TECENTRIQ 840 mg/14 ml infüzyonluk çözelti hazırlamak için konsantre Kısa Ürün Bilgisi
{ Atezolizumab }
1. BEŞERİ TIBBİ ÜRÜNÜN ADI
TECENTRIQ 840 mg/14 mL infüzyonluk çözelti hazırlamak için konsantre Steril
2. KALİTATİF VE KANTİTATİF BİLEŞİM
Etkin madde
14 mL konsantre bir flakon içinde 840 mg atezolizumab içerir.
Dilüsyondan sonra 1 mL solüsyon yaklaşık olarak 3,2 mg atezolizumab içerir (bkz. Bölüm 6.6).
Atezolizumab, rekombinant DNA teknolojisiyle Çin hamsteri yumurtalık hücrelerinde üretilen, bir Fc bölgesi değiştirilmiş, hümanize IgG1 anti-programlı ölüm-ligandı 1 (PD-L1) monoklonal antikorudur.
Yardımcı maddeler
Yardımcı maddeler için Bölüm 6.1'e bakınız.
3. FARMASÖTİK FORMU
İnfüzyonluk çözelti hazırlamak için steril konsantre Berrak, renksiz ila hafif sarımsı sıvı
4. KLİNİK ÖZELLİKLER
4.1. Terapötik endikasyonlar
TECENTRIQ'in, tümörde PDL1 ekspresyonu %1 ve üzerinde olan (SP142 Ventana İmmünhistokimya ile), daha önce kemoterapi almamış metastatik veya rezeke edilemeyen üçlü negatif meme kanserli (ÜNMK) hastaların tedavisinde nab-paklitaksel ile kombine olarak kullanımı endikedir. Hastalar önceden adjuvan kemoterapi almışlarsa hastalıksız interval 12 ay ve üzeri olmalıdır.
4.2. Pozoloji ve uygulama şekli
TECENTRIQ, kanser tedavisinde deneyimli bir hekimin gözetimi altında uygulanmalıdır.
Daha önce tedavi almamış ÜNMK hastaları, geçerliliği gösterilmiş bir test ile doğrulanan PD-L1 tümör ekspresyonuna dayanarak tedavi için seçilmelidir (bkz. Bölüm 5.1).
Pozoloji/uygulama sıklığı ve süresi:
Önerilen TECENTRIQ dozu, 100 mg/m nab-paklitakseli takiben intravenöz yoldan uygulanan 840 mg'dır. Her 28 günlük siklusta, TECENTRIQ 1. ve 15. günlerde, nab-paklitaksel ise 1., 8. ve 15. günlerde uygulanır.
Tedavi süresi:
Klinik faydanın kaybedilmesine (bkz. Bölüm 5.1) veya yönetilemeyen toksisiteye kadar
hastaların TECENTRIQ ile tedavi edilmeleri önerilmektedir.
Geciken veya atlanan dozlar:
Planlanmış bir TECENTRIQ dozu atlanırsa mümkün olan en kısa sürede uygulanmalıdır; planlanan sonraki doza kadar beklenmemelidir. Uygulama planı, dozlar arasında uygun bir aralık korunacak şekilde ayarlanmalıdır.
Tedavi sırasında doz modifikasyonları:
TECENTRIQ için doz azaltımı önerilmez.
Doz gecikmesi veya kesilmesi (ayrıca bkz. Bölüm 4.4 ve 4.8):
Tablo 1: Belirtilen Advers İlaç Reaksiyonları için doz modifikasyon tavsiyesi
Advers reaksiyon | Şiddet | Tedavi modifikasyonu |
Pnömoni | 2. derece | TECENTRIQ tedavisine ara verilir.
Olay 12 hafta içinde 0. derece veya 1. dereceye iyileştiğinde ve kortikosteroidler günde ≤10 mg oral prednizon veya eşdeğerine düşürüldüğünde tedaviye devam edilebilir. |
3. veya 4. derece | TECENTRIQ tedavisi tamamen kesilir. | |
Hepatit | 2. derece: (ALT veya AST >3-5 x normalin üst sınırı [NÜS] veya kan bilirubin >1,5-3 x NÜS) | TECENTRIQ tedavisine ara verilir.
Olay 12 hafta içinde 0. derece veya 1. dereceye iyileştiğinde ve kortikosteroidler günde ≤10 mg oral prednizon veya eşdeğerine düşürüldüğünde tedaviye devam edilebilir. |
3. veya 4. derece: (ALT veya AST >5 x NÜS veya kan bilirubin >3 x NÜS) | TECENTRIQ tedavisi tamamen kesilir. | |
Kolit | 2. veya 3. derece ishal (başlangıca göre ≥4 dışkı/gün veya semptomatik kolit) | TECENTRIQ tedavisine ara verilir.
Olay 12 hafta içinde 0. derece veya 1. dereceye iyileştiğinde ve kortikosteroidler günde ≤10 mg oral prednizon veya eşdeğerine düşürüldüğünde tedaviye devam edilebilir. |
4. derece ishal veya kolit (yaşamı tehdit edici; acil müdahale endike) | TECENTRIQ tedavisi tamamen kesilir. |
Advers reaksiyon | Şiddet | Tedavi modifikasyonu |
Hipotiroidizm veya hipertiroidizm | Semptomatik | TECENTRIQ tedavisine ara verilir.
Hipotiroidizm: Semptomlar tiroid replasman tedavisi ile kontrol altına alındığında ve TSH düzeyleri düşmeye başladığında tedaviye devam edilebilir.
Hipertiroidizm: Semptomlar anti-tiroidilaçları ile kontrol altına alındığında ve tiroid fonksiyonu iyileşmeye başladığında tedaviye devam edilebilir. |
Adrenal yetmezlik | Semptomatik | TECENTRIQ tedavisine ara verilir.
Semptomlar 12 hafta içinde 0. derece veya 1. dereceye iyileştiğinde ve kortikosteroidler günde ≤10 mg oral prednizon veya eşdeğerine düşürüldüğünde ve hastanın durumu replasman tedavisinde stabil hale geldiğinde tedaviye devam edilebilir. |
Hipofizit | 2. veya 3. derece | TECENTRIQ tedavisine ara verilir.
Semptomlar 12 hafta içinde 0. derece veya 1. dereceye iyileştiğinde ve kortikosteroidler günde ≤10 mg oral prednizon veya eşdeğerine düşürüldüğünde ve hastanın durumu replasman tedavisinde stabil hale geldiğinde tedaviye devam edilebilir. |
4. derece | TECENTRIQ tedavisi tamamen kesilir. | |
Tip 1 diabetes mellitus | 3. veya 4. derece hiperglisemi (açlık glikozu >250 mg/dL veya 13,9 mmol/L) | TECENTRIQ tedavisine ara verilir
İnsülin replasman tedavisinde metabolik kontrol elde |
Advers reaksiyon | Şiddet | Tedavi modifikasyonu |
|
| edildiğinde tedaviye devam edilebilir. |
İnfüzyonla ilişkili reaksiyonlar | 1. veya 2. derece | İnfüzyon hızı azaltılır veya kesilir.
Olay düzeldikten sonra tedavi sürdürülebilir. |
3. veya 4. derece | TECENTRIQ tedavisi tamamen kesilir. | |
Döküntü / Ciddi cilt advers reaksiyonları | 3. derece
veya şüpheli Stevens-Johnson sendromu (SJS) veya toksik epidermal nekroliz (TEN) | TECENTRIQ tedavisine ara verilir.
Semptomlar 12 hafta içinde Derece 0 veya Derece 1'e iyileştiğinde ve kortikosteroidler günde ≤ 10 mg prednizon veya eşdeğerine düşürüldüğünde tedaviye devam edilebilir. |
| 4. derece
veya doğrulanmış Stevens- Johnson sendromu (SJS) veya toksik epidermal nekroliz (TEN) | TECENTRIQ tedavisi tamamen kesilir. |
Miyastenik sendrom/ miyastenia gravis, Guillain-Barré sendromu ve meningoensefalit | Tüm dereceler | TECENTRIQ tedavisi tamamen kesilir. |
Pankreatit | Serum amilaz veya lipaz düzeylerinde 3. veya 4. derece yükselme (> 2 x NÜS) veya 2. veya 3. derece pankreatit | TECENTRIQ tedavisine ara verilir.
Serum amilaz ve lipaz düzeyleri 12 hafta içinde 0. derece veya 1. dereceye iyileştiğinde veya pankreatit semptomları düzeldiğinde ve kortikosteroidler günde ≤10 mg oral prednizon veya eşdeğerine düşürüldüğünde TECENTRIQ ile tedaviye devam edilebilir. |
4. derece veya herhangi bir derecede nükseden pankreatit | TECENTRIQ tedavisi tamamen kesilir. | |
Miyokardit | 2. derece veya üzeri | TECENTRIQ tedavisi tamamen kesilir. |
Nefrit | 2. derece: (kreatinin seviyesi >1,5 ila 3 x | TECENTRIQ tedavisine ara verilir. |
Advers reaksiyon | Şiddet | Tedavi modifikasyonu |
| başlangıç veya >1,5 ila 3 x NÜS) |
Olay 12 hafta içinde 0. derece veya 1. dereceye iyileştiğinde ve kortikosteroidler günde ≤10 mg oral prednizon veya eşdeğerine düşürüldüğünde tedaviye devam edilebilir. |
3. veya 4. derece: (kreatinin seviyesi >3 x başlangıç veya >3 x NÜS) | TECENTRIQ tedavisi tamamen kesilir. | |
Miyozit | 2. veya 3. derece | TECENTRIQ tedavisine ara verilir. |
4. derece veya tekrarlayan 3. derece miyozit | TECENTRIQ tedavisi tamamen kesilir. | |
İmmünite ile ilişkili diğer advers reaksiyonlar | 2. veya 3. derece | Olay 12 hafta içinde 0. derece veya 1. dereceye iyileşene ve kortikosteroidler günde ≤10 mg oral prednizon veya eşdeğerine düşürülene kadar TECENTRIQ tedavisine ara verilir. |
4. veya 3. derece nükseden advers olaylar | TECENTRIQ tedavisi tamamen kesilir. (replasman hormonlarıyla kontrol altına alınan endokrinopatiler hariç) |
Not: Toksisite dereceleri, Ulusal Kanser Enstitüsü Advers Olaylar için Ortak Terminoloji Kriterleri, Versiyon 4.0'a (NCI-CTCAE v.4) uygundur.
