TARCEVA ROCHE 25 mg 30 film kaplı tablet Kısa Ürün Bilgisi
{ Erlotinib }
1. BEŞERİ TIBBİ ÜRÜNÜN ADI
TARCEVA 25 mg film kaplı tablet
2. KALİTATİF VE KANTİTATİF BİLEŞİM
Etkin madde
Bir film kaplı tablet 25 mg erlotinibe eşdeğer miktarda 27,32 mg erlotinib hidroklorür içerir.
Yardımcı maddeler
Laktoz monohidrat 27,43 mg (inek sütünden elde edilmektedir) Sodyum nişasta glikolat 8,00 mg
Sodyum laurilsülfat 1,00 mg Yardımcı maddeler için Bölüm 6.1'e bakınız.
3. FARMASÖTİK FORMU
Film kaplı tablet.
Bir yüzüne ‘'T 25'' oyulmuş, sarımsı beyaz renkte, yuvarlak, bikonveks tabletler halindedir.
4. KLİNİK ÖZELLİKLER
4.1. Terapötik endikasyonlar
Küçük hücreli dışı akciğer kanseri (KHDAK):
TARCEVA, epidermal büyüme faktörü reseptörü (EGFR) gen exon 19 delesyonu ve/veya exon 21(L858R) mutasyonu akredite bir laboratuvarda gösterilen, metastatik non-skuamöz küçük hücreli dışı akciğer kanseri hastalarının birinci basamak tedavisinde ve yukarıda tanımlanan mutasyon ve delesyonu olan non-skuamöz küçük hücreli dışı akciğer kanseri hastalarında bir basamak kemoterapi sonu progresyonunda ikinci basamak tedavisinde progresyona kadar kullanımı endikedir.
4.2. Pozoloji ve uygulama şekli
TARCEVA tedavisi, antikanser terapilerin kullanımında deneyimli olan bir hekim
tarafından başlatılmalıdır.
Doktor tarafından başka şekilde tavsiye edilmediği takdirde; Standart doz:
Küçük Hücreli Dışı Akciğer Kanseri:
İleri veya metastatik evre küçük hücreli dışı akciğer kanseri (KHDAK) olan birinci basamak kemoterapi almamış hastalarda TARCEVA tedavisine başlamadan önce EGFR mutasyon testi yapılmalıdır.
Önerilen günlük TARCEVA dozu yemeklerden en az bir saat önce veya en az iki saat sonra alınmak üzere 150 mg'dır.
Uygulama şekli:
Ağızdan alınır.
Özel popülasyonlara ilişkin ek bilgiler:
Doz ayarlaması gerektiğinde, dozu 50 mg'lık adımlarla düşürmeniz tavsiye edilmektedir (Bkz. Bölüm 4.4).
CYP3A4 substratları ve düzenleyicileri ile eş zamanlı kullanımında doz ayarlaması gerekebilir (Bkz. Bölüm 4.5).
Karaciğer yetmezliği:
Erlotinib birincil olarak karaciğer aracılığıyla metabolize edilir ve safra ile itrah edilir. Hafif derece karaciğer fonksiyon bozukluğu (Child-Pugh skoru 7-9) olan hastalar karaciğer fonksiyonu yeterli olan hastalar ile karşılaştırıldığında, erlotinib atılımı benzer olmasına rağmen, karaciğer yetmezliği olan hastalarda TARCEVA uygulanırken dikkatli olunmalıdır. Eğer ciddi advers olaylar gelişirse, doz azaltımı veya TARCEVA'ya ara verilmesi düşünülmelidir. Ciddi karaciğer fonksiyon bozukluğu olan hastalarda (AST/SGOT ve ALT/SGPT değerleri > 5 x normal üst sınır) erlotinibin güvenliliği ve etkililiği çalışılmamıştır (Bkz. Bölüm 4.4 ve 5.2). Total bilirubini normal üst limitten 3 kat yüksek olan hastalarda TARCEVA kullanılmamalıdır.
Böbrek yetmezliği:
TARCEVA'nın güvenlilik ve etkililiği böbrek yetmezliği bulunan hastalarda (serum kreatinin konsantrasyonu > 1,5 x normal üst sınır) araştırılmamıştır. Farmakokinetik çalışmalara göre hafif veya orta derece böbrek yetmezliği hastalarında doz ayarlaması gerekmemektedir (Bkz. Bölüm 5.2). İleri derece böbrek yetmezliği hastalarında TARCEVA kullanımı önerilmemektedir.
Pediyatrik popülasyon:
TARCEVA'nın güvenlilik ve etkililiği 18 yaşın altındaki hastalarda araştırılmamıştır. Pediyatrik hastalarda kullanımı önerilmemektedir.
Geriyatrik popülasyon:
TARCEVA'nın güvenlilik ve etkililiği yaşlı hastalarda araştırılmamıştır.
Sigara içenler:
Sigara içmenin erlotinib maruziyetini %50-60 azalttığı gösterilmiştir. Halihazırda sigara içen küçük hücreli dışı akciğer kanseri (KHDAK) hastalarında tolere edilebilen maksimum TARCEVA dozu 300 mg'dır.
Sigara içmeye devam eden hastalarda, başarısız kemoterapi sonrası ikinci basamak tedavide önerilen 150 mg doz ile karşılaştırıldığında 300 mg doz kullanımı etkililikte artış göstermemiştir. Güvenlilik verileri 300 mg ve 150 mg dozları arasında karşılaştırılabilirdir ancak, daha yüksek erlotinib dozu alan hastalarda döküntü, interstisyel akciğer hastalığı ve diyare insidansında sayısal bir artış olmuştur. Halihazırda sigara içen hastalara sigarayı bırakmaları önerilmelidir (bkz. Bölüm 4.4, 4.5, 5.1 ve 5.2).
4.3. Kontrendikasyonlar
TARCEVA, erlotinib veya Bölüm 6.1'de listelenen yardımcı maddelerden herhangi birine karşı aşırı duyarlılık durumunda kontrendikedir.
4.4. Özel kullanım uyarıları ve önlemleri
EGFR mutasyon durumunun saptanması
TARCEVA'nın lokal ileri ya da metastatik KHDAK'de birinci basamak tedavide kullanımı değerlendirilirken, hastanın EGFR mutasyon durumunun belirlenmesi önemlidir.
Doku örneğinden elde edilen tümör DNA'sını veya kan (plazma) örneğinden elde edilen dolaşımdaki serbest DNA'yı (cfDNA) kullanan, önceden belirlenmiş pozitiflik limitleri olan ve EGFR mutasyon durumunun belirlenmesi konusunda faydası kanıtlanmış, valide, sağlam, güvenilir ve duyarlı bir test uygulanmalıdır.
Eğer plazma bazlı cfDNA kullanılmış ve sonuç aktive edici mutasyonlar yönünden negatif bulunmuşsa, plazma bazlı testle yalancı negatif sonuçlar çıkması muhtemel olduğundan mümkünse doku testi de yapılmalıdır.
Sigara içenler
Sigara içmeyenlere kıyasla sigara içenlerde erlotinib plazma konsantrasyonlarının düşük olması sebebiyle, halihazırda sigara içenlere sigarayı bırakmaları önerilmelidir. Konsantrasyondaki düşüş derecesinin klinik olarak anlamlı olması beklenmektedir (bkz. Bölüm 4.2, 4.5, 5.1 ve 5.2).
İnterstisyel Akciğer Hastalığı
Küçük hücreli dışı akciğer kanseri (KHDAK) tedavisi için TARCEVA almakta olan hastalarda çok seyrek olarak, bazıları ölümcül olabilen, interstisyel akciğer hastalığı (İAH) benzeri olgular bildirilmiştir. KHDAK'deki BR.21 isimli faz III çalışmada ciddi interstisyel akciğer hastalığı benzeri olguların görülme sıklığı plasebo ve TARCEVA gruplarında %0,8 olmuştur.
KHDAK için randomize kontrollü klinik çalışmaların (kontrol grubu olmaması nedeniyle faz I ve tek kollu faz II çalışmaları hariç) meta analizinde, İAH benzeri olguların insidansı TARCEVA grubunda %0,9 ve kontrol grubundaki hastalarda %0,4 olmuştur.
İnterstisyel akciğer hastalığı benzeri olgu bulunduğundan şüphe edilen hastalarda bildirilen tanılara bazı örnekler, pnömoni, radyasyon pnömonisi, aşırı duyarlılık
pnömonisi, interstisyel pnömoni, interstisyel akciğer hastalığı, obliteratif bronşiyolit, pulmoner fibrozis, akut solunum sıkıntısı sendromu (ASSS), akciğer infiltrasyonu, alveolittir. Bu semptomlar, TARCEVA tedavisine başladıktan birkaç gün sonra ile birkaç ay arasında ortaya çıkmıştır. Eş zamanlı veya öncesindeki kemoterapi, öncesinde radyoterapi, daha önceden mevcut olan parankimal akciğer hastalığı, metastatik akciğer hastalığı veya pulmoner enfeksiyonlar gibi, karıştırıcı faktörler sık görülmüştür. Japonya'da yapılan çalışmalarda daha yüksek interstisyel akciğer hastalığı insidansı (%1,5 mortalite oranı ile yaklaşık olarak %5) görülmüştür.
