistemik tedavi aldı. Buna ek olarak, sunitinib hastalarının %24'ü plasebo hastalarının ise
%22'si somatostatin analogları almıştır.
Plaseboya kıyasla sunitinib için araştırmacılar tarafından değerlendirilen PFS'de klinik olarak anlamlı bir avantaj gözlenmiştir. Medyan PFS, sunitinib kolunda 11,4 ay ve plasebo kolunda 5,5 ay olmuştur [risk oranı (nispi risk) : 0,418 (%95 GA 0,263-0,662), p = 0,0001].
Hastalığın progresyonunu belirlemek için RECIST'in araştırmacı tümör ölçümleri uygulanmasına dayanan türetilmiş tümör yanıt değerlendirmeleri yapıldığında benzer sonuçlar gözlemlenmiştir (Tablo 3). Değerlendirilen temel özelliklerin tüm alt gruplarında, önceki alınan sistemik tedavilerin sayısına göre bir analizi de içerecek şekilde sunitinib lehine bir risk oranı gözlenmiştir. Sunitinib kolunda 29 hasta ve plasebo kolunda 24 hasta önceden sistemik tedavi almadı; bu hastalardan PFS için risk oranı 0,365 (%95 GA: 0,156- 0,857), p = 0,0156 idi. Benzer şekilde, sunitinib kolundaki 57 hastada (28'i önceden bir sistemik terapi ve 29'u önceden 2 veya daha fazla sistemik terapi almış) ve plasebo kolundaki 61 hastada (25'i önceden bir sistemik terapi ve 36'sı önceden 2 veya daha fazla sistemik terapi almış), PFS için risk oranı 0,456 (%95 GA: 0,264-0,787), p = 0,0036 idi.
Progresyon kararının, araştırmacı tarafından bildirilen tümör ölçümlerine dayandığı, PFS olayı olarak muamele edilen çalışma sonlandırma haricindeki nedenlerle sansürlenmiş tüm hastalarda PFS duyarlılık analizi yapıldı. Bu analiz, sunitinibin tedavi etkisinin konservatif bir tahminini sağlamış ve 0,507 değerinde bir risk oranı (%95 GA: 0,350-0,733), p = 0,000193 ortaya koyarak birincil analizi desteklemiştir.
pNET ile ilgili pivot çalışma, bağımsız bir “ İlaç İzleme Komitesiâ€'nin önerisi üzerine erken sonlandırılmıştır ve birincil sonlanım noktası, her ikisi de tedavi etkisinin tahminlerini etkilemiş olan araştırmacı değerlendirmesine dayandırılmıştır.
PFS'nin araştırmacı temelli değerlendirmesinde yanlılığı bertaraf edebilmek için kör bağımsız merkez incelemesi yapıldı; bu inceleme araştırmacı değerlendirmesini desteklemiştir. Tablo 3'te gösterilmiştir.