TECENTRIQ ile tedavi edilen hastalara ilacın riskleri hakkında bilgi veren Hasta Uyarı Kartları verilmelidir.
Uygulama şekli:
TECENTRIQ intravenöz kullanım içindir. TECENTRIQ infüzyonları intravenöz puşe veya bolus şeklinde uygulanmamalıdır.
İlk TECENTRIQ dozu 60 dakika uygulanmalıdır. İlk infüzyon iyi tolere edilirse, sonraki tüm infüzyonlar 30 dakikada uygulanabilir.
Tıbbi ürünün uygulanmadan önceden seyreltilmesi ve kullanımına ilişkin talimatlar için Bölüm
6.6'ya bakınız.
Özel popülasyonlara ilişkin ek bilgiler:
Karaciğer yetmezliği:
Popülasyon farmakokinetik analizine göre hafif veya orta derecede karaciğer bozukluğu olan hastalarda doz ayarlaması gerekli değildir. Şiddetlikaraciğer bozukluğu olan hastalara ilişkin
veri mevcut değildir (bkz. Bölüm 5.2).
Böbrek yetmezliği:
Popülasyon farmakokinetik analizine göre hafif veya orta derecede böbrek bozukluğu olan hastalarda doz ayarlaması gerekli değildir (bkz. Bölüm 5.2). Şiddetli böbrek yetmezliği olan hastalardan elde edilen veriler, bu popülasyon hakkında sonuca varmak için çok sınırlıdır.
Pediyatrik popülasyon:
TECENTRIQ'in 18 yaşından küçük çocuklarda ve adolesanlarda güvenliliği ve etkililiği belirlenmemiştir. Mevcut veriler Bölüm 4.8, 5.1 ve 5.2'de açıklanmıştır, ancak pozoloji konusunda herhangi bir öneri yapılamaz.
Geriyatrik popülasyon:
Popülasyon farmakokinetik analizine göre ≥ 65 yaşındaki hastalarda TECENTRIQ doz ayarlaması gerekli değildir (bkz. Bölüm 4.8 ve 5.1).
Doğu İşbirliği Onkoloji Grubu (ECOG) performans durumu skoru ≥ 2:
ECOG performans durumu skoru ≥ 2 olan hastalar ÜNMK klinik çalışmasına dahil edilmemiştir (bkz. Bölüm 4.4 ve 5.1).
4.3. Kontrendikasyonlar
TECENTRIQ'in etkin maddesi atezolizumaba veya Bölüm 6.1'de listelenen yardımcı maddelerden herhangi birine aşırı duyarlılığı olan hastalarda kontrendikedir.
4.4. Özel kullanım uyarıları ve önlemleri
TECENTRIQ ile tedavi sırasında oluşan immünite ile ilişkili advers reaksiyonların çoğu ilacın kesilmesi ve kortikosteroidlerin ve/veya destekleyici tedavinin başlatılmasıyla geri döndürülebilir olmuştur. Birden fazla vücut sistemini etkileyen immünite ile ilişkili advers reaksiyonlar görülmüştür ve bu reaksiyonlar TECENTRIQ'in son dozundan sonra da oluşabilir.
İmmünite ile ilişkili şüpheli advers reaksiyonlar için etiyolojiyi doğrulamak veya diğer nedenleri dışlamak için yeterli değerlendirme yapılmalıdır. Advers etkilerin şiddetine bağlı olarak, TECENTRIQ tedavisine ara verilir ve kortikosteroid uygulanır. Olay ≤ 1. dereceye iyileştiğinde kortikosteroid kullanımı 1 ay boyunca azaltılarak kesilir. İmmünite ile ilişkili istenmeyen reaksiyonların kortikosteroid kullanımı ile kontrol edilemediği hastalarda, klinik çalışmalardan elde edilen sınırlı verilere dayanarak, diğer sistemik immunosupresan ajanların kullanımı düşünülebilir.
Herhangi bir ≥ 3. derece toksisite ikinci defa ortaya çıkarsa ve replasman hormonlar ile kontrol edilen endokrinopatiler hariç herhangi bir 4. derece immünite ile ilişkili advers reaksiyon görülürse TECENTRIQ tedavisi tamamen kesilir (bkz. Bölüm 4.2 ve 4.8).
İmmünite ile ilişkili pnömoni:
TECENTRIQ ile yürütülen klinik çalışmalarda ölümcül vakalar da dahil olmak üzere pnömoni vakaları gözlenmiştir (bkz. Bölüm 4.8). Hastalar pnömoni belirtileri ve semptomları için izlenmeli ve immün ilişkili pnömonit dışındaki nedenler hariç bırakılmalıdır.
2. derece pnömoni durumunda TECENTRIQ ile tedaviye ara verilmeli ve günde 1-2 mg/kg prednizon veya eşdeğeri ile tedavi başlatılmalıdır. Semptomlar ≤1. dereceye iyileşirse
kortikosteroidler ≥1 ayda azaltılmalıdır. Olay 12 hafta içinde ≤1. dereceye iyileşirse ve kortikosteroidler günde ≤10 mg oral prednizon veya eşdeğerine düşürülürse TECENTRIQ ile tedaviye devam edilebilir. 3. veya 4. derece pnömoni durumunda TECENTRIQ ile tedavi kalıcı olarak bırakılmalıdır.
İmmünite ile ilişkili hepatit:
TECENTRIQ ile yürütülen klinik çalışmalarda bazıları ölümcül sonuçlara yol açan hepatit vakaları gözlenmiştir (bkz. Bölüm 4.8). Hastalar hepatit belirtileri ve semptomları için izlenmelidir.
Aspartat aminotransferaz (AST), alanin aminotransferaz (ALT) ve bilirubin, TECENTRIQ ile tedaviye başlamadan önce ve tedavi sırasında periyodik olarak ve klinik değerlendirmeye göre endike olduğu gibi izlenmelidir.
2. derece anormallik (ALT veya AST >3-5 x NÜS veya kan bilirubin >1,5-3 x NÜS) 5-7 günden uzun süre devam ederse TECENTRIQ ile tedaviye ara verilmeli ve günde 1-2 mg/kg prednizon veya eşdeğeri ile tedavi başlatılmalıdır. Olaylar ≤1. dereceye iyileşirse kortikosteroidler ≥1 ayda azaltılmalıdır.
Olay 12 hafta içinde ≤1. dereceye iyileşirse ve kortikosteroidler günde ≤10 mg oral prednizon veya eşdeğerine düşürülürse TECENTRIQ ile tedaviye devam edilebilir. 3. veya 4. derece olaylarda (ALT veya AST >5 x NÜS veya kan bilirubin >3 x NÜS) TECENTRIQ ile tedavi kalıcı olarak bırakılmalıdır.
İmmünite ile ilişkili kolit:
TECENTRIQ ile yürütülen klinik çalışmalarda ishal veya kolit vakaları gözlenmiştir (bkz. Bölüm 4.8). Hastalar kolit belirtileri ve semptomları için izlenmelidir.
2. veya 3. derece ishal (başlangıca göre ≥4 dışkı/gün artış) veya kolit (semptomatik) durumunda TECENTRIQ ile tedaviye ara verilmelidir. 2. derece ishal veya kolit durumunda semptomlar
>5 gün devam ederse veya nüksederse, günde 1-2 mg/kg prednizon veya eşdeğeri ile tedavi başlatılmalıdır. 3. derece ishal veya kolit durumunda intravenöz kortikosteroidlerle (1-2 mg/kg/gün metilprednizolon veya eşdeğeri) tedavi başlatılmalı ve iyileşme sonrasında oral kortikosteroidlere (günde 1-2 mg/kg prednizon veya eşdeğeri) geçilmelidir. Semptomlar ≤1. dereceye iyileşirse kortikosteroidler ≥1 ayda azaltılmalıdır. Olay 12 hafta içinde ≤1. dereceye iyileşirse ve kortikosteroidler günde ≤10 mg oral prednizon veya eşdeğerine düşürülürse TECENTRIQ ile tedaviye devam edilebilir. 4. derece (yaşamı tehdit edici; acil müdahale endike) ishal veya kolit durumunda TECENTRIQ ile tedavi kalıcı olarak bırakılmalıdır.