Dispne, öksürük ve ateş gibi ani başlangıçlı yeni ve/veya ilerleyici açıklanamayan pulmoner semptomlar gelişen hastalarda, TARCEVA tedavisi tanısal değerlendirmeler tamamlanana dek kesilmelidir. İnterstisyel akciğer hastalığı tanısı konacak olursa, TARCEVA tedavisi kesilmelive gereken uygun tedaviye başlanmalıdır (Bkz. Bölüm 4.8).
Diyare, dehidratasyon, elekrolit dengesizliği, böbrek yetmezliği
TARCEVA kullanmakta olan hastaların yaklaşık %50'sinde diyare (bazı nadir vakalarda ölümle sonuçlanabilen) gözlenmiş olup, orta ve şiddetli diyarenin loperamid ile tedavi edilmesi gerekir. Bazı olgularda dozun düşürülmesi gerekli olabilir. Klinik çalışmalarda 50 mg'lik adımlar şeklinde doz düşürülmesi yapılmıştır. 25 mg'lık adımlar şeklinde doz azaltma üzerine çalışılmamıştır. Şiddetli veya inatçı diyare, bulantı, iştahsızlık ve dehidratasyon ile birlikte kusma görülmesi halinde TARCEVA tedavisi kesilmeli ve dehidratasyonu tedavi etmek için gerekli önlemler alınmalıdır (Bkz. Bölüm 4.8). Hipokalemi ve akut renal yetmezliği vakaları (bazıları ölümcül olabilen) seyrek olarak bildirilmiştir. Bazı renal yetmezlikler eşzamanlı kemoterapi uygulaması ile iç içe girerken, bazıları da diyareye, kusma ve/veya iştahsızlığa bağlı dehidratasyona sekonder olmuştur. Daha şiddetli veya inatçı diyare vakalarında veya dehidratasyona yol açan vakalarda, özellikle kötüleştiren risk faktörü (özellikle beraber kullanılan kemoterapi ve diğer ilaçlar, semptomlar veya hastalıklar veya ileri yaş dahil diğer yatkınlık durumları) bulunan hasta gruplarında, TARCEVA tedavisi kesilmelidir ve hastayı intravenöz olarak yoğun bir şekilde rehidrate etmek için gerekli önlemler alınmalıdır. Ek olarak, dehidratasyon riski bulunan hastalarda, böbrek fonksiyonları ve potasyum dahil serum elektrolitleri periyodik olarak izlenmelidir.
Hepatit, hepatik yetmezlik
TARCEVA tedavisi sırasında, ölümcül de olabilen seyrek hepatik bozukluk vakaları bildirilmiştir. Karıştırıcı faktörler önceden var olan karaciğer hastalığı veya eşlik eden hepatotoksik medikasyonları içermektedir. Bu yüzden, bu hastalarda periyodik karaciğer fonksiyon testleri düşünülmelidir. Karaciğer fonksiyonlarındaki değişiklikler ciddi olduğunda TARCEVA dozlamasına ara verilmelidir (Bkz. Bölüm 4.8). TARCEVA'nın ciddi hepatik disfonksiyonu olan hastalarda kullanılması önerilmemektedir.
Gastrointestinal perforasyon
TARCEVA kullanan hastalarda yaygın olmayan (bazı vakalarda ölümle sonuçlanabilen) şekilde görülen gastrointestinal perforasyonun gelişme riski yüksektir. Antianjiyogenik ilaçlar, kortikosteroid, NSAİİ ve/veya taksan bazlı kemoterapi ile eşzamanlı tedavi alan veya daha önceden peptik ülserasyon veya divertiküler hastalık geçmişi olan hastalarda
risk yüksektir. Gastrointestinal perforasyon gelişen hastalarda, TARCEVA tedavisi kalıcı olarak kesilmelidir (Bkz. Bölüm 4.8).
Büllöz veya eksfoliyatif deri hastalıkları
Steven Johnson sendromu/toksik epidermal nekrolizi işaret eden çok seyrek vakaları içeren, bazıları ölümcül olabilen büllöz, kabartılı ve eksfolyatif deri rahatsızlıkları bildirilmiştir (Bkz. Bölüm 4.8). Hastalar ciddi büllöz, kabartılı ve eksfoliyatif deri rahatsızlıkları geliştirirse, TARCEVA tedavisine ara verilmelidir veya kesilmelidir. Büllöz ve eksfolyatif deri rahatsızlıkları bulunan hastalar deri enfeksiyonuna karşı test edilmeli ve lokal tedavi kılavuzlarına göre tedavi edilmelidir.
Oküler hastalıklar
Hastalarda göz enflamasyonu, lakrimasyon, ışığa hassasiyet, bulanık görme, gözde ağrı ve/veya kızarıklık gibi akut ya da kötüleşen keratit oluşumuna işaret eden semptomların görülmesi halinde derhal göz hekimine başvurulmalıdır. Eğer ülseratif keratit teşhis edilmişse TARCEVA tedavisine ara verilmelidir veya kesilmelidir. Eğer keratit teşhis edilmişse tedaviye devam edilmesinin risk-yarar değerlendirmesi dikkatlice yapılmalıdır. TARCEVA keratit, ülseratif keratit ve şiddetli göz kuruluğu öyküsü olanlarda dikkatle kullanılmalıdır. Kontakt lens, keratit ve ülserasyon için risk faktörüdür. TARCEVA kullanımı sırasında çok seyrek korneal perforasyon veya ülserasyon vakaları bildirilmiştir (Bkz. Bölüm 4.8).
Diğer tıbbi ürünlerle etkileşim
CYP3A4'ün potent indükleyicileri TARCEVA'nın etkililiğini azaltabilirken CYP3A4'ün potent inhibitörleri toksisitede artışa yol açabilir. Bu tip tedavi ajanlarının birlikte kullanımından kaçınılmalıdır (Bkz. Bölüm 4.5).
Diğer etkileşim şekilleri
Erlotinib 5'ten yüksek pH seviyesinde çözünürlükte azalma ile karakterizedir. Proton pompası inhibitörleri, H antagonistleri ve antasidler gibi üst gastro-intestinal sistemin pH'sını değiştiren tıbbi ürünler, erlotinibin çözünürlüğünü dolayısıyla biyoyararlanımını değiştirebilir. Bu ürünlerle birlikte kullanıldığında TARCEVA dozunun artırılması, maruziyet düşüşünü telafi etmeyecektir. Erlotinibin proton pompası inhibitörleriyle birlikte kullanımından kaçınılmalıdır. Erlotinibin, H antagonistleri ve antasidlerle birlikte kullanımındaki etki bilinmemektedir, ancak düşük biyoyararlanım beklenmektedir. Dolayısıyla, bu kombinasyonların birlikte kullanılmasından kaçınılmalıdır (Bkz. Bölüm 4.5). TARCEVA tedavisi sırasında antasid kullanımı gerekliyse, günlük TARCEVA dozundan en az 4 saat önce veya 2 saat sonra alınmalıdır.
Diğer
TARCEVA, daha önce herhangi bir EGFR yolağı inhibitörü kullanmış hastalarda kullanılmaz.
Yardımcı maddeler
Her bir TARCEVA tablet, laktoz monohidrat içerir. Nadir kalıtımsal galaktoz intoleransı, Lapp-laktoz yetmezliği veya glukoz-galaktoz malabsorpsiyon problemi olan hastaların bu ilacı kullanmamaları gerekir.
Her bir TARCEVA 25 mg film kaplı tablet, 0,30 mg – 0,42 mg arası miktarda sodyum içerir. Bu durum, kontrollü sodyum diyetinde olan hastalar için göz önünde bulundurulmalıdır.
4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri
Etkileşim çalışmaları yalnızca yetişkinler üzerinde gerçekleştirilmiştir. Erlotinib ve diğer CYP substratları
Erlotinib CYP1A1'in potent bir inhibitörü, CYP3A4 ve CYP2C8'in orta derecede
inhibitörü ve in vitro UGT1A1 ile glukoronidasyonun güçlü bir inhibitörüdür. CYP1A1'in güçlü inhibisyonunun fizyolojik bağlamı CYP1A1'in insan dokularındaki çok sınırlı ekspresyonu sebebiyle bilinmemektedir.
Erlotinib, CYP1A2'nin orta düzey inhibitörü olan siprofloksasin ile birlikte uygulandığında, erlotinibe maruziyet [EAA] %39 oranda anlamlı düzeyde artmış, öte yandan maksimum konsantrasyon (C) seviyelerinde istatistiksel olarak anlamlı herhangi bir değişim bulunmamıştır. Benzer şekilde, aktif metabolite maruziyet sırasıyla EAA ve C seviyeleri için yaklaşık %60 ve %48 oranında artmıştır. Söz konusu artışların klinik anlamlılığı saptanmamıştır. Siprofloksasin veya güçlü CYP1A2 inhibitörleri (örn. fluvoksamin) erlotinib ile kombine edildiğinde dikkatli olunmalıdır. Erlotinib kaynaklı advers reaksiyonların gözlenmesi halinde, erlotinibin dozu azaltılabilir.
TARCEVA'nın ön tedavisi veya eş zamanlı uygulaması, prototip CYP3A4 substratları midazolam ve eritromisinin klerensini değiştirmemiştir, ancak midazolamın oral biyoyararlanımını %24'e kadar azalttığı görülmüştür. Başka bir klinik çalışmada, erlotinibin eş zamanlı uygulanan CYP3A4/2C8 substratı paklitakselin farmakokinetiğini etkilemediği gösterilmiştir. Bu nedenle diğer CYP3A4 substratlarının klerensi ile anlamlı etkileşimlerin olması pek mümkün değildir.