Tablo 3 - Faz III çalışmasından elde edilen pNET etkinliği sonuçları
Etkililik parametresi | SUTENT (n = 86) | Plasebo (n = 85) | HR (%95 GA) | p-değeri |
Araştırmacı değerlendirmesine göre progresyonsuz sağkalım [medyan, aylar (%95 GA)] | 11,4 (7,4–19,8) | 5,5 (3,6–7,4) | 0,418 (0,263–0,662) |
0,0001 |
Araştırıcı tümör değerlendirmelerine RECIST uygulanmasına dayanan türetilmiş tümör yanıtı değerlendirmesi ile progresyonsuz sağkalım [medyan, aylar (%95 GA)] |
12,6 (7,4–16,9) |
5,4 (3,5–6,0) |
0,401 (0,252–0,640) |
0,000066 |
Tümör değerlendirmelerinin kör bağımsız merkez gözden geçirilmesiyle progresyonsuz sağkalım [medyan, aylar (%95 GA)], |
12,6 (11,1–20,6) |
5,8 (3,8–7,2) |
0,315 (0,181–0,546) |
0,000015 |
Genel sağkalım [5 yıl takip] [medyan, aylar (%95 GA)] | 38,6 (25,6–56,4) | 29,1 (16,4–36,8) | 0,730 (0,504–1,057) | 0,0940 |
Objektif yanıt oranı [%, (%95 GA)] | 9,3 (3,2–15,4) | 0 | NA | 0,0066 |
Şekil 1 – pNET Faz 3 çalışmasında, Kaplan Meier Progresyonsuz Sağkalım Grafiği
Progression Free Survival Probability (%)
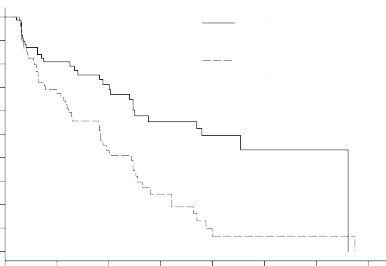
![]()
Progresyonsuz Sağkalım Olasılığı (%)
Time (Months)
![]()
Süre (Ay)
![]()
OS verileri, çalışma sonunda olgunlaşmamıştır. Sunitinib kolu için [20,6 ay (%95 GA 20,6, NR) verileri, plasebo kolu için NR (%95 GA 15,5, NR) verileri ile karşılaştırıldığında risk oranı: 0,409 (%95 GA: 0,187-0,894), p-değeri = 0,0204]'dır. Sunitinib kolunda 9 ve plasebo kolunda 21 ölüm meydana gelmiştir.
Hastalık progresyonu sırasında hastalar körleştirilmemiştir ve plasebo alan hastalara, ayrı bir uzantı çalışmasında açık etiketli SUTENT'e erişim sunulmuştur. Çalışmanın erken dönemde sonlanması nedeniyle, kalan hastalar körleştirilmemiştir ve bu hastalara, ayrı bir uzantı çalışmasında açık etiketli SUTENT'e erişim sağlanmıştır. Plasebo kolundaki 85 hastadan (%69,4) 59'u, hastalığın ilerlemesi veya çalışma sonlanmasındaki körleme kalktıktan sonra açık etiketli sunitinibe geçti. Uzatma çalışmasında 5 yıllık izlem sonrasında gözlemlenen OS, 0,730 (%95 GA 0,504-1,057) risk oranını gösterdi.
Avrupa Kanser Araştırma ve Tedavisi Organizasyonu Yaşam Kalitesi Anketinden (EORTC QLQC-30) alınan sonuçlar; toplamda genel sağlık ile ilişkili yaşam kalitesinin ve beş fonksiyon alanının (fiziksel, rol, bilişsel, duygusal ve sosyal), sınırlı advers semptomatik etkilerle, plaseboya karşı sunitinib tedavisi alan hastalarda korunduğunu göstermiştir.
Progresif, ileri/metastatik, iyi diferansiye edilmiş, rezeke edilemeyen pNET'li hastalarda sunitinibin etkililiğini ve güvenilirliğini değerlendiren Faz IV çok uluslu, çok merkezli, tek kollu, açık etiketli bir çalışma gerçekleştirildi.
Yüz altı hasta (hiçbir tedavi almamış kohortunda 61 hasta ve sonraki basamak kohortunda 45 hasta) günde bir kez 37,5 mg oral yoldan sunitinib ile kesintisiz günlük dozlama programı ile tedavi gördü.
Araştırmacı tarafından değerlendirilen progresyonsuz sağkalım hem genel popülasyonda (%95 GA: 10,9-16,7) hem de hiçbir tedavi almamış kohortta (%95 GA: 7,4-16,8) 13,2 ay idi.
Özel popülasyonlara ilişkin ek bilgiler:
Pediyatrik popülasyon:
Pediyatrik hastalarda sunitinib kullanımına ilişkin deneyimler kısıtlıdır (Bkz. Bölüm
4.2).