İmmünite ile ilişkili endokrinopatiler:
TECENTRIQ ile yürütülen klinik çalışmalarda hipotiroidizm, hipertiroidizm, adrenal yetmezlik, hipofizit ve diyabetik ketoasidoz dahil olmak üzere tip 1 diabetes mellitus vakaları gözlenmiştir (bkz. Bölüm 4.8).
Hastalar endokrinopatilerin klinik belirtileri ve semptomları için izlenmelidir. Tiroid fonksiyonu TECENTRIQ ile tedavi öncesinde ve tedavi sırasında periyodik olarak izlenmelidir. Başlangıçta anormal tiroid fonksiyon testleri olan hastaların uygun şekilde tedavi edilmesi düşünülmelidir.
Anormal tiroid fonksiyonu olan asemptomatik hastalar TECENTRIQ alabilir. Semptomatik
hipotiroidizm durumunda TECENTRIQ ile tedaviye ara verilmeli ve tiroid hormonu replasmanı gerektiğinde başlatılmalıdır. İzole hipotiroidizm kortikosteroidler kullanılmadan replasman tedavisi ile yönetilebilir. Semptomatik hipertiroidizm durumunda TECENTRIQ ile tedaviye ara verilmeli ve bir anti-tiroid tıbbi ürün gerektiği gibi başlatılmalıdır. Semptomlar kontrol altına alındığında ve tiroid fonksiyonu iyileştiğinde TECENTRIQ ile tedaviye devam edilebilir.
Semptomatik adrenal yetmezlik durumunda TECENTRIQ ile tedaviye ara verilmeli ve intravenöz kortikosteroid (günde 1-2 mg/kg metilprednizolon veya eşdeğeri) ile tedavi başlatılmalıdır. Semptomlar iyileştiğinde günde 1-2 mg/kg oral prednizon veya eşdeğeri ile tedavi uygulanmalıdır. Semptomlar ≤1. dereceye iyileşirse kortikosteroidler ≥1 ayda azaltılmalıdır. Olay 12 hafta içinde ≤1. dereceye iyileşirse ve kortikosteroidler günde ≤10 mg oral prednizon veya eşdeğerine düşürülürse ve hastanın durumu replasman tedavisinde stabilse (gerektiyse) tedaviye devam edilebilir.
2. veya 3. derece hipofizit için TECENTRIQ ile tedaviye ara verilmeli ve intravenöz kortikosteroidler (1 ila 2 mg/kg/gün metilprednizolon veya eşdeğeri) ile tedavi başlatılmalı ve ihtiyaca göre hormon replasman tedavisi başlatılmalıdır. Belirtiler düzeldiğinde 1-2 mg/kg/gün prednizon veya eşdeğeri ile tedavi uygulanmalıdır. Semptomlar ≤1. dereceye kadar yükselirse, kortikosteroidler ≥ 1 ay boyunca azaltılarak kesilebilir. Olay, 12 hafta içinde ≤1. dereceye yükselir ve kortikosteroidler günde ≤ 10 mg prednizona veya eşdeğerine düşürülürse ve hasta yedek tedavide (eğer gerekliyse) stabil kalırsa, tedaviye devam edilebilir. 4. derece hipofizit için TECENTRIQ tedavisine ara verilmelidir.
Tip 1 diabetes mellitus için insülin tedavisi başlatılmalıdır. ≥3. derece hiperglisemi (açlık glikozu >250 mg/dL veya 13,9 mmol/L) durumunda TECENTRIQ ile tedaviye ara verilmelidir. İnsülin replasman tedavisinde metabolik kontrol elde edilirse TECENTRIQ ile tedaviye devam edilebilir.
İmmünite ile ilişkili meningoensefalit:
TECENTRIQ ile yürütülen klinik çalışmalarda meningoensefalit gözlenmiştir (bkz. Bölüm 4.8). Hastalar menenjit veya ensefalitin klinik belirtileri ve semptomları için izlenmelidir.
Herhangi bir derece menenjit veya ensefalit durumunda TECENTRIQ ile tedavi kalıcı olarak bırakılmalıdır. İntravenöz kortikosteroidler (günde 1-2 mg/kg metilprednizolon veya eşdeğeri) ile tedavi başlatılmalı ve hastanın durumu iyileştiğinde 1-2 mg/kg oral prednizon veya eşdeğerine geçilmelidir.
İmmünite ile ilişkili nöropatiler:
TECENTRIQ tedavisi gören hastalarda yaşamı tehdit edici olabilen miyastenik sendrom/miyastenia gravis veya Guillain-Barré sendromu gözlenmiştir. Hastalar motor ve duyusal nöropati semptomları için izlenmelidir.
Herhangi bir derece miyastenik sendrom/miyastenia gravis veya Guillain-Barré sendromu durumunda TECENTRIQ ile tedavi kalıcı olarak bırakılmalıdır. Günde 1-2 mg/kg dozda oral prednizon veya eşdeğeriyle sistemik kortikosteroidlerin başlatılması düşünülmelidir.
İmmünite ile ilişkili pankreatit:
TECENTRIQ ile yürütülen klinik çalışmalarda serum amilaz ve lipaz düzeylerinde artışlar dahil olmak üzere pankreatit gözlenmiştir (bkz. Bölüm 4.8). Hastalar akut pankreatiti
düşündüren belirtiler ve semptomlar için yakından izlenmelidir.
Serum amilaz veya lipaz düzeylerinde ≥3. derece artış (>2 x NÜS) veya 2. veya 3. derece pankreatit durumunda TECENTRIQ ile tedaviye ara verilmeli ve intravenöz kortikosteroidler (günde 1-2 mg/kg metilprednizolon veya eşdeğeri) ile tedavi başlatılmalıdır. Semptomlar iyileştiğinde günde 1-2 mg/kg oral prednizon veya eşdeğeri ile tedavi uygulanmalıdır. Serum amilaz ve lipaz düzeyleri 12 hafta içinde ≤1. dereceye iyileştiğinde veya pankreatit semptomları düzeldiğinde ve kortikosteroidler günde ≤10 mg oral prednizon veya eşdeğerine düşürüldüğünde TECENTRIQ ile tedaviye devam edilebilir. 4. derece veya herhangi bir derecede nükseden pankreatit durumunda TECENTRIQ ile tedavi kalıcı olarak bırakılmalıdır.
İmmünite ile ilişkili miyokardit:
TECENTRIQ ile yürütülen klinik çalışmalarda ölümcül vakalar da dahil olmak üzere miyokardit gözlemlenmiştir (bkz. Bölüm 4.8). Hastalar miyokarditi düşündüren belirtiler ve semptomlar için yakından izlenmelidir. Miyokardit ayrıca miyozitin klinik bir belirtisi olabilir ve buna uygun olarak tedavi edilmelidir.
Kardiyak veya kardiyopulmoner semptomları olan hastalar, uygun önlemlerin erken evrede başlatılabilmesi için potansiyel miyokardit açısından değerlendirilmelidir. Miyokardit şüphesi varsa, TECENTRIQ tedavisine ara verilmeli, günde 1 ila 2 mg/kg vücut ağırlığı/gün prednizon veya eşdeğeriyle sistemik kortikosteroidler başlanmalı ve güncel klinik kılavuzlara göre tanısal çalışmayla birlikte derhal kardiyoloji konsültasyonu yapılmalıdır. Miyokardit tanısı konulduğunda, derece ≥ 2 miyokardit için atezolizumab tedavisi kalıcı olarak kesilmelidir (bkz. Bölüm 4.2).
İmmünite ile ilişkili nefrit:
TECENTRIQ ile yapılan klinik çalışmalarda nefrit gözlenmiştir (bkz. Bölüm 4.8). Hastalar böbrek fonksiyonundaki değişiklikler açısından izlenmelidir.
2. derece nefrit durumunda TECENTRIQ tedavisine ara verilmeli ve günde 1 ila 2 mg/kg vücut ağırlığı/gün prednizon veya eşdeğeriyle sistemik kortikosteroidler başlanmalıdır. Olay 12 hafta içinde derece ≤ 1'e yükselirse ve kortikosteroidler günde ≤ 10 mg prednizon veya eşdeğerine düşürülürse atezolizumab ile tedaviye devam edilebilir. 3. veya 4. derece nefrit için TECENTRIQ ile tedavi kalıcı olarak bırakılmalıdır.
İmmünite ile ilişkili miyozit:
Atezolizumab ile ölümcül vakalar dahil miyozit vakaları gözlenmiştir (bkz. Bölüm 4.8). Hastalar miyozit belirti ve semptomları açısından izlenmelidir. Olası miyoziti olan hastalar miyokardit belirtileri açısından izlenmelidir.
Bir hastada miyozit belirti ve semptomları gelişirse, yakından takip edilmeli ve hasta zaman kaybetmeden değerlendirme ve tedavi için bir uzmana yönlendirilmelidir. 2. veya 3. derece miyozit durumunda TECENTRIQ tedavisine ara verilmeli ve kortikosteroid tedavisi (1-2 mg/kg vücut ağırlığı/gün prednizon veya eşdeğeri) başlatılmalıdır. Semptomlar derece ≤ 1'e yükselirse, klinik olarak belirtildiği gibi kortikosteroidler azaltılmalıdır. Olay 12 hafta içinde derece ≤ 1'e yükselirse ve kortikosteroidler günde ≤ 10 mg oral prednizon veya eşdeğerine düşürülürse TECENTRIQ ile tedaviye devam edilebilir. 4. veya 3. derece tekrarlayan miyozit durumunda veya başlangıçtan sonraki 12 hafta içinde kortikosteroid dozu günde ≤ 10 mg prednizon eşdeğerine düşürülemediğinde TECENTRIQ ile tedavi kalıcı olarak kesilmelidir.