Glukoronidasyonun inhibisyonu, UGT1A1'in substratları olan ve yalnızca bu yolla atılan tıbbi ürünlerle etkileşimlere neden olabilir. Düşük UGT1A1 ekspresyon seviyeleri bulunan veya genetik glukoronidasyon bozukluklarına (örn. Gilbert hastalığı) sahip olan hastalar, artmış bilirubin serum konsantrasyonu ortaya koyabilir ve bu hastalar dikkatle tedavi edilmelidir.
Erlotinib insanlarda karaciğerde hepatik sitokromlar, birincil olarak CYP3A4 ve daha az ölçüde CYP1A2 ile metabolize edilmektedir. Bağırsakta CYP3A4 ile akciğerde CYP1A1 ile ve tümör dokusunda CYP1B1 tarafından gerçekleştirilen ekstrahepatik metabolizma da erlotinibin metabolik klerensine ayrıca katkıda bulunmaktadır. Bu enzimler tarafından metabolize edilen veya bu enzimlerin inhibitörü veya indükleyicisi olan ilaçlarla potansiyel etkileşimler ortaya çıkabilir.
CYP3A4 aktivitesinin potent inhibitörleri, erlotinib metabolizmasını azaltır ve erlotinib plazma konsantrasyonlarını arttırırlar. CYP3A4 metabolizmasının ketokonazol ile inhibisyonu (5 gün süreyle, ağızdan günde iki kez 200 mg) artmış bir erlotinibe maruz kalma (medyan erlotinib maruziyetinde %86 artış [EAA - eğri altı alan]) ve yalnızca erlotinibe kıyasla C değerinde %69'luk bir artışa yol açmıştır. Bu nedenle, erlotinib
azol antifungalleri (başka deyişle ketakonazol, itrakonazol, vorikonazol) proteaz inhibitörleri, eritromisin veya klaritromisin gibi güçlü CYP3A4 inhibitörleri ile kombine edildiğinde dikkatli olunmalıdır. Gerekli ise, özellikle toksisite görülüyorsa, erlotinib dozu azaltılmalıdır.
CYP3A4 aktivitesinin güçlü indükleyicileri, erlotinib metabolizmasını arttırır ve erlotinib plazma konsantrasyonlarını anlamlı düzeyde düşürürler. Bir klinik çalışmada erlotinib ve güçlü bir CYP3A4 indükleyicisi olan rifampisinin (7 gün süreyle, ağızdan günde 1 kez 600 mg) eş zamanlı kullanımı medyan erlotinib EAA seviyelerinde
%69'luk düşüş ile sonuçlanmıştır. Rifampisinin 450 mg tek TARCEVA dozu ile eşzamanlı uygulaması, erlotinibe ortalama maruziyeti (EAA) rifampisin tedavisi olmaksızın uygulanan tek doz 150 mg TARCEVA dozu ile gözlenen seviyenin %57,5'i ile sonuçlanmıştır. Bu nedenle, TARCEVA'nın CYP3A4 indükleyicileriyle eş zamanlı uygulamasından kaçınılmalıdır. TARCEVA'nın rifampisin gibi güçlü bir CYP3A4 indükleyicisi ile eşzamanlı uygulanması gerektiği hastalarda güvenlilik (böbrek ve karaciğer fonksiyonları ve serum elektrolitleri dahil) yakından izlenerek dozda 300 mg'a kadar artış yapılması değerlendirilmelidir ve 2 haftadan uzun süreyle iyi tolere edilmesi halinde, yakın güvenlilik izlemesi ile 450 mg'a kadar bir artış daha yapılması dikkate alınabilir. Örneğin fenitoin, karbamazepin, barbitüratlar veya St. John's Wort (hypericum perforratum) gibi diğer indükleyicilerle de maruziyet düzeyinde azalma oluşabilir. Söz konusu etkin maddeler erlotinib ile kombine edildiğinde dikkat edilmelidir. Mümkün olduğu zamanlarda güçlü CYP3A4 indükleyici aktivitesi bulunmayan alternatif tedavi seçenekleri değerlendirilmelidir.
Erlotinib ve kumarin türevi antikoagülanlar
TARCEVA alan hastalarda yükselmiş Uluslararası Normalleştirilmiş Oran (INR - International Normalized Ratio) ve bazı durumlarda öldürücü bulunmuş kanama olgularına sebep olan varfarin dahil kumarin türevi antikoagülanlar ile etkileşimler bildirilmiştir. Kumarin türevi antikoagülan ilaçları kullanmakta olan hastalar protrombin zamanı veya INR değişiklikleri açısından düzenli olarak izlenmelidir.
Erlotinib ve statinler
TARCEVA ile bir statin kombinasyonu, seyrek görülen rabdomiyoliz dahil statin kaynaklı miyopati potansiyelini artırabilir.
Erlotinib ve sigara içenler
Bir farmakokinetik etkileşim çalışmasının sonuçları sigara kullananlarda TARCEVA uygulaması sonrasında EAA, C ve plazma konsantrasyonunda sigara kullanmayanlara kıyasla 24 saatte sırasıyla anlamlı 2,8-, 1,5- ve 9 katı azalma olduğunu göstermiştir. Bu nedenle halihazırda sigara içmekte olan hastalar TARCEVA ile tedavi başlatılmadan önce mümkün olan en kısa sürede sigarayı bırakmaları için teşvik edilmelidir, çünkü aksi takdirde plazma erlotinib konsantrasyonları azalmaktadır.
CURRENTS çalışmasından elde edilen veriler, sigara içen hastalarda önerilen 150 mg doz ile karşılaştırıldığında 300 mg'lık yüksek erlotinib dozunun fayda gösterdiğine dair herhangi bir kanıt göstermemiştir. Güvenlilik verileri 300 mg ve 150 mg dozları arasında karşılaştırılabilirdir ancak, daha yüksek erlotinib dozu alan hastalarda döküntü, interstisyel akciğer hastalığı ve diyare insidansında sayısal bir artış olmuştur (bkz. Bölüm 4.2, 4.4, 5.1 ve 5.2).
Erlotinib ve p-glikoprotein inhibitörleri
Erlotinib, P-glikoprotein etkin maddesi taşıyıcısı için bir substrattır. Pgp inhibitörleri (ör: siklosporin ve verapamil) ile eşzamanlı uygulama erlotinib dağılımını ve /veya eliminasyonunu değiştirebilir. Bu etkileşimin sonucunda, ör. MSS toksisitesi açısından neler olduğu saptanmamıştır. Bu tür durumlarda dikkatli olunmalıdır.
Erlotinib ve pH değiştiren tıbbi ürünler
Erlotinib 5'ten yüksek pH seviyesinde çözünürlükte azalma ile karakterizedir. Üst sindirim kanalının pH'sını değiştiren ilaçlar, erlotinib çözünürlüğünü ve buna bağlı olarak biyoyararlanımını değiştirebilir. Erlotinibin bir proton pompası inhibitörü olan omeprazol ile birlikte uygulanması erlotinib maruziyetini [EAA] ve maksimum konsantrasyonunu [C] sırasıyla %46 ve %61 azaltmıştır. T ve yarı ömründe herhangi bir değişiklik olmamıştır. TARCEVA ve bir H2-reseptör antagonisti olan ranitidinin 300 mg'ı ile beraber kullanılması, erlotinibe maruziyeti [EAA] ve C'ı sırasıyla %33 ve %54 oranında azaltmıştır. Bu tip ajanlarla eşzamanlı uygulandığında Tarceva'nın dozunun arttırılmasının maruziyetteki bu kaybı telafi etmesi pek mümkün değildir. Bununla birlikte, TARCEVA 150 mg günde iki kere, ranitidinden 2 saat önce veya 10 saat sonrasında ayrı saatlere bölünerek kullanılırsa, erlotinib maruziyeti [EAA] ve C sırasıyla sadece %15 ve %17 oranında azalmıştır. Antasitlerin erlotinib emilimi üzerindeki etkisi araştırılmamıştır, ancak emilim bozularak plazma seviyelerinde düşüşe yol açabilir. Özet olarak, erlotinibin proton pompası inhibitörleri ile kombinasyonundan kaçınılmalıdır. TARCEVA ile tedavi sırasında antasitlerin kullanımı düşünülüyorsa, bu ilaçların TARCEVA'nın günlük dozundan en az 4 saat önce veya 2 saat sonra alınması gerekir. Ranitidin kullanımı düşünülüyorsa, ayrı saatlere bölünerek kullanılmalı, başka deyişle TARCEVA ranitidin dozu alınmadan en az 2 saat önce veya alındıktan 10 saat sonra alınmalıdır.
Erlotinib ve Gemsitabin
Bir Faz Ib çalışmada, gemsitabinin erlotinib farmakokinetiği üzerinde anlamlı herhangi bir etkisine veya erlotinibin gemsitabin farmakokinetiği üzerinde anlamlı herhangi bir etkisine rastlanmamıştır.
Erlotinib ve Karboplatin/Paklitaksel
Erlotinib, platin konsantrasyonlarını artırır. Bir klinik çalışmada karboplatin ve paklitaksel ile birlikte erlotinib kullanımı, toplam platin EAA değerinde %10,6'lık bir artışa yol açmıştır. İstatistiksel olarak anlamlı olmasına rağmen, bu farkın klinik açıdan önemli olduğu düşünülmemektedir. Klinik uygulamada karboplatin maruziyetini artıran, böbrek yetmezliği gibi diğer kofaktörler olabilir. Erlotinib farmakokinetiği üzerine karboplatin veya paklitakselin anlamlı etkisi bulunmamaktadır.