Refrakter solid tümörleri olup, büyük bölümü primer beyin tümörü tanısı olan 35 hastada; 30'u pediyatrik hastalardan (3-17 yaş) ve 5'i genç erişkin hastalardan (18-21 yaş) oluşan, oral sunitinibe ilişkin, bir Faz I doz artış çalışması gerçekleştirilmiştir. Çalışmanın birinci kısmında doz kısıtlayıcı kardiyotoksisite gözlenmiş ve bu nedenle önceden potansiyel kardiyotoksik tedaviler (antrasiklinler dahil) veya kardiyak radyasyon uygulanan hastalar dışlanacak şekilde düzeltme yapılmıştır. Daha önce kanser tedavisi alan fakat kardiyak toksisite açısından risk faktörleri bulunmayan hastaların yer aldığı çalışmanın ikinci kısmında, 4/2 şemasında günlük 15 mg/m dozunda (MTD) sunitinib genellikle tolere edilebilir ve klinik açıdan kontrol altına alınabilir olmuştur. Olguların hiçbirinde tam yanıt veya kısmi yanıt elde edilmemiştir. 6 hastada (%17) stabil hastalık gözlenmiştir. GİST'i olan bir hasta, 15 mg/m doz düzeyinde dahil edilmiş ve yarara ilişkin kanıt gözlenmemiştir. Gözlenen advers ilaç reaksiyonları genel olarak erişkinlerde görülenlere benzer bulunmuştur (Bkz. Bölüm 4.8).
HGG veya epandimomu bulunan, 27'si pediyatrik hastadan (3-16 yaş) ve 2'si genç erişkin hastadan (18-19 yaş) oluşan 29 hastada, Faz II açık etiketli bir çalışma gerçekleştirilmiştir. Çalışma, hastalık kontrolününolmamasınedeniyleplanlıbir ara dönem analizi sırasında
kapatılmıştır. Medyan PFS, HGG grubunda 2,3 ay ve epandimoma grubunda 2,7 ay olmuştur. Medyan genel OS, HGG grubunda 5,1 ay ve epandimoma grubunda 12,3 ay olmuştur. En yaygın (≥% 10) tedaviye bağlı advers olayların, her iki grupta kombine olarak nötrofil sayısının azalması (6 hasta [%20,7]) ve intrakraniyal hemoraji (3 hasta [%10,3]) olduğu bildirilmiştir (Bkz. Bölüm 4.8).
Günde 15 mg/m ila 30 mg/m arasında değişen dozlarda 4/2 şemasına göre sunitinib alan, yaşları 13-16 arasında değişen GİST'i olan 6 pediyatrik hastada oral sunitinibin bir Faz
½ çalışmasından elde edilen kanıtlar ve mevcut yayınlanmış veriler (GİST'i olan 20 pediyatrik veya genç erişkin hasta) sunitinib tedavisinin, 26 hastadan 18'inde (%69,2) hem imatinib yetersizliğinden veya intoleransından sonra (21 hastadan 16'sında stabil hastalık) hem de novo/cerrahi ameliyat sonrasında (5 hastadan 2'sinde stabil hastalık) hastalık stabilizasyonu sağladığı gözlenmiştir. Faz ½ çalışmasında, 6 hastanın 3'ünde stabil hastalık ve 3'ünde hastalık ilerlemesi gözlenmiştir (sırasıyla 1 hastaya neo adjuvan ve 1 hastaya adjuvan imatinib verilmiştir). Aynı çalışmada, 6 hastanın 4'ünde (%66,7) Grade 3-4 tedaviye bağlı advers olaylar (Grade 3 hipofosfatemi, nötropeni ve trombositopeninin her biri 1 hastada ve 1 hastada Grade 4 nötropeni) görülmüştür. Ek olarak, yayınlar, 5 hastada gözlenen şu Grade 3 advers ilaç reaksiyonları bildirmiştir: Yorgunluk (2), gastrointestinal advers ilaç reaksiyonları (diyare dahil) (2), hematolojik advers ilaç reaksiyonları (anemi dahil) (2), kolesistit (1), hipertiroidizm (1) ve mukozit (1).