İmmünite ile ilişkili şiddetli kutanöz advers reaksiyonlar:
TECENTRIQ ile tedavi edilen hastalarda Stevens-Johnson sendromu (SJS) ve toksik epidermal nekroliz (TEN) vakaları dahil olmak üzere immünite ile ilişkili şiddetli kutanöz advers reaksiyonlar (SCAR'lar) bildirilmiştir. Hastalar, şüpheli şiddetli cilt reaksiyonlar açısından izlenmeli ve diğer sebepler dışlanmalıdır. Şüpheli SCAR'lar varlığında hastalar ileri tanı ve tedavi için bir uzmana yönlendirilmelidir.
Advers reaksiyonun derecesine bağlı olarak, 3. derece cilt reaksiyonu durumunda TECENTRIQ ile tedaviye ara verilmeli ve günde 1-2 mg/kg prednizon veya eşdeğeri ile sistemik kortikosteroidler başlanmalıdır. Olay 12 hafta içinde derece ≤ 1'e iyileşir ve kortikosteroidler günde ≤ 10 mg prednizon veya eşdeğerine düşürülürse TECENTRIQ ile tedaviye devam edilebilir. 4. derece cilt reaksiyonları için TECENTRIQ tedavisi kalıcı olarak bırakılmalı ve kortikosteroidler uygulanmalıdır.
SJS veya TEN şüphesi olan hastalarda TECENTRIQ tedavisine ara verilmelidir. Doğrulanmış SJS veya TEN durumunda TECENTRIQ tedavisi kalıcı olarak kesilmelidir.
Daha önce diğer immün sistemi uyarıcı antikanser ajanlarla tedavi sırasında ciddi veya yaşamı tehdit eden bir kutanöz advers reaksiyon deneyimleyen bir hastada TECENTRIQ kullanımı düşünülürken dikkatli olunmalıdır.
İmmünite ile ilişkili diğer advers reaksiyonlar:
TECENTRIQ'in etki mekanizması göz önüne alındığında, enfektif olmayan sistit dahil olmak üzere bağışıklıkla ilgili diğer potansiyel advers reaksiyonlar meydana gelebilir.
Diğer nedenleri dışlamak için bağışıklıkla ilgili tüm şüpheli advers reaksiyonlar değerlendirilmelidir. Hastalar, bağışıklıkla ilgili advers reaksiyonların belirti ve semptomları açısından izlenmeli ve reaksiyonun ciddiyetine bağlı olarak, klinik olarak belirtildiği gibi tedavi değişiklikleri ve kortikosteroidlerle gözetim altında tutulmalıdır (bkz. Bölüm 4.2 ve Bölüm 4.8).
İnfüzyon ile ilişkili reaksiyonlar:
TECENTRIQ ile yürütülen klinik çalışmalarda infüzyon ile ilgili reaksiyonlar gözlenmiştir (bkz. Bölüm 4.8).
1. veya 2. derece infüzyon ile ilişkili reaksiyon görüldüğünde infüzyon hızı düşürülmeli veya tedaviye ara verilmelidir. 3. veya 4. derece infüzyon ile ilgili reaksiyon görüldüğünde TECENTRIQ tedavisi tamamen sonlandırılmalıdır. 1. veya 2. derece infüzyon ile ilişkili reaksiyon görülen hastalar yakından izlenerek TECENTRIQ almaya devam edebilir; bu hastalarda antipiretik ve antihistaminiklerle premedikasyon değerlendirilebilir.
Hastalık spesifik önlemler:
Metastatik ÜNMK'de TECENTRIQ'in nab-paklitaksel ile kombinasyon halinde kullanımı
TECENTRIQ ve nab-paklitaksel ile tedavi sırasında ortaya çıkan nötropeni ve periferik nöropatiler, nab-paklitakselin kesilmesiyle geri dönüşümlü olabilir. Doktorlar, bu ilacın özel önlemleri ve kontrendikasyonları için nab-paklitaksel Kısa Ürün Bilgisine (KÜB) başvurmalıdır.
Klinik çalışmalardan dışlanan hastalar:
Otoimmün hastalık geçmişi, pnömoni geçmişi, aktif beyin metastazı, HIV, hepatit B veya hepatit C enfeksiyonu, önemli kardiyovasküler hastalık ve yetersiz hematolojik ve uç organ işlevi olan hastalar TECENTRIQ ile yürütülen klinik çalışmalara alınmamıştır. Kayıttan önceki 28 gün içinde canlı, atenüe aşı uygulanan hastalar; 4 hafta içinde sistemik immün sistemi uyarıcı ajan alan hastalar veya çalışmaya girişten 2 hafta önce sistemik immunosupresif tıbbi ürün kullanan hastalar; çalışma tedavisinin başlamasından önceki 2 hafta içinde terapötik oral veya intravenöz antibiyotik kullanan hastalar klinik çalışmalara alınmamıştır.
Hasta Uyarı Kartı:
TECENTRIQ reçete eden hekimin hasta ile TECENTRIQ tedavisinin risklerini konuşması gerekmektedir. TECENTRIQ ile tedavi edilen hastalara ilacın riskleri hakkında bilgi veren Hasta Uyarı Kartları verilmeli ve kartı her zaman yanlarında taşımaları söylenmelidir.
Biyoteknolojik ürünlerin takip edilebilirliğinin sağlanması için uygulanan ürünün ticari ismi ve seri numarası mutlaka hasta dosyasına kaydedilmelidir.
4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri
TECENTRIQ ile herhangi bir resmi farmakokinetik ilaç etkileşimi çalışması yapılmamıştır. TECENTRIQ dolaşımdan katabolizma ile temizlendiği için metabolik ilaç-ilaç etkileşimleri beklenmemektedir.
TECENTRIQ ile tedaviye başlamadan önce, TECENTRIQ'in farmakodinamik aktivitesine ve etkililiğine yapabilecekleri potansiyel etkiler nedeniyle sistemik kortikosteroidlerin veya immunosupresanların kullanılmasından kaçınılmalıdır. Bununla birlikte, sistemik kortikosteroidler veya diğer immunosupresif maddeler, TECENTRIQ tedavisine başladıktan sonra immünite ile ilişkili advers reaksiyonların tedavisinde kullanılabilir (bkz. Bölüm 4.4).
Özel popülasyonlara ilişkin ek bilgiler:
TECENTRIQ ile herhangi bir farmakokinetik ilaç etkileşimi çalışması yapılmamıştır.
Pediyatrik popülasyon:
TECENTRIQ ile pediyatrik popülasyonda herhangi bir farmakokinetik ilaç etkileşimi çalışması yapılmamıştır.
4.6. Gebelik ve laktasyon
:Gebelik kategorisi: D
Çocuk doğurma potansiyeli bulunan kadınlar/doğum kontrolü (kontrasepsiyon):
Çocuk doğurma potansiyeline sahip kadınlar TECENTRIQ ile tedavi sırasında ve tedaviden 5 ay sonraya kadar etkili bir doğum kontrol yöntemi kullanmalıdır.
Gebelik dönemi:
Atezolizumabın fetüs üzerinde zararlı farmakolojik etkileri bulunmaktadır. Atezolizumab ile gelişimsel çalışmalar ve üreme çalışmaları yapılmamıştır. Hayvan çalışmalarıyla, PD-L1/PD-1 yolak inhibisyonunun fare veya sıçan gebelik modellerinde immünite ile ilişkili, fetüs ölümüyle sonuçlanan fetüs gelişiminin reddine sebep olduğu gösterilmiştir (bkz. Bölüm 5.3). İnsanlara
yönelik potansiyel risk bilinmemektedir ancak hayvan çalışmalarından alınan sonuçlar, etki mekanizmasına bağlı olarak, gebelik döneminde atezolizumab uygulamasının artmış düşük ve ölü doğum oranları dahil olmak üzere fötal zarara sebep olabileceğini göstermektedir.
Atezolizumab bir insan G1 immünoglobülinidir (IgG1) ve IgG1'in plasenta bariyerini aştığı bilinmektedir. Bu nedenle, atezolizumabın anneden gelişmekte olan fetüse geçme potansiyeli bulunmaktadır.
Gebe kadınların klinik durumu atezolizumab ile tedavi gerektirmedikçe gebelik sırasında TECENTRIQ kullanılmamalıdır.
Laktasyon dönemi:
Atezolizumabın anne sütüne geçip geçmediği bilinmemektedir. Atezolizumab bir monoklonal antikordur ve ilk gelen sütte bulunması ve daha sonra da az miktarda sütte bulunması beklenmektedir. Yeni doğanlar ve infantlar üzerindeki risk dışlanamaz. Emzirmenin çocuk için faydaları ve tedavinin anne için faydaları dikkate alınarak emzirmenin kesilmesi veya TECENTRIQ tedavisinin kesilmesi kararlaştırılmalıdır.