Erlotinib ve Kapesitabin
Kapesitabin, erlotinib konsantrasyonlarını artırabilir. Tek ajan olarak erlotinib uygulanan çalışmadan elde edilen değerlerle karşılaştırıldığında, kapesitabin ile birlikte uygulanan erlotinib çalışmasında, erlotinib EAA'sı istatistiksel olarak anlamlı derecede artmaktadır ve C düzeyi sınırda bir artış göstermektedir. Kapesitabin farmakokinetiği üzerinde erlotinibin anlamlı etkisi bulunmamaktadır.
Erlotinib ve proteazom inhibitörleri
Çalışma mekanizmasına bağlı olarak, bortezomib dahil proteazom inhibitörlerinin erlotinib gibi EGFR inhibitörlerinin etkisini değiştirmesi beklenebilir. Bu değişim, proteazomdan EGFR degradasyonunu gösteren sınırlı klinik veriler ve preklinik çalışmalarla desteklenmektedir.
Özel popülasyonlara ilişkin ek bilgiler:
Özel popülasyonlara ilişkin etkileşim çalışması yapılmamıştır.
Pediyatrik popülasyon:
TARCEVA'nın 18 yaşın altındaki hastalarda etkileşim çalışması yapılmamıştır.
4.6. Gebelik ve laktasyon
Gebelik kategorisi: D
Çocuk doğurma potansiyeli bulunan kadınlar/ Doğum kontrolü(Kontrasepsiyon) Doğurganlık potansiyeline sahip kadınların TARCEVA kullanırken gebe kalmaktan kaçınmaları konusunda uyarılmaları gereklidir. Tedavi sırasında ve tedavinin tamamlanmasından sonraki en az iki hafta boyunca, yeterli doğum kontrol yöntemleri kullanılmalıdır.
Gebelik dönemi
Gebe kadınlarda TARCEVA kullanımı ile ilgili yeterince veri bulunmamaktadır. Hayvanlar üzerinde yapılan çalışmalar teratojenisite veya abnormal parturisyon ile ilgili bir sonuç göstermemiştir. Buna karşın, gebelik üzerinde olası bir advers olay, tavşan ve sıçanlarda artan embriyo/fetal letalite görüldüğünden gözardı edilememektedir (bkz. Bölüm 5.3). İnsanlar için potansiyel risk bilinmemektedir. TARCEVA kesinlikle gerekli olmadıkça gebelik döneminde kullanılmamalıdır. Gebe kadınlarda tedavi, ancak anne için beklenen faydaların, fetüs için doğabilecek risklerden daha üstün olması halinde sürdürülmelidir.
Laktasyon dönemi
Erlotinibin anne sütüne geçip geçmediği bilinmemektedir. TARCEVA'nın süt üretimindeki etkisini veya anne sütünde bulunmasını değerlendiren herhangi bir çalışma yapılmamıştır. Bebek için potansiyel zarar bilinmediğinden, anneler TARCEVA kullanırken ve son dozu aldıktan en az 2 hafta sonraya kadar emzirmemeleri konusunda uyarılmalıdır.
Üreme yeteneği/ Fertilite
Hayvanlarda yapılan çalışmalarda fertilite bozukluğu görülmemiştir. Ancak, hayvanlardaki çalışmalar, üreme parametreleri üzerinde etkileri olduğu gösterildiğinden fertilite üzerine advers etkiler göz ardı edilemez (bkz. Bölüm 5.3). İnsanlar için potansiyel risk bilinmemektedir.
4.7. Araç ve makine kullanımı üzerindeki etkiler
4.7. Araç ve makine kullanımı üzerindeki etkiler
4.8. İstenmeyen etkiler
TARCEVA'nın güvenlilik değerlendirmesi, en az bir doz 150 mg TARCEVA monoterapi alan 1500 hastadan ve gemsitabinle kombinasyon halinde 100 mg veya 150 mg TARCEVA alan 300 hastanın verilerine dayanmaktadır.
TARCEVA ile monoterapi veya kombinasyon halinde kemoterapi alan hastalarda ortaya çıkan advers reaksiyonların insidansı Ulusal Kanser Enstitüsü - Ortak Toksisite Kriterleri (NCI-CTC) Derecesine göre Tablo 1'de özetlenmektedir. Listelenen istenmeyen etkiler TARCEVA ile tedavi edilen hastalarda plasebo grubuna göre daha sık (≥%3) ve TARCEVA grubunda hastaların en az %10'unda ortaya çıkan advers reaksiyonlardır. Diğer klinik çalışmalarda ortaya çıkan istenmeyen etkiler Tablo 2'de özetlenmektedir.
Klinik araştırmalarda ortaya çıkan istenmeyen etkiler Tablo 1'de MedDRA organ sistemine göre sıralanmıştır. İstenmeyen etkileri sıklıklarına göre sıralamak için şu terimler kullanılmıştır: çok yaygın (1/10); yaygın (1/100, <1/10); yaygın olmayan (1/1.000, <1/100); seyrek (1/10.000, <1/1.000); çok seyrek (<1/10.000); bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor).
Her bir sıklık gruplamasında, advers reaksiyonlar azalan ciddiyet sırasına göre sunulmaktadır.
Küçük Hücreli Dışı Akciğer Kanseri (KHDAK) (TARCEVA, monoterapi olarak kullanılır.)
EGFR Mutasyonlu Hastaların Birinci Basamak Tedavisi
154 hastada gerçekleştirilen açık etiketli, randomize, Faz III ML20650 çalışmasında EGFR aktive edici mutasyonları olan KHDAK hastalarının birinci basamak tedavisinde TARCEVA'nın güvenliliği 75 hastada değerlendirilmiştir. Bu hastalarda yeni güvenlilik sinyalleri gözlenmemiştir.
ML20650 çalışmasında TARCEVA ile tedavi edilen hastalarda en sık görülen yan etkiler döküntü ve diyare olmuştur (sırasıyla %80 ve %57), çoğu Derece 1/2'dir ve girişim olmadan yönetilebilmiştir. Derece 3 döküntü ve diyare, hastaların sırasıyla %9'u ve %4'ünde görülmüştür. Derece 4 döküntü veya diyare görülmemiştir. Hem döküntü hem de diyare, hastaların %1'inde TARCEVA'nın bırakılmasına neden olmuştur. Döküntü ve diyare için doz modifikasyonları (kesilmeler veya azaltmalar), sırasıyla hastaların %11'i ve %7'sinde gerekli olmuştur.
İdame Tedavi
Diğer iki çift kör, randomize, plasebo kontrollü Faz III çalışmalarında BO18192 (SATURN) ve BO25460 (IUNO); TARCEVA, birinci basamak kemoterapiden sonra idame olarak uygulanmıştır. Bu çalışmalar, birinci basamak standart platin bazlı
kemoterapiyi takiben ileri evre, tekrarlayan veya metastatik KHDAK'li toplam 1532 hastada yürütülmüş ve yeni güvenlilik sinyalleri tanımlanmamıştır.
BO18192 ve BO25460 çalışmalarında TARCEVA ile tedavi edilen hastalarda en sık görülen advers reaksiyonlar döküntü (BO18192: tüm dereceler %49,2, derece 3: %6,0; BO25460: tüm dereceler %39,4, derece 3: %5,0) ve diyaredir (BO18192: tümü dereceler %20,3, derece 3: %1,8 BO25460: tüm dereceler %24,2, derece 3: %2,5). Her iki çalışmada da Derece 4 döküntü veya diyare gözlenmemiştir. BO18192 çalışmasında hastaların sırasıyla %1'inde ve <%1'inde döküntü ve diyare TARCEVA tedavisinin kesilmesine neden olurken, BO25460 çalışmasında hiçbir hasta döküntü veya diyare nedeniyle TARCEVA tedavisini bırakmamıştır. BO18192 çalışmasında hastaların sırasıyla %8,3 ve %3'ünde ve BO25460 çalışmasında hastaların sırasıyla
%5,6 ve %2,8'inde döküntü ve diyare için doz modifikasyonları (kesilmeler veya azaltmalar) gerekli olmuştur.
İkinci ve İleri Basamak Tedavi
Randomize, çift-kör bir çalışmada (BR.21; TARCEVA ikinci seçenek tedavi olarak uygulanmıştır), döküntü (%75) ve diyare (%54) en yaygın rapor edilen advers reaksiyonlar olmuştur. Çoğu şiddet açısından Derece 1 veya Derece 2 düzeyinde olmuş ve müdahaleye gerek kalmaksızın düzelmişlerdir. Derece 3/4 döküntü ve diyare TARCEVA ile tedavi edilen hastaların sırasıyla %9'unda ve %6'sında ortaya çıkmıştır ve her biri hastaların %1'inin çalışmadan ayrılması ile sonuçlanmıştır. Döküntü ve diyare için hastaların sırasıyla %6 ve %1'inde doz düşüşüne ihtiyaç olmuştur. BR.21 çalışmasında, döküntünün başlamasına kadar geçen medyan süre 8 gün ve diyarenin başlamasına kadar geçen medyan süre 12 gün olarak bulunmuştur.
Genel olarak, döküntü gün ışığı alan bölgelerde ortaya çıkabilen veya kötüleşebilen hafif veya orta şiddette eritematöz ve papulopustüler döküntü olarak kendini belli etmektedir. Gün ışığına maruz kalan hastalarda, koruyucu giysiler ve/veya güneş koruyucu preparat (örn. mineral içeren) kullanımı önerilebilir.