GİST'i olan pediyatrik hastalarda (6-17 yaş grubu) sunitinibin farmakokinetik (PK) ve kilit güvenlilik ile etkililik sonlanım noktalarının ekstrapolasyonu amacıyla, bir popülasyon PK ve farmakokinetik/farmakodinamik (PK/PD) analizi gerçekleştirilmiştir. Bu analizde GİST veya solid tümörleri olan erişkinlerden ve solid tümörleri olan pediyatrik hastalardan toplanan veriler temel alınmıştır. Modelleme analizleri doğrultusunda, küçük yaş ve düşük vücut ölçümlerinin plazma ilaç maruziyetine güvenlilik ve etkililik yanıtlarını olumsuz etkilemediği belirlenmiştir. Sunitinib yarar/riskinin, küçük yaş veya düşük vücut ölçümlerinden olumsuz etkilenmediği ve başlıca plazma ilaç maruziyetine bağlı olduğu görülmüştür.
Avrupa İlaç Ajansı, böbrek veya renal pelvis karsinomunun (nefroblastom, nefroblastomatoz, berrak hücreli sarkom, mezoblastik nefrom, renal medüller karsinom ve böbrek rabdoid tümörü hariç olmak üzere) tedavisinde çocuk nüfusun tüm alt gruplarında SUTENT ile yapılan çalışmaların sonuçlarını sunma yükümlülüğünden feragat etti (çocuklarda kullanım hakkında bilgi için bölüm 4.2'ye bakın).
Avrupa İlaç Ajansı, gastroenteropankreatik nöroendokrin tümörlerin (nöroblastom, nöroganglioblastoma, feokromasitom hariç) tedavisinde çocuk nüfusunun tüm alt gruplarında SUTENT ile yapılan çalışmaların sonuçlarını sunma yükümlülüğünden feragat etti (bkz. çocuklarda kullanım hakkında bilgi için bölüm 4.2).
5.2. Farmakokinetik özellikler
Genel özellikler
Sunitinibin farmakokinetiği 135 sağlıklı gönüllüde ve solid tümörlü 266 hastada değerlendirilmiştir. Her iki grupta da farmakokinetik benzerdir.
Emilim:
Sunitinib ağızdan uygulanmasını takiben 6-12 saat (T) içinde maksimum konsantrasyona (C) ulaşır. Yiyeceklerin sunitinibin biyoyararlanımına herhangi bir etkisi yoktur. Sunitinib ve primer aktif metabolitin kararlı durum konsantrasyonlarına 10-14 gün içinde ulaşılır. 14. gün itibariyle sunitinib ve aktif metabolitinin kombine plazma konsantrasyonları 62,9-101 ng/ml olup, bu konsantrasyonlar klinik verilerde öngörülen in vitro olarak reseptör fosforilasyonunu inhibe edecek ve in vivo olarak tümör stazı/büyümesini azaltacak hedef konsantrasyonlardır. Primer aktif metabolit toplam maruziyetin %23-%37'sini oluşturmaktadır. Tekrarlayan günlük uygulamalarda veya test edilen doz rejimlerinin tekrarlayan kürlerinde sunitinib veya primer aktif metabolitinin farmakokinetiğinde anlamlı değişiklikler olmamıştır.
Dağılım:
Sunitinib ve primer aktif metabolitinin in vitro çalışmalarda konsantrasyondan bağımsız olarak plazma proteinine bağlanma derecesi sırasıyla %95 ve %90 olmuştur. Sunitinib için dağılım hacmi (V), dokulara dağılımı gösterecek şekilde, büyüktür (2230 litre).
Metabolik etkileşimler
Test edilen tüm sitokrom P450 izoformları (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5, ve CYP4A9/11) için hesaplanan in vitro Ki değerleri; sunitinib ve primer aktif metabolitinin, bu enzimlerle metabolize olan diğer etkin maddelerin metabolizmasını indükleme ihtimalinin az olacağını göstermektedir.
Biyotransformasyon:
Sunitinib asıl olarak, bir sitokrom P450 enzimi olan CYP3A4 tarafından metabolize edilir. Primer aktif metaboliti desetil sunitinibtir ve bu metabolit tekrardan CYP3A4 tarafından metabolize edilir.