Üreme yeteneği/fertilite:
Atezolizumabın fertilite üzerindeki olası etkilerine ilişkin veri bulunmamaktadır. Atezolizumabın doğurganlık üzerindeki etkisini değerlendirme amaçlı reprodüktif ve gelişimsel toksisite çalışmaları yapılmamıştır. Bununla birlikte, 26 haftalık tekrarlanan doz toksisitesi çalışmasına dayalı olarak, atezolizumabın, önerilen dozu alan hastalarda eğri altı alanı (EAA)'nın yaklaşık 6 katı tahmini bir EAA'da adet döngüleri üzerinde bir etkiye sahip olduğu ve bu etkinin geri dönüşümlü olduğu görülmüştür (bkz. Bölüm 5.3). Erkek üreme organları üzerinde etki görülmemiştir.
4.7. Araç ve makine kullanımı üzerindeki etkiler
TECENTRIQ'in araç ve makine kullanma yeteneği üzerinde düşük düzeyde etkisi vardır. Yorgunluk hisseden hastalara semptomlar hafifleyene kadar araç ve makine kullanmamaları tavsiyesinde bulunulmalıdır (bkz. Bölüm 4.8).
4.8. İstenmeyen etkiler
Güvenlilik profilinin özeti:
TECENTRIQ monoterapisinin güvenliliği, birden fazla tümör tipinde 4349 hastadan toplanan verilere dayanmaktadır. En yaygın advers reaksiyonlar, (>%10) yorgunluk (%30,1), iştah azalması (%21,3), bulantı (%20), döküntü (%19,3), ateş (%19,0), öksürük (%18,6), ishal (%18),
dispne (%17,2), artralji (%16,7), asteni (%13,2), kaşıntı (%13,2), sırt ağrısı (%12,8), kusma (%12,5), idrar yolu enfeksiyonu (%11,5) ve baş ağrısı (%10,3) olmuştur. TECENTRIQ monoterapi çalışmalarının tanımı için, TECENTRIQ 1200 mg/20 mL infüzyonluk çözelti hazırlamak için konsantre'nin Kısa Ürün Bilgisi'ne bakınız.
Diğer tıbbi ürünlerle kombinasyon halinde verilen TECENTRIQ'in güvenliliği, birden fazla tümör tipinde 4535 hastada değerlendirilmiştir. En yaygın advers reaksiyonlar (≥ %20) anemi (%36,8), nötropeni (%36,6), bulantı (%35,5), yorgunluk (%33,1), alopesi (%28,1), döküntü
(%27,8), ishal (27,6), trombositopeni (%27,1), kabızlık (%25,8), iştah azalması (%24,7) ve periferik nöropati (%24,4)dir.
Ciddi advers reaksiyonlarla ilgili daha fazla bilgi için bkz. Bölüm 4.4.
Advers reaksiyonların tablo halinde listesi
Advers İlaç Reaksiyonları (ADR), TECENTRIQ monoterapisi ve kombinasyon terapisi için Tablo 2'de MedDRA sistem organ sınıfına (SOC) ve sıklık kategorilerine göre aşağıda listelenmiştir.
Aşağıdaki sıklık kategorileri kullanılmıştır:
Çok yaygın (≥1/10), yaygın (≥1/100 ila <1/10), yaygın olmayan (≥1/1000 ila <1/100), seyrek (≥1/10.000 ila <1/1000), çok seyrek (<1/10.000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). Her bir sıklık grubunda advers reaksiyonlar azalan ciddiyet sırasına göre verilmektedir.
Tablo 2: TECENTRIQ ile tedavi edilen hastalarda meydana gelen advers reaksiyonların özeti
| TECENTRIQ monoterapisi | TECENTRIQ kombinasyon tedavisi |
Enfeksiyonlar ve enfestasyonlar | ||
Çok yaygın | İdrar yolları enfeksiyonu | Akciğer enfeksiyonu |
Yaygın |
| Sepsis |
Kan ve lenf listemi hastalıkları | ||
Çok yaygın |
| Anemi, trombositopeni, nötropeni, lökopeni |
Yaygın | Trombositopeni | Lenfopeni |
Bağışıklık sistemi hastalıkları | ||
Yaygın | İnfüzyonla ilişkili reaksiyon | İnfüzyonla ilişkili reaksiyon |
Endokrin hastalıklar | ||
Çok yaygın |
| Hipotiroidizm |
Yaygın | Hipotiroidizm, hipertiroidizm | Hipertiroidizm |
Yaygın olmayan | Diabetes mellitus, adrenal yetmezlik |
|
Seyrek | Hipofizit |
|
Metabolizma ve beslenme hastalıkları | ||
Çok yaygın | İştah kaybı | İştah kaybı |
Yaygın | Hipokalemi, hiponatremi, hiperglisemi | Hipokalemi, hiponatremi, hipomagnezemi |
Sinir sistemi hastalıkları | ||
Çok yaygın | Baş ağrısı | Periferal nöropati, başağrısı |
Yaygın |
| Bayılma, baş dönmesi |
Yaygın olmayan | Guillain-Barré sendromu, meningoensefalit |
|
Seyrek | Miyastenik sendrom |
|
Göz hastalıkları | ||
Seyrek | Üveit |
|
Kardiyak hastalıklar | ||
Seyrek | Miyokardit |
|
Vasküler hastalıklar | ||
Çok yaygın |
| Hipertansiyon |
Yaygın | Hipotansiyon |
|
Solunum, göğüs bozuklukları ve mediastinal hastalıklar | ||
Çok yaygın | Dispne, öksürük | Dispne, öksürük, nazofarenjit |
Yaygın | Pnömoni, hipoksi, nazofarenjit | Disfoni |
Gastrointestinal hastalıklar | ||
Çok yaygın | Mide bulantısı, kusma, ishal | Mide bulantısı, kusma, ishal, kabızlık |
Yaygın | Kolit, karın ağrısı, disfaji, ağız ve yutak ağrısı | Stomatit, disguzi |
Yaygın olmayan | Pankreatit |
|
Hepatobiliyer hastalıkları | ||
Yaygın | AST artışı, ALT artışı, hepatit | AST artışı, ALT artışı |
Deri ve deri altı doku hastalıkları | ||
Çok yaygın | Döküntü, prürit | Döküntü, prürit, alopesi |
Yaygın | Kuru cilt |
|
Yaygın olmayan | Ciddi cilt advers reaksiyonları, sedef hastalığı | Ciddi cilt advers reaksiyonları, sedef hastalığı |
Seyrek | Pemfigoid | Pemfigoid |
Kas-iskelet bozuklukları, bağ doku ve kemik hastalıkları | ||
Çok yaygın | Artralji, sırt ağrısı | Artralji, kas-iskelet ağrısı, sırt ağrısı |
Yaygın | Kas-iskelet ağrısı |
|
Yaygın olmayan | Miyozit |
|
Böbrek ve idrar yolu hastalıkları | ||
Yaygın | Kan kreatininin artması | Proteinüri, kan kreatininin artması |
Yaygın olmayan | Nefrit |
|
Bilinmiyor | Bulaşıcı olmayan sistit |
|
Genel bozukluklar ve uygulama bölgesine ilişkin hastalıklar | ||
Çok yaygın | Ateş, yorgunluk, asteni | Ateş, yorgunluk, asteni, periferik ödem |
Yaygın | Grip benzeri hastalık, titreme |
|
Araştırmalar | ||
Yaygın |
| Kan alkalin fosfataz artışı |
anormal, tri-iyodotironin serbest azalmış, tri-iyodotironin serbest artmış, sessiz tiroidit, kronik tiroidit raporlarını içerir.
Hipoksi, oksijen satürasyonunda azalma, pOazalması raporlarını içerir.
Seçilen advers reaksiyonların açıklaması:
Aşağıdaki veriler, klinik açıdan anlamlı advers reaksiyonlarla ilgili olarak TECENTRIQ monoterapisine maruziyeti yansıtır (bkz. Bölüm 5.1). Kombinasyon tedavisi olarak verildiğinde TECENTRIQ için seçilen advers reaksiyonlarla ilgili ayrıntılar, yalnızca
TECENTRIQ monoterapisine kıyasla klinik açıdan anlamlı farklılıkların bildirilmesi durumunda sunulmaktadır. Bu advers reaksiyonlar için yönetim kılavuzları Bölüm 4.2 ve 4.4'te tanımlanmıştır.
İmmünite ile ilişkili pnömoni:
Pnömoni, TECENTRIQ monoterapisi gören hastaların %3'ünde (130/4.349) meydana gelmiştir. 130 hasta içinde iki ölümcül olay olmuştur. Başlangıca kadar geçen medyan süre 4 ay (aralık: 3 gün – 29,8 ay) olmuştur. Medyan süre 1,6 ay (aralık: 1 gün – 27,8+ ay; "+" sansürlenmiş bir değeri gösterir) olmuştur. Pnömoni, 29 hastada (%0,7) TECENTRİQ'in bırakılmasına yol açmıştır. Kortikosteroid kullanımı gerektiren pnömoni, TECENTRIQ monoterapisi gören hastaların %1,7'sinde (76/4.349) meydana gelmiştir.
İmmünite ile ilişkili hepatit:
Hepatit, TECENTRIQ monoterapisi gören hastaların %1,7'sinde (75/4.349) meydana gelmiştir. 75 hasta içinde iki ölümcül olay olmuştur. Başlangıca kadar geçen medyan süre 1,6 ay (aralık: 7 gün – 18,8 ay) olmuştur. Medyan süre 2,1 ay (aralık: 1 gün – 22+ ay; "+" sansürlenmiş bir değeri gösterir) olmuştur. Hepatit, 13 hastada (%0,3) atezolizumabın bırakılmasına yol açmıştır. Kortikosteroid kullanımı gerektiren hepatit, TECENTRIQ monoterapisi gören hastaların %0,5'inde (22/4.349) meydana gelmiştir.