Tablo 1: BR.21 (TARCEVA ile tedavi) ve PA.3 (TARCEVA+gemsitabin ile tedavi) çalışmalarında hastaların %10'unda görülen ve plasebo grubuna göre daha sık (≥ 3%) görülen yan etkiler
| Tarceva (BR.21) N = 485 | Tarceva (PA.3) N = 259 | Sıklık kategorisi ve en yüksek insidans | ||||
NCI-CTC Derecesi | Tüm Derecel er |
3 |
4 | Tüm Derecel er |
3 |
4 | |
MedDRA Tercih edilen terimi |
% |
% |
% |
% |
% |
% | |
|
|
|
|
|
|
|
|
Enfeksiyonlar ve enfestasyonlar Enfeksiyon* |
24 |
4 |
0 |
31 |
3 |
<1 |
çok yaygın |
Metabolizma ve beslenme hastalıkları Anoreksiya Kilo azalması |
52 - |
8 - |
1 - |
- 39 |
- 2 |
- 0 |
çok yaygın çok yaygın |
Göz hastalıkları Kuru göz sendromu (keratokonjonktivit sikka) Konjonktivit |
12 12 |
0 <1 |
0 0 |
- - |
- - |
- - |
çok yaygın çok yaygın |
Psikiyatrik hastalıklar Depresyon |
- |
- |
- |
19 |
2 |
0 |
çok yaygın |
Sinir sistemi hastalıkları Nöropati Baş ağrısı |
- - |
- - |
- - |
13 15 |
1 <1 |
<1 0 |
çok yaygın çok yaygın |
Solunum, göğüs bozuklukları ve mediastinal hastalıklar Dispne Öksürük |
41 33 |
17 4 |
11 0 |
- 16 |
- 0 |
- 0 |
çok yaygın çok yaygın |
Gastrointestinal hastalıklar Diyare** Bulantı Kusma Stomatit Karın ağrısı Dispepsi Flatulans |
54 33 23 17 11 - - |
6 3 2 <1 2 - - |
<1 0 <1 0 <1 - - |
48 - - 22 - 17 13 |
5 - - <1 - <1 0 |
<1 - - 0 - 0 0 |
çok yaygın çok yaygın çok yaygın çok yaygın çok yaygın çok yaygın çok yaygın |
| Tarceva (BR.21) N = 485 | Tarceva (PA.3) N = 259 | Sıklık kategorisi ve en yüksek insidans | ||||
NCI-CTC Derecesi | Tüm Derecel er |
3 |
4 | Tüm Derecel er |
3 |
4 | |
MedDRA Tercih edilen terimi |
% |
% |
% |
% |
% |
% | |
Deri ve deri altı doku hastalıkları Döküntü*** Kaşıntı Kuru cilt Alopesi |
75 13 12 - |
8 <1 0 - |
<1 0 0 - |
69 - - 14 |
5 - - 0 |
0 - - 0 |
çok yaygın çok yaygın çok yaygın çok yaygın |
|
|
|
|
|
|
|
|
Genel bozukluklar ve uygulama bölgesine ilişkin hastalıklar Yorgunluk Pireksi Titreme |
52 - - |
14 - - |
4 - - |
73 36 12 |
14 3 0 |
2 0 0 |
çok yaygın çok yaygın çok yaygın |
* Nötropeni ile birlikte olan veya olmayan ciddi enfeksiyonlar pnömoni, sepsis ve deri altı dokusu iltihabını içermektedir.
** Dehidrasyon, hipokalemi ve renal yetmezliğe yol açabilir.
*** Dermatit akneiform dahil döküntü
- Eşik altında kalan yüzdeye denk gelir.
Tablo 2: Sıklığa Göre Advers Etkilerin Özeti
Sistem organ sınıfı | Çok yaygın (≥1/10) | Yaygın (≥1/100 ila <1/10) | Yaygın olmayan (≥1/1.000 ila <1/100) | Seyrek (≥1/10.000 ila <1/1.000) | Çok seyrek (<1/10.000) |
Göz hastalıkları |
| - Kirpik değişiklikleri |
| ||
Solunum, göğüs bozuklukla rı ve mediastinal hastalıkları |
| - Epistaksis | - İnterstisyel akciğer hastalığı (İAH) |
|
|
Gastrointes tinal hastalıkları | Diyare | -Gastrointestinal kanama | - Gastrointestinal perforasyonlar |
|
|
Hepato- | - Karaciğer |
|
| - Karaciğer |
|
Keratit
4.9. Doz aşımı ve tedavisi
Semptomlar:
Sağlıklı kişilerde 1.000 mg'a varan ve kanser hastalarında 1.600 mg'a varan tek oral dozlar tolere edilmiştir. Sağlıklı kişilerde tekrarlanan günde iki kez 200 mg dozu, doz uygulamasının henüz birkaç gün sonrasından itibaren kötü tolere edilmiştir. Bu çalışmaların verilerine dayanarak, önerilen dozun üzerinde diyare, döküntü ve olası karaciğer transaminazları yükselmesi gibi şiddetli advers olaylar ortaya çıkabilir.
Yönetim:
Doz aşımından şüphelenilmesi durumunda, TARCEVA kesilmeli ve semptomatik tedavi başlatılmalıdır.
5. FARMAKOLOJİK ÖZELLİKLER
5.1. Farmakodinamik özellikler
Farmakoterapötik grup: Antineoplastik ilaç, protein kinaz inhibitörü, epidermal büyüme faktörü reseptörü (EGFR) tirozin kinaz inhibitörleri
ATC kodu: L01EB02
Etki mekanizması:
Erlotinib epidermal büyüme faktör reseptörü/insan epidermal büyüme faktör tip 1 reseptörünün (EGFR, aynı zamanda HER1 olarak bilinen) inhibitörüdür. EGFR'nin intrasellüler fosforilizasyonunu etkili bir şekilde inhibe eder. EGFR/HER1 reseptörü normal hücre ve kanser hücrelerinin hücre yüzeylerinde eksprese edilir. Klinik dışı modellerde, EGFR fosfotirozininin inhibisyonu hücre stazı ve/veya ölümü ile sonlanmaktadır.
EGFR mutasyonları antiapopitotik ve proliferatif sinyal yollarında konstitütif aktivasyona yol açabilir. Erlotinibin bu EGFR mutasyon pozitif tümörlerde EGFR aracılı sinyalleri engelleyen güçlü etkisi, EGFR'nin mutant kinaz bölgesindeki ATP bağlayan bölgesine erlotinibin sıkıca bağlanmasına atfedilmiştir. Aşağı akım sinyalin engellenmesi nedeniyle, hücre çoğalması durmakta ve intrinsik apopitotik yolla hücre ölümü başlamaktadır. EGFR'yi aktive eden bu mutasyonların ekspresyonunun tetiklendiği fare modellerinde tümör regresyonu gözlenmiştir.
Klinik etkililik
EGFR aktive eden mutasyonları olan hastalarda küçük hücreli dışı akciğer kanserinin (KHDAK) birinci basamak tedavisi (monoterapi olarak TARCEVA uygulaması)
EGFR aktive eden mutasyonları olan KHDAK hastalarının birinci basamak tedavisinde TARCEVA ilacının etkinliği faz III, randomize, açık etiketli çalışmada gösterilmiştir (ML20650, EURTAC). Bu çalışma metastatik veya lokal olarak ileri evre (evre IIIB ve IV) KHDAK olan, daha önce ilerlemiş hastalığı için kemoterapi veya sistemik antitümör tedavisi almamış, EGFR tirozin kinaz bölgesinde mutasyonları (ekson 19 delesyonu veya ekson 21 mutasyonu) bulunan beyaz ırktan hastalarda gerçekleştirilmiştir. Hastalar günlük TARCEVA 150 mg veya dört döngü platin bazlı ikili kemoterapi almak üzere 1:1 oranında randomize edilmişlerdir.
Birincil sonlanım noktası, araştırmacının değerlendirdiği progresyonsuz sağkalım (PFS) olan çalışmaya ait etkililik sonuçları Tablo 3'te verilmektedir.