Sunitinibin potent CYP3A4 indükleyicileri veya inhibitörleri ile birlikte kullanımı ile sunitinibin plazma düzeyi değişebileceğinden, birlikte kullanımından kaçınılmalıdır (Bkz. Bölüm 4.4 ve 4.5).
Eliminasyon:
Atılım primer olarak feçes yoluyla (%61) gerçekleşir. Renal eliminasyon metabolitler ile birlikte uygulanan dozun %16'sıdır. Sunitinib ve primer aktif metaboliti plazmada, idrarda ve feçeste görülen ilaç bağlantılı esas bileşiklerdir ve sırasıyla %91,5, %86,4 ve
%73,8 oranında görülmektedir. Minör metabolitler idrar ve feçeste görülmüş, ancak genellikle plazmada görülmemişlerdir. Total oral klerens (CL/F) 34-62 litre/saat olmuştur. Sağlıklı gönüllülere tek doz oral uygulamanın ardından sunitinibin terminal yarılanma ömrü yaklaşık olarak 40-60 saat iken, primer aktif desetil metabolitininki 80- 110 saat olmuştur.
Doğrusallık / Doğrusal olmayan durum:
25-100 mg'lık doz aralığında plazma konsantrasyon-zaman eğrisi altındaki alan (EAA) ve C dozla orantılı olarak artar. Günlük tekrarlayan uygulamalarda sunitinib miktarı 3-4 katına çıkarken, primer metabolitinin miktarı 7-10 katına çıkar.
BCRP inhibitörü olan ilaçlarla birlikte kullanım
In vitro ortamda sunitinib, efluks taşıyıcı BCRP'nın substratıdır. A6181038 çalışmasında bir BCRP inhibitörü olan gefitinibin, sunitinib veya toplam ilacın (sunitinib + metabolit) Cve EAA değerleri üzerinde klinik olarak anlamlı bir etkisi olmamıştır (Bkz. Bölüm 4.5). Bu çalışma mRHK'lı hastalarda sunitinibin gefitinib ile birlikte kullanıldığında güvenlilik/tolere edilebilirliği, maksimum tolere edilen dozu ve antitümör aktivitesinin araştırıldığı çok merkezli, açık etiketli, Faz 1/2 bir çalışmadır. Gefitinib (günlük 250 mg) ve sunitinib (4 hafta kullanım sonrası 2 hafta ara verecek şekilde günlük 37,5 mg [Kohort 1, n=4] veya 50 mg [Kohort 2, n=7]) birlikte uygulandığındaki famakokinetikleri sekonder çalışma objektifi olarak değerlendirilmiştir. Sunitinib farmakokinetik parametrelerindeki değişiklikler klinik olarak anlamlı bulunmamış ve bir ilaç-ilaç etkileşimi olduğunu göstermemiştir; ancak göreceli düşük hasta sayısı (N=7+4) ve farmakokinetik parametrelerdeki hastalar arası varyasyonun orta-yüksek seviyede olması gözönüne alındığında, bu çalışmanın farmakokinetik ilaç-ilaç etkileşimi sonuçlarını değerlendirirken dikkatli olunması gerekmektedir.
Hastalardaki karakteristik özellikler
Karaciğer yetmezliği:
Sunitinib ve aktif metaboliti esas olarak karaciğer tarafından metabolize edilir. Hafif (Child-Pugh Sınıf A) veya orta (Child-Pugh Sınıf B) karaciğer yetmezliği olan hastalarda sunitinibin tek bir dozuna sistemik maruz kalım, normal karaciğer fonksiyonu olan deneklerle karşılaştırıldığında benzer olmuştur. Sunitinib, ciddi (Child- Pugh Sınıf C) karaciğer yetersizliği olan hastalarda çalışılmamıştır.
Çalışmalara ALT veya AST değerleri >2,5xULN (normalin en üst sınırı) veya karaciğer metastazı > 5,0 x ULN olan hastalar dahil edilmemiştir.