İmmünite ile ilişkili kolit:
Kolit, TECENTRIQ monoterapisi gören hastaların %1,1'inde (50/4.349) meydana gelmiştir. Başlangıca kadar geçen medyan süre 5,1 ay (aralık: 15 gün – 17,2 ay) olmuştur. Medyan süre 1,2 ay (aralık: 1 gün – 35,9+ ay; "+" sansürlenmiş bir değeri gösterir) olmuştur. Kolit, 17 hastada (%0,4) atezolizumabın bırakılmasına yol açmıştır. Kortikosteroid kullanımı gerektiren kolit TECENTRIQ monoterapisi gören hastaların %0,6'sında (24/4.349) meydana gelmiştir.
İmmünite ile ilişkili endokrinopatiler:
Tiroid bozuklukları
Hipotiroidizm, TECENTRIQ monoterapisi gören hastaların % 7,6'sında (331/4.349) meydana gelmiştir. Başlangıca kadar geçen medyan süre 4,3 ay (aralık: 1 gün – 34,5 ay) olmuştur.
Hipertiroidizm, TECENTRIQ monoterapisi gören hastaların %2,1'inde (93/4.349) meydana gelmiştir. Başlangıca kadar geçen medyan süre 2,6 ay (aralık: 1 gün – 24,3 ay) olmuştur.
Adrenal yetmezlik
Adrenal yetmezlik, TECENTRIQ monoterapisi gören hastaların %0,5'inde (21/4.349) meydana gelmiştir. Başlangıca kadar geçen medyan süre 6,1 ay (aralık: 2 gün – 21,4 ay) olmuştur. Adrenal yetmezlik nedeniyle 5 (%0,1) hastada TECENTRİQ kullanımı bırakılmıştır. Kortikosteroid kullanımı gerektiren adrenal yetmezlik TECENTRIQ monoterapisi gören hastaların %0,4'ünde (17/4.349) meydana gelmiştir.
Hipofizit
Hipofizit, TECENTRIQ monoterapisi gören hastaların %0,1'inden azında (4/4.349) meydana gelmiştir. Başlangıca kadar geçen medyan süre 6,1 ay (aralık: 23 gün - 13,7 ay'dır) olmuştur. Üç hastada (<%0,1) kortikosteroid kullanımı gerekmiştir ve 1 (<%0,1) hastada TECENTRIQ tedavisi durdurulmuştur.
Hipofizit, TECENTRIQ ile kombinasyon halinde nab-paklitaksel ve karboplatin tedavisi gören hastaların %0,4'ünde (2/473) meydana gelmiştir. Başlangıca kadar geçen medyan süre 5,2 ay (aralık: 5,1 - 5,3 ay) olmuştur. Her iki hastada da kortikosteroid kullanımı gerekmiştir.
Diabetes mellitus
Diabetes mellitus, TECENTRIQ monoterapisi gören hastaların % 0,5'inde (20/4.349) meydana gelmiştir. Medyan süre 5,5 ay (aralık: 4 gün – 29 ay) olmuştur. Diabetes mellitus, <%0,1 (3/4.349) hastada TECENTRIQ'in bırakılmasına yol açmıştır.
İmmünite ile ilişkili meningoensefalit:
Menenjit, TECENTRIQ monoterapisi gören hastaların %0,4'ünde (18/4.349) meydana gelmiştir. Başlangıca kadar geçen süre 16 gün (aralık: 1 gün – 12,5 ay) olmuştur. Medyan süre 22 gün (aralık: 6 gün ila 14,5+ ay; +, sansürlenmiş bir değeri gösterir) olmuştur.
Kortikosteroid kullanımını gerektiren menenjit TECENTRIQ tedavisi gören hastaların %0,2'sinde (10/4.349) meydana gelmiştir ve 8 (%0,2) hastada TECENTRIQ'in bırakılmasına yol açmıştır.
İmmünite ile ilişkili nöropatiler:
Guillain-Barré sendromu ve demiyalizan polinöropati, TECENTRIQ monoterapisi gören hastaların %0,1'inde (6/4.349) meydana gelmiştir. Başlangıca kadar geçen medyan süre 4,1 ay (aralık: 17 gün – 8,1 ay) olmuştur. Medyan süre 8 ay (aralık: 19 gün – 24,5 ay+, "+" sansürlenmiş bir değeri gösterir). Bir (<%0,1) hasta, Guillain-Barré sendromu nedeniyle TECENTRIQ kullanımını bırakmıştır. Kortikosteroid kullanımı gerektiren Guillain-Barré sendromu TECENTRIQ tedavisi gören hastaların %0,1'inden azında (3/4.349) meydana gelmiştir.
Miyastenik sendrom:
Miyestenia gravis, TECENTRIQ monoterapisi gören hastaların <%0,1'inde (1/4.349) meydana gelmiştir. Başlangıca kadar geçen süre 1,2 aydır.
İmmünite ile ilişkili pankreatit:
Yüksek amilaz ve yüksek lipaz dahil olmak üzere pankreatit, TECENTRIQ monoterapisi gören hastaların %0,7'sinde (32/4.349) meydana gelmiştir. Başlangıca kadar geçen medyan süre 5,5 ay (aralık: 1 gün – 24,8 ay) olmuştur. Medyan süre 24 gün (aralık: 3+ gün – 22,4+ ay; "+" sansürlenmiş bir değeri gösterir) olmuştur. Pankreatit, 3 hastada (<%0,1) TECENTRIQ'in bırakılmasına yol açmıştır. Kortikosteroid kullanımı gerektiren pankreatit olguları TECENTRIQ monoterapisi gören hastaların %0,1'inde (5/4.349) meydana gelmiştir.
İmmünite ile ilişkili miyokardit:
Miyokardit, TECENTRIQ monoterapisi gören hastaların <%0,1'inde (3/4.349) meydana gelmiştir. Başlangıca kadar geçen medyan süre 2,1 ay (aralık: 1,5 ay – 4,9 ay) olmuştur. Medyan süre 14 gün (aralık: 14 gün – 2,8 ay) olmuştur. İki hastada (<%0,1) kortikosteroid kullanımı gerekmiştir ve 2 (<%0,1) hastada TECENTRIQ tedavisi durdurulmuştur.
İmmünite ile ilişkili nefrit:
Nefrit, TECENTRIQ tedavisi gören hastaların %0,2'sinde(10/4.349) meydana gelmiştir. Başlangıca kadar geçen medyan süre 5 ay (aralık: 2 gün – 17,5 ay) olmuştur. Nefrit 5 hastada (%0,1) TECENTRIQ'in bırakılmasına neden olmuştur. Dört hastada (<%0,1) kortikosteroid kullanımına gerek duyulmuştur.
İmmünite ile ilişkili miyozit:
Miyozit, TECENTRIQ monoterapisi gören hastaların %0,5'inde (20/4.349) meydana gelmiştir. Başlangıca kadar geçen medyan süre 3,3 aydır (aralık: 12 gün - 11 ay). Medyan süre 5,7 ay (aralık: 2 gün - 36,9 +ay; + sansürlenmiş bir değeri gösterir). Miyozit 2 hastada (<%0,1) TECENTRIQ'in bırakılmasına neden olmuştur. 7 hastada (%0,2) kortikosteroid kullanımına gerek duyulmuştur.
İmmünite ile ilişkili şiddetli kutanöz advers reaksiyonlar:
TECENTRIQ monoterapisi gören hastaların %0,6'sında (28/4.349) şiddetli kutanöz advers reaksiyonlar meydana gelmiştir. 28 hastadan birinde ölümcül olay meydana gelmiştir. Başlangıca kadar geçen medyan süre 5,2 aydır (aralık: 4 gün - 15,5 ay). Medyan süre 2,4 ay (aralık: 1 gün - 37,5 +ay; + sansürlenmiş bir değeri gösterir). Şiddetli kutanöz advers reaksiyonlar 3 hastada (< %0,1) TECENTRIQ tedavisinin bırakılmasına neden olmuştur. %0,2 hastada (9/4.349) kortikosteroid kullanımına gerek duyulmuştur.
İmmünojenite:
Çoklu faz II ve III çalışmalarında, hastaların %13,1 ile %54,1'i tedaviyleortaya çıkananti-ilaç antikorları (ADA) geliştirmiştir. Tedavi sonucu oluşmuş anti-ilaç antikoru (ADA) gelişen hastaların başlangıçta genel olarak sağlık durumu ve hastalık karakteristikleri açısından daha zayıf olduğu görülmüştür. Başlangıçtaki bu sağlık ve hastalık karakteristiklerindeki dengesizlikler, farmakokinetik, etkililik ve güvenlilik analizlerinin yorumlanmasında karışıklık yaratabilmektedir. Anti-ilaç antikorlarının (ADA) etkililiğe etkisini araştırmak için başlangıçtaki sağlık ve hastalık karakteristiklerindeki dengesizlikleri ayarlayan keşif analizleri yapılmıştır. Bu analizlerde ADA geliştiren hastaların, ADA geliştirmeyen hastalara kıyasla etkililik faydasında azalma olasılığı gözardı edilmemiştir. Anti-ilaç antikorlarının başlangıca kadar geçen medyan süresi 3 ila 5 hafta olmuştur.