Şekil 1: ML20650 (EURTAC) çalışmasında araştırmacı tarafından değerlendirilen PFS için Kaplan-Meier eğrisi (Nisan 2012 kesimli)
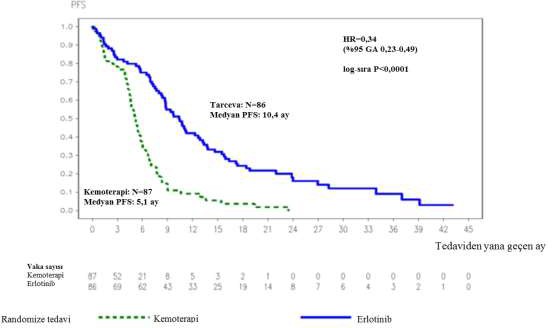
HR: Risk oranı, GA: Güven aralığı, PFS: Progresyonsuz sağkalım
Tablo 3: ML20650 (EURTAC) çalışmasında TARCEVA'nın kemoterapi ile karşılaştırmalı etkililik sonuçları
|
| TARCEVA | Kemoterapi | Risk Oranı (95% GA) | p-değeri |
Önceden |
| n=77 | n=76 |
|
|
planlanmış | |||||
Birincil sonlanım noktası: Progresyonsuz Sağkalım (PFS, ay olarak medyan)* Araştırmacı tarafından değerlendirilen **
Bağımsız inceleme ** |
|
|
|
| |
ara analiz |
|
|
|
| |
(%35 OS |
|
|
|
| |
matürite) (n=153) | 9,4 | 5,2 | 0,42 [0,27-0,64] | p<0,0001 | |
Kesim tarihi: Ağu | 10,4 | 5,4 | 0,47 [0,27-0,78] | p=0,003 | |
2010 |
|
|
|
| |
En İyi Genel Yanıt Oranı | %54,5 | %10,5 |
| p<0,0001 | |
| (CR/PR) | ||||
| Genel Sağkalım (OS) (ay) | 22,9 | 18,8 | 0,80 | p=0,4170 |
| [0,47-1,37] | ||||
Araştırma |
| n=86 | n=87 |
|
|
amaçlı analiz (%40 OS matürite) (n=173) | |||||
PFS (ay olarak medyan), Araştırmacı tarafından değerlendirilen |
9,7 |
5,2 | 0,37 [0,27-0,54] |
p<0,0001 | |
En İyi Genel Yanıt Oranı (CR/PR) | %58,1 | %14,9 |
| p<0,0001 | |
Kesim tarihi: Oca 2011 |
OS (ay) |
19,3 |
19,5 | 1,04 [0,65-1,68] |
p=0,8702 |
Güncellenm iş analiz (%62 OS |
| n=86 | n=87 |
|
|
PFS (ay olarak medyan) | 10,4 | 5,1 | 0,34 [0,23-0,49] | p<0,0001 |
matürite) |
|
|
|
|
|
(n=173) |
|
|
|
|
|
Kesim tarihi: Nisan | OS*** (ay) | 22,9 | 20,8 | 0,93 [0,64-1,36] | p=0,7149 |
2012 |
|
|
|
|
|
CR=komple yanıt; PR=kısmi yanıt
* Hastalığın ilerlemesi veya ölüm riskinde %58'lik bir düşüş gözlemlenmiştir
** Araştırmacı ve IRC arasındaki genel uyum oranı %70'tir.
*** Kemoterapi kolundaki hastaların daha sonra bir EGFR tirozin kinaz inhibitörü ile tedavi alan %82'si ve bu hastaların 2'si dışında tümünün daha sonra TARCEVA kullanması ile yüksek bir çapraz geçiş oranı gözlemlenmiştir.
Birinci basamak kemoterapi sonrası KHDAK'nin idame tedavisi (monoterapi olarak TARCEVA uygulaması)
KHDAK için birinci basamak kemoterapiden sonra idame olarak TARCEVA'nın etkililiği ve güvenliliği, randomize, çift kör, plasebo kontrollü bir çalışmada araştırılmıştır (BO18192, SATURN). Bu çalışma, 4 kür platin bazlı ikili kemoterapiden sonra progresyon göstermemiş, lokal olarak ilerlemiş veya metastatik KHDAK'li 889 hastada gerçekleştirilmiştir. Hastalar, hastalık progresyonuna kadar günde bir kere oral olarak TARCEVA 150 mg veya plasebo almak üzere 1:1 oranında randomize edilmişlerdir. Çalışmanın birincil sonlanım noktası, tüm hastalarda progresyonsuz sağkalımdır (PFS). Temel demografik ve hastalık özellikleri, iki tedavi kolu arasında iyi dengelenmiştir. ECOG PS>1 olan, önemli hepatik veya renal komorbiditesi olan hastalar çalışmaya dahil edilmemiştir.
Bu çalışmada, genel popülasyonda, birincil PFS sonlanım noktası (HR= 0,71 p< 0,0001) ve ikincil OS sonlanım noktası (HR= 0,81 p=0,0088) için fayda görülmüştür. Ancak en büyük fayda, önemli bir PFS faydası (HR=0,10, %95 GA, 0,04 - 0,25; p<0,0001) ve 0,83'lük bir genel sağkalım HR'si (%95 GA, 0,34 -2,02) gösteren, EGFR aktive edici mutasyonları olan hastalarda (n= 49) önceden tanımlanmış bir keşif analizinde gözlenmiştir. EGFR mutasyonu pozitif alt gruptaki plasebo hastalarının
%67'si, EGFR-TKİ'lerle ikinci veya daha ileri basamak tedavisi almıştır.
BO25460 (IUNO) çalışması, tümörleri EGFR aktive edici bir mutasyon (ekson 19 delesyonu veya ekson 21 L858R mutasyonu) bulundurmayan ve dört kür platin bazlı kemoterapiden sonra hastalık progresyonu göstermemiş ileri evre KHDAK'li 643 hastada gerçekleştirilmiştir.
Çalışmanın amacı, erlotinib uygulanan birinci basamak idame tedavisinin genel sağkalımını, hastalığın ilerlemesi sırasında uygulanan erlotinib ile karşılaştırmaktır. Çalışma birincil sonlanım noktasını karşılamamıştır. Tümörü EGFR aktive edici bir mutasyon bulundurmayan hastalarda birinci basamak idame tedavisinde TARCEVA'nın genel sağkalımı, ikinci basamak tedavi olarak uygulanan TARCEVA'dan üstün değildir (HR= 1,02, %95 GA, 0,85 – 1,22, p=0,82). PFS'nin ikincil sonlanım noktası, idame tedavisinde TARCEVA ve plasebo arasında fark göstermemiştir (HR=0,94, %95 GA, 0,80 - 1,11; p=0,48).
BO25460 (IUNO) çalışmasından elde edilen verilere göre, EGFR aktive edici mutasyonu olmayan hastalarda birinci basamak idame tedavisi için TARCEVA kullanımı önerilmemektedir.
En az bir başarısız kemoterapi sonrası KHDAK tedavisi (monoterapi olarak TARCEVA uygulaması)
İkinci-üçüncü basamak tedavi olarak TARCEVA ilacının etkililik ve güvenliliği randomize, çift kör, plasebo kontrollü çalışmada (BR.21) gösterilmiştir. Bu çalışmada yer alan 731 hastada en az bir kemoterapi rejiminden sonra lokal olarak ilerlemiş veya metastatik KHDAK vardır. Hastalar günde bir kere oral olarak TARCEVA 150 mg veya plasebo almak üzere 2:1 oranında randomize edilmişlerdir. Çalışma sonlanım noktaları: genel sağkalım, progresyonsuz sağkalım (PFS), yanıt oranı, yanıt süresi, akciğer kanserine bağlı belirtilerin (öksürük, dispne ve ağrı) kötüleşmesine dek geçen süre ve güvenliliktir. Birincil sonlanım noktası sağkalımdır.
Demografik özellikler iki tedavi grubu arasında dengelidir. Hastaların üçte ikisi erkektir ve yaklaşık üçte birinde başlangıç Doğu Kooperatif Onkoloji Grubu (ECOG) performans durumu (PS) 2 olup, %9'unda başlangıç ECOG PS 3'tür. TARCEVA ve plasebo grubundaki hastaların sırasıyla %93 ve %92 kadarı daha önce platin içeren rejim ve sırasıyla %36 ve %37 kadarı taksan tedavisi almıştır.
TARCEVA grubunda plaseboya göre ölüm için ayarlanan tehlike oranı (HR) 0,73 (%95 GA, 0,60 – 0,87) (p =0,001) olarak saptanmıştır. 12. ayda yaşayan hastaların oranı TARCEVA ve plasebo grupları için sırasıyla %31,2 ve %21,5 olarak saptanmıştır. Medyan genel sağkalım TARCEVA grubunda 6,7 ay iken (%95 GA, 5,5 – 7,8 ay) plasebo grubunda 4,7 aydır (%95 GA, 4,1 – 6,3 ay).
Genel sağkalım üzerine etki farklı hasta alt kümelerinde incelenmiştir. Genel sağkalım üzerine TARCEVA etkisi şu hastalarda benzerdir: başlangıç performans durumu (ECOG) 2-3 (HR = 0,77, %95 GA, 0,6-1,0) veya 0-1 (HR = 0,73, %95 GA, 0,6-0,9),
erkek (HR = 0,76, %95 GA, 0,6-0,9) veya kadın (HR = 0,80, %95 GA, 0,6-1,1), < 65
yaş (HR = 0,75, %95 GA, 0,6-0,9) veya daha yaşlı hastalar (HR = 0,79, %95 GA, 0,6- 1,0), daha önce bir rejim alan hastalar (HR = 0,76, %95 GA, 0,6-1,0) veya daha önce birden fazla rejim alan (HR = 0,75, %95 GA, 0,6-1,0), beyazlar (HR = 0,79, %95 GA,
0,6-1,0) veya Asyalı hastalar (HR = 0,61, %95 GA, 0,4-1,0), adenokarsinomu olanlar (HR = 0,71, %95 GA, 0,6-0,9) veya skuamöz hücreli karsinomu olanlar (HR = 0,67,
%95 GA, 0,5-0,9). Şu hastalarda ise benzer değildir: diğer histolojileri olanlar (HR 1,04, %95 GA, 0,7-1,5), tanıda hastalığı evre IV olan hastalar (HR = 0,92, %95 GA, 0,7-1,2) veya tanıda hastalığı evre < IV olanlar (HR = 0,65, %95 GA, 0,5-0,8). Daha önce hiç sigara içmemiş olan hastalar, şu anda veya eskiden sigara içenlere nazaran (HR = 0,87, %95 GA, 0,71-1,05) erlotinibden daha fazla fayda sağlamıştır (sağkalım HR = 0,42, %95 GA, 0,28-0,64).