Böbrek yetmezliği:
Popülasyon farmakokinetik analizleri, kreatinin klerensi 42-347 ml/dak olan hastalarda, sunitinibin klerensinin değişmediğini göstermektedir. Tek doz SUTENT uygulamasından sonra, ciddi böbrek yetmezliği (kreatinin klerensi <30ml/dak) olan kişilerle normal renal fonksiyona (kreatinin klerensi >80ml/dak) sahip kişilerde sistemik maruziyet aynı olmuştur. Son evre böbrek yetmezliği hastalarında sunitinib ve primer metabolitinin hemodiyalizle eliminasyonu yapılamasa da, normal böbrek fonksiyonuna sahip kişilerle karşılaştırıldığında sistemik maruziyet sunitinib için %47, primer metaboliti için %31 daha az olmuştur.
Kilo ve performans durumu:
Demografik verinin popülasyon farmakokinetik analizi, vücut ağırlığı veya performans durumunda, başlangıç doz ayarlamalarının gerekli olmadığını gösterir.
Cinsiyet:
Eldeki veriler kadınlarda görünür sunitinib klerensinin (CL/F) erkeklere oranla %30 daha az olduğunu göstermektedir; ancak bu fark başlangıç dozunun değiştirilmesini gerektirmemektedir.
Pediyatrik popülasyon:
Pediyatrik hastalarda sunitinib kullanımına ilişkin deneyimler kısıtlıdır (Bkz. bölüm 4.2). GİST ve solid tümörleri olan erişkin hastaları ve solid tümörleri olan pediyatrik hastaları içeren toplu veritabanına ilişkin popülasyon PK analizleri tamamlanmıştır. Yaş ve vücut ölçümlerinin (toplam vücut ağırlığı veya vücut yüzey alanı) yanı sıra diğer eş değişkenlerin sunitinib ve metabolitlerinin önemli PK parametreleri üzerindeki etkisini değerlendirmek üzere adımsal eş değişken modelleme analizleri yapılmıştır. Test edilen yaş ve vücut ölçümü ile ilişkili eş değişkenlerden yaş, sunitinibin görünen klerensi üzerinde anlamlı bir eş değişken olarak belirlenmiştir (pediyatrik hasta ne kadar küçükse, görünen klerens o kadar düşüktür). Benzer şekilde, vücut yüzey alanı, aktif metabolitin görünen klerensi üzerinde anlamlı bir eş değişken olarak belirlenmiştir (vücut yüzey alanı ne kadar düşükse, görünen klerens o kadar düşüktür).
Ayrıca, 3 pediyatrik çalışmadan toplu veri setine ilişkin birleştirilmiş popülasyon PK analizine göre (2 pediyatrik solid tümör çalışması ve 1 pediyatrik GİST çalışması: 6-11 yaş ve 12-17 yaş) başlangıç vücut yüzey alanı (BSA), sunitinibin ve aktif metabolitinin görünen klirensi üzerinde anlamlı bir eş değişken olarak belirlenmiştir. Bu analize dayanarak, BSA değerleri 1,10 ve 1,87 m arasında olan pediyatrik hastalarda günlük yaklaşık 20 mg/m'lik bir doz, sunitinib ve aktif metabolitine plazma maruziyetinin (EAA'nın %75 ila %125'i arasında), GİST'i olan yetişkinlere, 4/2 şemasına göre (EAA 1233 ng.hr/mL) günlük 50 mg sunitinib uygulaması ile gözlenen plazma maruziyetine benzer olması beklenir. Pediyatrik çalışmalarda, 15 mg/m olan başlangıç sunitinib dozu (Faz I doz-artış çalışmasında tanımlanan MTD'ye göre, Bkz. Bölüm 5.1), GİST'i olan pediyatrik hastalarda 22,5 mg/m'ye ve ardından bireysel hasta güvenliliği/tolere edilebilirliğine bağlı olarak 30 mg/m'ye (toplam günlük 50 mg dozu aşmayacak şekilde) yükselmiştir. Ayrıca, GİST'i olan pediyatrik hastalarda yayınlanmış literatürlere göre, 16,6 mg/m ila 36 mg/m arasında değişmekte olan hesaplanmış başlangıç dozu, toplam günlük 50 mg dozu aşmayacak şekilde 40,4 mg/m‘ye kadar yükselmiştir.