TECENTRIQ monoterapisi (N=3.460) ve kombinasyon tedavisi (N=2.285) gören hastalardan elde edilen hasta havuzu verilerinden, ADA-pozitif popülasyonuna karşı ADA-negatif popülasyonundan elde edilen advers olayların sıklığı sırasıyla: Monoterapi için; 3. ve 4. derece yan etkiler %42,6'ya karşı %39,4, ciddi yan etkiler %39,6'ya karşı %33,3, tedavinin kesilmesine neden olan yan etkiler %8,5'e karşı %7,8 iken; Kombinasyon tedavisi için 3. ve 4. derece yan etkiler, %63,9'a karşı %60,9, ciddi yan etkiler %43,9'a karşı 35,6, tedavinin kesilmesine neden olan yan etkiler % 22,8'e karşı %18,4 olmuştur. Ancak mevcut verilerden
yola çıkarak olası ilaç advers reaksiyonlarının yolağı hakkında kesin sonuçlara varılamamaktadır.
Pediyatrik popülasyon
TECENTRIQ'in çocuklar ve adölesanlardaki güvenliliği bilinmemektedir. 69 pediyatrik hastada (<18 yaş) yapılan bir klinik çalışmada yeni bir güvenlilik sinyali oluşmamıştır ve güvenlilik profili erişkinlerinki ile karşılaştırılabilirdir.
Geriyatrik popülasyon
TECENTRIQ monoterapisi gören 65 yaş ve üzerindeki hastalar ile daha genç hastalar arasında genel olarak bir güvenlilik farklılığı gözlemlenmemiştir.
Şüpheli advers reaksiyonların raporlanması
Ruhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar/risk dengesinin sürekli olarak izlenmesine olanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu Türkiye Farmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e-posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks:0 312 218 35 99).
4.9. Doz aşımı ve tedavisi
Atezolizumab doz aşımına ilişkin bilgi mevcut değildir.
Doz aşımı durumunda, hastalar advers reaksiyon belirtileri veya semptomları bakımından yakından izlenmeli ve uygun semptomatik tedavi başlatılmalıdır.
5. FARMAKOLOJİK ÖZELLİKLER
5.1. Farmakodinamik özellikler
Farmakoterapötik grup: Antineoplastik ve İmmünomodülatör ajanlar, Monoklonal Antikorlar ve Antikör İlaç Konjugatları, PD-1/PDL-1 (Programlanmış hücre ölüm proteini 1 / ölüm ligandı 1) İnhibitörleri
ATC kodu: L01FF05
Etki mekanizması:
Hümanize IgG1 anti-programlı ölüm-ligandı 1(PD-L1), tümör hücreleri ve/veya tümör infiltre eden immün hücrelerinde eksprese olabilir ve tümör mikroortamında anti-tümör immün yanıtının inhibisyonuna katkıda bulunabilir. PD-L1'in T-hücrelerinde ve antijen sunan hücrelerde bulunan PD-1 ve B7.1 reseptörlerine bağlanması, sitotoksik T-hücre aktivitesini, T-hücre çoğalmasını ve sitokin üretimini baskılar.
Atezolizumab Fc bölgesi değiştirilmiş bir hümanize immünoglobülin G1 (IgG1) monoklonal antikorudur; doğrudan PD-L1'e bağlanır ve PD-1 ve B7.1 reseptörlerinin ikili blokajını sağlayarak, antikor bağımlı hücresel sitotoksisiteyi indüklemeden antitümör immün yanıtın yeniden aktive edilmesi de dahil, immün yanıtın PD-L1/PD-1 aracılı inhibisyonunu serbest bırakır. Atezolizumab, PD-L2/PD-1 etkileşimini koruyarak PD-L2/PD-1 aracılı inhibitör
sinyallerin devam etmesine izin verir.
Klinik etkililik ve güvenlilik:
1200 mg dozda üç haftada bir uygulanan TECENTRIQ ile yapılan çalışmaların tanımı için, TECENTRIQ 1200 mg/20 mL infüzyonluk çözelti hazırlamak için konsantre'nin Kısa Ürün Bilgisi'ne bakınız.
Üçlü negatif meme kanseri
IMpassion130 (WO29522): Daha önce metastatik hastalık için tedavi edilmemiş, lokal ileri veya metastatik ÜNMK hastalarında randomize faz III çalışma
Metastatik hastalık için daha önce kemoterapi almamış, rezeke edilemeyen, lokal ileri veya metastatik ÜNMK olan hastalarda, atezolizumab ile nab-paklitakselin etkililik ve güvenliliğini değerlendirmek için Faz III, çift kör, iki kollu, çok merkezli, uluslararası, randomize, plasebo kontrollü bir çalışma olan IMpassion130 yürütülmüştür. Taksan monoterapisi için uygun olan hastalar seçilmiştir (yani hızlı klinik ilerlemenin olmaması, yaşamı tehdit eden viseral metastazların veya hızlı semptom ve/veya hastalık kontrolü ihtiyacının olmaması). Son 12 ay içerisinde neoadjuvan veya adjuvan ortamında kemoterapi alan hastalar, otoimmün hastalık öyküsü olan, randomizasyondan önceki 4 hafta içinde bir canlı atenüe aşı, randomizasyondan önceki 4 hafta içinde sistemik immünostimülatör ajan uygulanmış veya randomizasyondan önceki 2 hafta içinde sistemik immunosupresif tıbbi ürün kullanmış hastalar, tedavi edilmemiş semptomatik veya kortikosteroide bağlı beyin metastazı öyküsü olan hastalar dahil edilmemiştir. Tümör değerlendirmeleri, 1. döngünün (1. gün) ardından ilk 12 ay süreyle 8 haftada (± 1 hafta) bir ve sonrasında 12 haftada (± 1 hafta) bir olmak üzere gerçekleştirilmiştir.
Toplamda 902 hasta çalışmaya dahil edilmiş ve karaciğer metastazı varlığı, taksan tedavisi öyküsü ve tümör infiltre edici immün hücrelerde (IC) PD-L1 ekspresyonu durumuna göre tabakalandırılmıştır (PD-L1 ile boyanmış tümör infiltre edici immün hücreler [IC] < %1 VENTANA PD-L1 (SP142) testi ile değerlendirilen tümör alanına karşı tümör alanının ≥ %1'i).
Hastalar, her 28 günlük siklusta 1. ve 15. günlerde intravenöz infüzyon yoluyla atezolizumab 840 mg veya plasebo ve her 28 günlük siklusta 1., 8. ve 15. günlerde intravenöz infüzyon yoluyla nab-paklitaksel (100 mg/m) almak üzere randomize edilmiştir. Hastalar, RECIST v1.1 uyarınca radyografik hastalıkta ilerleme veya kabul edilemez bir toksisite meydana gelene kadar tedavi görmüştür. Kabul edilemez toksisite nedeniyle nab-paklitaksel durdurulduğunda atezolizumab ile tedaviye devam edilebilir. Her tedavi kolunda medyan tedavi döngüsü sayısı atezolizumab için 7 ve nab-paklitaksel için 6'dır.
Çalışma popülasyonunun demografik ve başlangıç özellikleri, tedavi kolları arasında iyi dengelenmiştir. Hastaların çoğu kadındır (%99,6), %67,5'i beyaz, %17,8'i Asyalıdır. Medyan yaş 55' dir (aralık: 20-86). Başlangıçta ECOG performans skoru 0 (%58,4) veya 1'dir (%41,3). Genel olarak, dahil edilen hastaların %41'inde başlangıçta PD-L1 ekspresyonu ≥ %1 iken, %27'sinde karaciğer metastazları ve %7'sinde beyin metastazları mevcuttur. Yaklaşık olarak hastaların yarısı, (neo)adjuvan koşullarda bir taksan (%51) veya antrasiklin (%54) almıştır. PD-L1 ekspresyonu ≥ %1 olan hastalarda demografik bilgiler ve başlangıç tümör hastalığı, genel itibariyle daha geniş çalışma popülasyonunu temsil ediyordu.
Eş birincil etkililik sonlanım noktaları, ITT popülasyonunda ve RECIST v1.1 uyarınca PD-L1 ekspresyonu ≥ %1 olan hastalarda araştırmacı tarafından değerlendirilen, progresyonsuz sağkalımın (PS) yanı sıra ITT popülasyonunda ve PD-L1 ekspresyonu ≥ %1 olan hastalarda
genel sağkalımı (GS) içermiştir. İkincil etkililik sonlanım noktaları, RECIST v1.1 uyarınca objektif yanıt oranı (OYO) ve yanıt süresini (YS) içermiştir.
Medyan sağkalım takibi 13 aylık olan progresyonsuz sağkalım (PS) için son analiz sırasında PD-L1 ekspresyonu ≥%1 olan hastalar için IMpassion130'un PS, OYO ve YS sonuçları Tablo 3'de özetlenmiştir ve PS için Kaplan-Meier eğrileri PD-L1 ekspresyonu <%1 olan hastalar Şekil 1'de gösterilmektedir. Nab-paklitaksele atezolizumab eklendiğinde PS'de iyileşme gözlenmemiştir (TO 0,94, %95 GA 0,78, 1,13).
Nihai GS analizi, PD-L1 ekspresyonu ≥%1 olan ve medyan takip süresi 19,12 olan hastalarda yapılmıştır. GS sonuçları Tablo 3'de ve Kaplan-Meier eğrileri Şekil 1'de sunulmaktadır. PD-L1 ekspresyonu <%1 olan hastalar, nab-paklitaksele atezolizumab eklendiğinde gelişmiş GS gözlenmemiştir (TO 1,02, %95 GA 0,84, 1,24).