EGFR ekspresyon durumu bilinen hastaların %45 kadarında, EGFR pozitif tümörleri olanların tehlike oranı 0,68 (% 95 GA, 0,49-0,94) ve EGFR-negatif tümörü olanların tehlike oranı 0,93 (% 95 GA, 0,63-1,36) olarak saptanmıştır (EGFR pharmDx kit kullanılan IHC ile tanımlanmıştır ve yüzde ondan az boyalı tümör hücresi EGFR negatif
olarak tanımlanmıştır. Kalan %55 hastanın EGFR ekspresyon durumu bilinmemektedir ve HR 0,77 (%95 GA, 0,61-0,98) olarak saptanmıştır.
TARCEVA grubunda ortalama PFS 9,7 haftadır (%95 GA, 8,4 – 12,4 hafta) ve
plasebo grubunda 8,0 haftadır (%95 GA, 7,9 – 8,1 hafta).
Objektif yanıt oranı, Solid Tümörlerde Yanıt Değerlendirme Kriterleri'ne (RECIST) göre TARCEVA grubunda %8,9 olarak saptanmıştır (%95 GA, 6,4 – 12,0). İlk 330 hasta merkezi olarak değerlendirilmiştir (yanıt oranı %6,2); 401 hasta araştırmacı tarafından değerlendirilmiştir (yanıt oranı %11,2).
Medyan yanıt süresi 34,3 hafta olup, 9,7 ila 57,6+ hafta arasındadır. Tam yanıt, kısmi yanıt veya stabil hastalık yaşayan hasta oranı sırasıyla TARCEVA ve plasebo gruplarında %44,0 ve 27,5 olarak saptanmıştır (p = 0,004).
TARCEVA için sağkalım faydası, (RECIST'e göre) nesnel tümör yanıtı elde etmeyen hastalarda da gözlenmiştir. Bunun kanıtı, en iyi yanıtı stabil hastalık veya progresif hastalık olan hastalar arasında ölüm için HR'nin 0,82 (%95 GA, 0,68 – 0,99) olmasıyla ortaya konmuştur.
TARCEVA, plaseboya nazaran semptomlar üzerinde de fayda göstermiş olup öksürük, dispne ve ağrının kötüleşmesi için geçen süre anlamlı derecede uzamıştır.
Lokal ileri veya metastatik, sigara içen (yılda ortalama 38 paket) KHDAK hastalarında kemoterapi sonrası ikinci basamak tedavide iki TARCEVA dozunun karşılaştırıldığı (300 mg'a karşılık 150 mg) çift-kör, randomize Faz III çalışmada (MO22162, CURRENTS), 300 mg doz progresyonsuz sağkalım faydası göstermemiştir (sırasıyla 7 hafta ve 6.86 hafta).
Sekonder sonlanım noktalarının tümü primer sonlanım noktalarıyla tutarlıdır ve günde
300 mg ve 150 mg erlotinib ile tedavi edilen hastalar arasında sağkalım farkı görülmemiştir (HR 1,03, %95 GA 0,80 – 1,32). Güvenlilik verileri, 300 mg ve 150 mg dozları arasında karşılaştırılabilirdir ancak, daha yüksek erlotinib dozu alan hastalarda döküntü, interstisyel akciğer hastalığı ve diyare insidansında sayısal bir artış olmuştur. CURRENTS çalışmasından elde edilen veriler, sigara içen hastalarda önerilen 150 mg doz ile karşılaştırıldığında 300 mg dozla herhangi bir yarar görülmediği göstermiştir.
CURRENT çalışmasındaki hastalar EGFR mutasyon durumuna göre seçilmemiştir (bkz. Bölüm 4.2, 4.4, 4.5 ve 5.2).
5.2. Farmakokinetik özellikler
Genel özelliklerEmilim:
Oral uygulama sonrasında erlotinib ortalama doruk plazma düzeylerine oral dozdan yaklaşık 4 saat sonra ulaşır. Normal sağlıklı gönüllülerdeki bir çalışmada yaklaşık
%59'luk bir biyoyararlanım elde edilmiştir. Bir oral doz sonrasındaki biyoyararlanım, yiyeceklerle birlikte arttırılabilir.
Dağılım:
Erlotinib, 232 L'lik ortalama sanal dağılım hacmine sahiptir ve insan tümör dokularına dağılır. Günlük 150 mg oral TARCEVA dozu almakta olan 4 hastada (3'ü küçük hücreli dışı akciğer kanserli, biri de larinks kanserli) yapılan bir çalışmada, tedavinin 9. günündeki cerrahi eksizyonlardan alınan tümör örnekleri, tümördeki erlotinib konsantrasyonlarının ortalama 1.185 ng/g doku olduğunu ortaya koymuştur. Bu da kararlı durumda gözlenen doruk plazma konsantrasyonlarının genel ortalamasının
%63'üne karşılık gelmektedir (%5-161 aralığı). Primer aktif metabolitler ortalama 160 ng/g doku konsantrasyonlarında tespit edilmiş olup bu değer de kararlı durumda gözlenen doruk plazma konsantrasyonlarının %113'lük genel ortalamasına denk gelmektedir (%88-130). Plazma protein bağlanması yaklaşık olarak %95'tir. Erlotinib serum albumine ve alfa-1 asit glikoproteine (AAG) bağlanmaktadır.
Biyotransformasyon:
Erlotinib insanlarda karaciğerde hepatik sitokrom enzimleri tarafından, birincil olarak CYP3A4 ve daha az ölçüde CYP1A2 tarafından metabolize edilmektedir. CYP4A4'ün bağırsaktaki, CYP1A1'in akciğerdeki, CYP1B1'in tümör dokusundaki ekstrahepatik metabolizması erlotinibin metabolik klerensine potansiyel olarak yardım eder.
Tespit edilen 3 ana metabolik yol mevcuttur: 1) yan zincirlerden biri veya her ikisinin O-demetilasyonu ve bunu takiben karboksilik aside oksidasyon; 2) asetilen kısmının oksidasyonu ve takiben aril karboksilik aside hidrolizi; ve 3) fenil-asetilen kısmının aromatik hidroksilasyonu. Yan zincirlerden birinin O-demetilasyonu ile oluşan OSI 420 ve OSI 413 primer metabolitleri preklinik in vitro deneyler ve in vivo tümör modellerindeki erlotinib ile karşılaştırılabilir etkiye sahiptir. Bunlar plazmada erlotinibin
<%10'u oranında mevcut bulunup, erlotinib ile benzer farmakokinetik gösterirler.
Eliminasyon:
Erlotinib büyük oranda metabolitleri halinde birincil olarak feçes ile atılırken (>%90), renal eliminasyon bir oral dozun yalnızca küçük bir miktarına (yaklaşık %9) karşılık gelir. Tek ajan olarak TARCEVA verilen 591 hastadaki bir popülasyon farmakokinetik analizi, 36,2 saatlik ortalama yarı-ömürle, 4,47 L/saatlik ortalama görünen klerens ortaya koymuştur. Bu nedenle, kararlılık durumu plazma konsantrasyonlarına ulaşılmasının yaklaşık 7-8 gün içinde gerçekleşmesi beklenmektedir.
Doğrusallık/doğrusal olmayan durum:
Yeterli veri yoktur.
Hastalardaki karakteristik özellikler
Beklenen görünür klerens ile hasta yaşı, vücut ağırlığı, cinsiyet ve etnik özellikler arasında anlamlı bir ilişki gözlenmemiştir. Erlotinib farmakokinetiğini değiştiren hastaya ait faktörler, serum total bilirubin, albümin ve alfa-1 asit glikoprotein konsantrasyonları ve sigara kullanımının devam etmesidir. Artmış total bilirubin serum konsantrasyonları ve albümin ve alfa-1 asit glikoprotein konsantrasyonları daha yavaş hızda erlotinib klerensi ile birliktelik göstermiştir. Bu farklılıkların klinik relevansı belli değildir. Bununla birlikte, sigara içenlerde daha hızlı bir erlotinib klerensi gözlenmiştir. Bu durum, tek bir oral doz olarak 150 mg erlotinib alan, sigara içmeyen ve halihazırda
sigara içen sağlıklı bireylerde yapılan farmakokinetik çalışmada doğrulanmıştır. C'ın geometrik ortalaması sigara içmeyenlerde 1.056 ng/mL iken sigara içenlerde 689 ng/mL olmuştur ve sigara içenler için sigara içmeyenlere göre ortalama oran %65,2'dir (95% GA: 44,3 ila 95,9, p = 0,031). EAA için geometrik ortalama sigara içmeyenlerde 18726 ngxh/mL ve sigara içenlerde 6.718 ngxh/mL olmuştur ve ortalama oran %35,9'dur (95% GA: 23,7 ila 54,3, p < 0,0001). C için geometrik ortalama sigara içmeyenlerde 288 ng/mL ve sigara içenlerde 34,8 ng/mL olmuştur ve ortalama oran %12,1'dir (95% GA: 4,82 ila 30,2, p = 0,0001).