5.3. Klinik öncesi güvenlilik verileri
Maymun ve sıçanlarda 9 aya kadar devam eden tekrarlayan doz toksisite çalışmalarında primer hedef organ etkileri gastrointestinal sistem (maymunlarda emezis ve diyare), adrenal bez (sıçanlarda fibrozis görülen nekrozu takiben ve maymunlarda kortikal konjesyon ve/veya hemoraji), hemolenfopoetik sistem (kemik iliği hiposelülaritesi ve timusta lenfoid dokunun azalması, dalak ve lenf nodülü), ekzokrin pankreas (tek hücre nekrozuyla asinar hücre degranülasyonu), tükrük bezi (asinar hipertrofi), eklem (büyüme plağı kalınlaşması), uterus (atrofi), overler (azalmış foliküler gelişim) görülmüştür. Tüm bu bulgular klinik olarak anlamlı bir sunitinibin plazma maruziyeti seviyesinde görülmüştür. QT aralığında uzama, böbrekte mezangiyal matriks, gastrointestinal sistemde ve oral mukozada hemoraji ve testislerde (tübüler atrofi) ve anterior pitüiter hücreleri hipertrofisi diğer çalışmalarda görülen ilave etkiler arasındadır. Uterustaki (endometriyal atrofi) ve kemik büyüme plağındaki (fizeal kalınlaşma veya kıkırdakdisplazisi)değişimlersunitinibin farmakolojik etkisiyle
ilişkilendirilmiştir. Bu bulguların çoğu tedavi kesildiğinde 2-6 hafta içinde geri
dönüşümlü olmuştur.
Genotoksisite
Sunitinibin genotoksik potansiyeli in vitro ve in vivo olarak değerlendirilmiştir. Sunitinib, sıçan karaciğeri ile sağlanan metabolik aktivasyonu kullanan bakterilerde mutajenik değildi. Sunitinib in vitro olarak insan periferik kan lenfosit hücrelerinde yapısal kromozom aberasyonuna neden olmamıştır. İnsan periferik kan lenfositlerinde in vitro olarak metabolik aktivasyon varlığında ve yokluğunda poliploidi (sayısal kromozom aberasyonu) gözlenmiştir. Sunitinib, sıçan kemik iliğinde in vivo olarak klastojenik değildi. Esas aktif metabolit genotoksisite açısından değerlendirilmemiştir.
Karsinojenite
aylık oral gavaj doz-aralığı belirleme çalışmasında (0, 10, 25, 75 veya 200 mg/kg/gün dozlarında) devamlı günlük dozlama yapılan rasH2 transgenik farelerde test edilen en yüksek dozda (200 mg/kg/gün) dudenumun Brunner bezinin karsinoması ve hiperplazisi gözlenmiştir.
RasH2 transgenik farelerde günlük dozlama ile 6 aylık bir oral gavaj karsinojenite çalışması (0, 8, 25, 75 (50'ye azaltılan) mg/kg/gün) yapılmıştır. Günlük 25 mg/kg'lık ve daha fazla dozlarda 1-veya 6- aylık süreleri (günlük önerilen dozu kullanan hastaların EAA'sının 7,3 katı veya daha fazlası) takiben gastroduodenal karsinomalar, arka plan hemajiyosarkom insidansında artış, ve/veya gastrik mukozal hiperplazi gözlenmiştir.
6. FARMASÖTİK ÖZELLİKLER
6.1. Yardımcı maddelerin listesi
Kapsül içeriği:
Mannitol (E421) Kroskarmeloz Sodyum (E468) Povidon
Saf Su
Magnezyum Stearat (E572)
Kapsül kılıfı (Baskılı, karamel renkli, Boyut #2, Sert Jelatin Kapsül): Siyah demir oksit (E172i)
Sarı demir oksit (E172iii) Titanyum Dioksit (E171) Kırmızı Demir Oksit (E172ii) Jelatin (E441)
Beyaz Baskı Mürekkebi:
Şellak (E904) Dehidraft Alkol İzopropil Alkol Bütil Alkol
Propilen Glikol (E1520) Sodyum Hidroksit (E524) Povidon
Titanyum Dioksit (E171)
Ürün, sığır kaynaklı jelatin hammaddesi içermektedir.