Pivotal faz III KHDAK çalışmasında, halihazırda sigara içenlerde erlotinib için kararlı durum plazma konsantrasyonu 0,65 mcg/mL olmuştur ve bu değer sigarayı bırakanlarda veya hiç sigara içmemiş bireylerde görülen konstrasyonun iki katından daha azdır (1,28 mcg/mL, n=108). Bu etki ile birlikte görülen erlotinib plazma klerensinde %24'lük bir artış gözlenmiştir. Daha önce sigara içmiş olan KHDAK hastaları üzerinde yapılan faz I doz eskalasyon çalışmasında, kararlı durumdaki farmakokinetik analizleri TARCEVA dozu 150 mg'den maksimum tolere edilebilir doz olan 300 mg'ye artırıldığında erlotinib maruziyetinde doz orantılı artış göstermiştir. Bu çalışmadaki halihazırda sigara içenlerde uygulanan 300 mg dozda kararlı durum plazma konsantrasyonu 1,22 mcg/mL olmuştur (n=17) (bkz. Bölüm 4.2, 4.4, 4.5 ve 5.1).
Farmakokinetik çalışmaların sonuçlarına göre, plazma konsantrasyonlarını düşürebileceği için, halihazırda sigara içen hastalara TARCEVA tedavisi alırken sigarayı bırakmaları önerilmelidir.
Populasyon farmakokinetik analizinden elde edilen sonuçlara göre bir opioid varlığının maruziyeti %11 oranında artırdığı görülmüştür.
Pediyatrik populasyon:
Pediyatrik hastalara özgün çalışmalar bulunmamaktadır.
Geriyatrik populasyon:
Yaşlı hastalara özgün çalışmalar bulunmamaktadır.
Karaciğer yetmezliği:
Erlotinib birincil olarak karaciğer aracılığıyla metabolize edilir. Solid tümörleri olan ve orta derecede hepatik fonksiyon bozukluğu bulunan hastalarda (Child-Pugh skoru 7-9) erlotinib EAA ve C geometrik ortalaması sırası ile 27000 ngxh/mL ve 805 ng/mL olmuştur ve bu değerler primer karaciğer kanseri veya hepatik metastazları olanlar da dahil olmak üzere hepatik fonksiyonları yeterli olan hastalarda sırası ile 29300 ngxh/mL ve 1.090 ng/mL şeklindedir. C değerinin orta derecede hepatik fonksiyon bozukluğu olan hastalarda istatiksel olarak anlamlı derecede daha düşük olmasına karşın bu farkın klinik olarak anlamlı olmadığı düşünülmektedir. Şiddetli hepatik disfonksiyonun erlotinib farmakokinetiği üzerindeki etkisi ile ilgili veri bulunmamaktadır. Popülasyon farmakokinetik analizinde, total bilirubinin artmış serum konsantrasyonlarının daha düşük erlotinib klerensi hızı ile ilişkili olduğu görülmüştür.
Böbrek yetmezliği:
Erlotinib ve metabolitlerinin böbrekler tarafından atılımı önemli ölçüde değildir. Tek bir dozun %9'dan azı idrar ile atılmaktadır. Populasyon farmakokinetik analizinde,
erlotinib klerensi ve kreatinin klerensi arasında klinik olarak anlamlı bir ilişki görülmemiştir ancak kreatinin klerensi 15 mL/dk'den az olan hastalar ile ilgili bir veri bulunmamaktadır.
5.3. Klinik öncesi güvenlilik verileri
En az bir hayvan türü veya çalışmada gözlenmiş olan kronik doz verilmesine bağlı etkiler kornea (atrofi, ülserasyon), deri (foliküler dejenerasyon ve enflamasyon, kızarıklık ve alopesi), overler (atrofi), karaciğer (karaciğer nekrozu), böbrekler (renal papiller nekroz ve tübüler dilatasyon) ve gastrointestinal sistem (mide boşalmasında gecikme ve diyare) üzerine etkileri içermiştir. Kırmızı kan hücresi parametreleri düşmüş ve beyaz kan hücreleri öncelikle de nötrofiller, artmıştır. Alanin aminotransferaz (ALT), aspartat aminotransferaz (AST) ve bilirubinde tedavi ile ilişkili artışlar meydana gelmiştir. Bu bulgular, klinik olarak anlamlı maruziyetlerin altındaki maruziyetlerde görülmüştür.
Etki mekanizmasına dayanarak, erlotinibin teratojenik olma potansiyeli bulunmaktadır. Sıçanlarda ve farelerde maksimum tolere edilebilir doz ve/veya maternal olarak toksik dozlarda yapılan reproduktif toksikoloji testlerinden elde edilen veriler reproduktif (sıçanlarda embriyotoksisite, tavşanlarda ise embriyo resorpsiyon ve fetotoksisite) ve gelişimsel (yavru büyümesinde düşüş ve sıçanlarda sağkalım) toksisite göstermiştir ancak bu teratojenik değildir ve fertiliteyi olumsuz bozmamıştır. Bu bulgular klinik olarak anlamlı tüm maruziyetlerde görülmüştür.
Konvansiyonel genotoksisite çalışmalarında erlotinib negatif sonuç göstermiştir. Sıçanlarda ve farelerde erlotinib ile yapılan iki yıllık karsinojenisite çalışmaları insan terapötik maruziyetini aşan maruziyetlere kadar negatif olmuştur (C ve/veya EAA'ya dayanarak, sırası ile 2 kata ve 10 kata kadar daha fazla).
Sıçanlarda UV irradyasyonu sonrası hafif derecede fototoksik deri reaksiyonu görülmüştür.
6. FARMASÖTİK ÖZELLİKLER
6.1. Yardımcı maddelerin listesi
Tablet çekirdeği:
Laktoz monohidrat (inek sütünden elde edilmektedir) Mikrokristalin selüloz (E460)
Sodyum nişasta glikolat Tip A Sodyum laurilsülfat Magnezyum stearat (E470 b)
Film kaplama karışımı: Hipromelloz (E464) Hidroksipropil selüloz (E463) Titanyum dioksit (E171) Makrogol
6.2. Geçimsizlikler
Yeterli veri yoktur.
6.3. Raf ömrü
48 ay
6.4. Saklamaya yönelik özel tedbirler
30°C'nin altındaki oda sıcaklığında saklanmalıdır.
6.5. Ambalajın niteliği ve içeriği
30 tabletlik PVC blister
6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler
Kullanılmamış/Son kullanma tarihi geçmiş ürünlerin imhası
Farmasötik ürünlerin çevreye bırakılmasından kaçınılmalıdır. İlaçlar, atık suları ve evsel atık ile imha edilmemelidir. Varsa bulunduğunuz yerdeki donanımlı atık toplama sistemlerini kullanınız.
Kullanılmamış olan ürünler ya da atık materyallar “Tıbbi Atıkların Kontrolü yönetmeliği†ve “Ambalaj Atıklarının Kontrolü yönetmelikleriâ€ne uygun olarak imha edilmelidir.
 HIV ve Aids
HIV, Human Immunodeficiency Virus’dür (İnsanlarda Bağışıklık Sistemini Bozan
Virüsdür). Bu virüs AIDS hastalığına sebep olur.
HIV ve Aids
HIV, Human Immunodeficiency Virus’dür (İnsanlarda Bağışıklık Sistemini Bozan
Virüsdür). Bu virüs AIDS hastalığına sebep olur. |
 Tiroid Kanseri
En sık görülen tiroid kanseri türü olan papiller tiroid kanseri, tüm tiroid kanserlerinin yaklaşık %70'ini oluşturur.
Tiroid Kanseri
En sık görülen tiroid kanseri türü olan papiller tiroid kanseri, tüm tiroid kanserlerinin yaklaşık %70'ini oluşturur. |
İLAÇ EŞDEĞERLERİ
| Eşdeğer İlaç Adı | Barkodu | İlaç Fiyatı |
|---|---|---|
| DIYAGLIP | 8699717090736 | 652.40TL |
| ERTINOB | 8699262090878 | |
| ERTIVEC | 8699828091554 | 18,499.17TL |
| ETINIB | 8699717090705 | 21,894.43TL |
| LUNGERLO | 8699514095699 | 18,500.12TL |
| Diğer Eşdeğer İlaçlar |
 |
Artrit Artrit, oldukça yaygın bir hastalıktır ancak iyi anlaşılamamıştır. Aslında “artrit” tek bir hastalığın adı değildir; eklem ağrısı veya eklem hastalıklarını adlandırmanın gayri resmi yoludur. |
 |
En Yaygın Alerji Türleri Bağışıklık sistemi, polen, arı zehiri veya evcil hayvan gibi yabancı bir maddeye veya çoğu insanda reaksiyona neden olmayan bir yiyeceğe tepki gösterdiğinde alerjiler meydana gelir. |
 Kanseri") |
Rahim Boyu ( Serviks ) Kanseri Rahim boynu (serviks) kanseri 35 yaş altı kadınlarda görülen vakalarda meme kanserinden sonra ikinci sırayı alır.Serviks kanserinin gelişmesi yıllarca sürebilir. |
İLAÇ GENEL BİLGİLERİ
Roche Müstahzarları Sanayi A.Ş.
| Geri Ödeme Kodu | A11816 |
| Satış Fiyatı | 7606.47 TL [ 24 Mar 2025 ] |
| Önceki Satış Fiyatı | 7606.47 TL [ 17 Mar 2025 ] |
| Original / Jenerik | Original İlaç |
| Reçete Durumu | Normal Reçeteli bir ilaçdır. |
| Barkodu | 8699505092003 |
| Etkin Madde | Erlotinib |
| ATC Kodu | L01EB02 |
| Birim Miktar | 25 |
| Birim Cinsi | MG |
| Ambalaj Miktarı | 30 |
| Antineoplastik ve İmmünomodülatör Ajanlar |
| İthal ( ref. ülke : Portekiz ) ve Beşeri bir ilaçdır. |