6.2. Geçimsizlikler
Geçerli değildir.
6.3. Raf ömrü
36 ay
6.4. Saklamaya yönelik özel tedbirler
25°C'nin altında oda sıcaklığında saklanmalıdır.
6.5. Ambalajın niteliği ve içeriği
Her biri 7 kapsül içeren Aluminyum/PVC/Aclar® blisterlerde ambalajlanmıştır. Paket
büyüklüğü 14 (7×2) veya 28 (7×4) kapsül halindedir.
Tüm ambalaj boyutları pazarlanmayabilir.
6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler
Kullanılmamış olan ürünler ya da atık materyaller “Tıbbi Atıkların Kontrolü Yönetmeliği†ve “Ambalaj Atıklarının Kontrolü Yönetmeliğiâ€ne uygun olarak imha edilmelidir.
 Diyabet Hastalığı
Diyabet, insülin hormonu ile ilgili problemlerden kaynaklanan bir hastalıktır.
Diyabet Hastalığı
Diyabet, insülin hormonu ile ilgili problemlerden kaynaklanan bir hastalıktır. |
 Şizofrenlik
Şizofrenliğin psikiatrik teşhisi hakkında çok fazla anlaşmazlık vardır. Bu
sayfadaki bilgiler, şizofrenliğin teşhisi, nedenleri ve tedavisi hakkındaki faklı teoriler
hakkında bilgi verecektir.
Şizofrenlik
Şizofrenliğin psikiatrik teşhisi hakkında çok fazla anlaşmazlık vardır. Bu
sayfadaki bilgiler, şizofrenliğin teşhisi, nedenleri ve tedavisi hakkındaki faklı teoriler
hakkında bilgi verecektir. |
İLAÇ EŞDEĞERLERİ
| Eşdeğer İlaç Adı | Barkodu | İlaç Fiyatı |
|---|---|---|
| SUNIKSA | 8699717150089 | 23,255.08TL |
| SUNO | 8699541153706 | 11,682.81TL |
| SUNUBIS | 8699540023260 | 11,682.81TL |
| SUTENT | 8699532158710 | |
| SUTESGO | 8699525159809 | 11,682.81TL |
| Diğer Eşdeğer İlaçlar |
 |
Deri Kanseri Deri kanseri çok rastlanan bir hastalıktır. Üç ana türü bulunur ;genelde kemirici ülser olarak bilinen bazal hücreli karsinom, yassı hücreli karsinom ve kötü huylu tümör. |
 |
HIV ve Aids HIV, Human Immunodeficiency Virus’dür (İnsanlarda Bağışıklık Sistemini Bozan Virüsdür). Bu virüs AIDS hastalığına sebep olur. |
 |
Asperger Sendromu Asperger sendromu, otistik gurubun bir bölümü olan bir özürdür. Bu genelde, gurubun daha ”yüksek” tarafında yer aldığı düşünülen kişilere uygun bir tanıdır. |
İLAÇ GENEL BİLGİLERİ
Pfizer İlaçları Ltd.Şti.
| Geri Ödeme Kodu | A10553 |
| Satış Fiyatı | TL |
| Önceki Satış Fiyatı | |
| Original / Jenerik | Original İlaç |
| Reçete Durumu | Normal Reçeteli bir ilaçdır. |
| Barkodu | 8699532157553 |
| Etkin Madde | Sunitinib Maleat |
| ATC Kodu | L01XE04 |
| Birim Miktar | 50 |
| Birim Cinsi | MG |
| Ambalaj Miktarı | 14 |
| Antineoplastik ve İmmünomodülatör Ajanlar > Diğer Kanser İlaçları > Sunitinib |
| İthal ( ref. ülke : Fransa ) ve Beşeri bir ilaçdır. |