PD-L1 ekspresyonu ≥%1 olan hastalarda, önceki (neo)adjuvan tedavi, BRCA1/2 mutasyonu ve başlangıçta asemptomatik beyin metastazları araştırılarak keşif amaçlı alt grup analizleri yapılmıştır. Daha önce (neo) adjuvan tedavi almış hastalarda (n=242) birincil (nihai) PS için tehlike oranı 0,79 ve final GS için 0,77 iken, daha önce (neo)adjuvan tedavi almamış hastalarda (n=127), birincil (nihai) PS için tehlike oranı, final GS için 0,44 ve 0,54 olarak saptanmıştır.
IMpassion130 çalışmasında, test edilen 614 hastanın 89'u (%15) patojenik BRCA1/2 mutasyonları taşımaktaydı. PD-L1+/BRCA1/2 mutant alt grubundan 19 hasta atezolizumab artı nab-paklitaksel ve 26 plasebo artı nab-paklitaksel almıştır. Araştırmacı analize dayanarak ve küçük örnek boyutunun kabul edilmesiyle, BRCA1/2 mutasyonunun varlığı, atezolizumab ve nab-paklitakselin PS klinik yararını etkilemiyor gibi görünmektedir.
Tedavi edilen hasta sayısı az olmasına rağmen, başlangıçta asemptomatik beyin metastazı olan hastalarda etkililik kanıtı yoktu; medyan PS, atezolizumab artı nab-paklitaksel kolunda (n=15) 2,2 ay iken, plasebo artı nab-paklitaksel kolunda (n=11) 5,6 aydır (TO 1,40; %95 GA 0,57,
3,44).
Tablo 3: PD-L1 ekspresyonu ≥ %1 olan hastalarda etkililik özeti (IMpassion130)
Anahtar etkililik sonlanım noktaları | Atezolizumab+ nab-paklitaksel | Plasebo+ nab-paklitaksel |
Birincil etkililik sonlanım noktaları | n=185 | n=184 |
Araştırmacı tarafından değerlendirilen PS (RECIST v1.1)-Primer analiz | ||
Olay sayısı (%) | 138 (%74,6) | 157 (%85,3) |
Medyan PS süresi (ay) | 7,5 | 5 |
%95 GA | (6,7; 9,2) | (3,8; 5,6) |
Tabakalandırılmış tehlike oranı‡ (%95 GA) | 0,62 (0,49; 0,78) | |
p değeri | <0,0001 | |
12-aylık PS (%) | 29,1 | 16,4 |
Araştırmacı tarafından değerlendirilen PS (RECIST v1.1)- Güncellenmiş keşif analizi | ||
Olay sayısı (%) | 149 (%80,5) | 163 (%88,6) |
Medyan PS süresi (ay) | 7,5 | 5,3 |
%95 GA | (6,7; 9,2) | (3,8; 5,6) |
Tabakalandırılmış tehlike oranı‡ (%95 GA) | 0,63 (0,5; 0,8) | |
p değeri | <0,0001 | |
12-aylık PS (%) | 30,3 17,3 | |
GS | ||
Ölüm sayısı(%) | 120 (%64,9) | 139 (%75,5) |
Medyan olay süresi (ay) | 25,4 | 17,9 |
%95 GA | (19,6, 30,7) | (13,6, 20,3) |
Tabakalandırılmış tehlike oranı‡ (%95 GA) |
|
0,67 (0,53, 0,86) |
İkincil ve araştırmaya yönelik sonlanım noktaları | ||
Araştırmacı tarafından değerlendirilen OYO (RECIST 1.1) | n=185 | n=183 |
Yanıt veren hasta sayısı (%) | 109 (%58,9) | 78 (%42,6) |
%95 GA | (51,5; 66,1) | (35,4; 50,1) |
Tam yanıt sayısı (%) | 19 (%10,3) | 2 (%1,1) |
Kısmi yanıt sayısı (%) | 90 (%48,6) | 76 (%41,5) |
Stabil hastalık sayısı | 38 (%20,5) | 49 (%26,8) |
Araştırmacı tarafından değerlendirilen YS | n=109 | n=78 |
Ay olarak medyan | 8,5 | 5,5 |
%95 GA | (7,3; 9,7) | (3,7; 7,1) |
‡ Karaciğer metastazı varlığı ve taksan tedavisi öyküsüne göre tabakalandırılmıştır.
PS= Progresyonsuz sağkalım; RECIST= Solid Tümörlerde Yanıt Değerlendirme Kriterleri v1.1.; GA= Güven aralığı; OYO= Objektif yanıt oranı; YS= Yanıt süresi; GS= Genel sağkalım
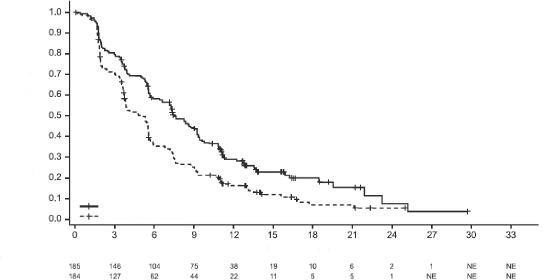
Şekil 1: PD-L1 ekspresyonu ≥ %1 olan hastalarda Progresyonsuz Sağkalım için Kaplan-Meier Eğrisi (IMpassion130)
Şekil 2: PD-L1 ekspresyonu ≥ %1 olan hastalarda Genel Sağkalım için Kaplan-Meier Eğrisi (IMpassion130)
EORTC QLQ-C30 ile ölçülen hasta tarafından bildirilen genel sağlık durumu/sağlıkla ilişkili yaşam kalitesinin (HRQoL) bozulmasına kadar geçen süre (başlangıç puanından sürekli ≥10 puanlık bir düşüş) her tedavi grubunda benzerdir ve bu tüm hastaların karşılaştırılabilir bir süre boyunca temel HRQoL'lerini koruduğunu göstermektedir.
5.2. Farmakokinetik özellikler
Genel özelliklerAtezolizumaba maruziyet 1 mg/kg - 20 mg/kg doz aralığında 3 haftada bir uygulanan sabit doz
1200 mg doz ile orantılı olarak artmıştır. 472 hastayı içeren bir popülasyon analizi, aşağıdaki doz aralığı için atezolizumab farmakokinetiğini birinci derece eliminasyonla bir doğrusal iki bölmeli düzenleme modeli ile 1 - 20 mg/kg olarak açıklamıştır. Üç haftada bir uygulanan 1200 mg atezolizumab dozu ve 2 haftada bir uygulanan 840 mg atezolizumab dozu ve 4 haftada bir uygulanan 1680 mg atezolizumab dozunun farmakokinetik özellikleri aynıdır. Bu üç doz rejimiyle karşılaştırılabilir toplam maruziyetlere ulaşılması beklenmektedir. Bir popülasyon farmakokinetik analizi, 6-9 hafta tekrarlı dozlamadan sonra kararlı durumun elde edildiğini öne sürmektedir. Eğri altındaki alanda, maksimum konsantrasyon ve en düşük konsantrasyonda sistemik birikim sırasıyla 1,91; 1,46 ve 2,75 kat olmuştur.
Emilim:
Atezolizumab intravenöz infüzyon şeklinde uygulanır. Diğer uygulama yollarıyla yapılan çalışmalar olmamıştır.
Dağılım:
Popülasyon farmakokinetik analizi, bir hastada merkezi kompartman dağılım hacminin 3,28 L ve kararlı durumunda hacmin 6,91 L olduğunu göstermektedir.
Biyotransformasyon:
Atezolizumabın metabolizması doğrudan araştırılmamıştır. Antikor klerensi esas olarak katabolizmayla gerçekleşir.
Eliminasyon:
Popülasyon farmakokinetik analizi, atezolizumabın klerensinin 0,2 L/gün ve tipik terminal eliminasyon yarı ömrünün 27 gün olduğunu göstermektedir.
Özel popülasyonlara ilişkin ek bilgiler:
Popülasyon farmakokinetiği ve maruziyet-yanıt analizlerine göre aşağıdaki faktörlerin atezolizumabın farmakokinetiği üzerinde bir etkisi yoktur: Yaş, (21-89 yaş), bölge, etnik köken, böbrek bozukluğu, hafif karaciğer bozukluğu, PD-L1 ekspresyonu düzeyi veya ECOG performans durumu. Vücut ağırlığı, cinsiyet, pozitif ADA durumu, albümin seviyeleri ve tümör yükünün atezolizumab farmakokinetiği üzerindeki etkisi istatistiki olarak anlamlı ancak klinik olarak anlamlı değildir. Doz ayarlaması önerilmemektedir.
Pediyatrik popülasyon:
Pediyatrik (<18 yaş, n=69) ve genç erişkin (18-30 yaş, n=18) hastalarda yürütülen erken faz, çok merkezli, açık etiketli bir çalışmadan elde edilen farmakokinetik sonuçlar, atezolizumab klerensi ve dağılım hacminin, normal vücut ağırlığına göre normalize edildiğinde 15 mg/kg alan pediyatrik hastalar ve her 3 haftada bir 1200 mg atezolizumab alan genç erişkin hastalar arasında karşılaştırılabilir olduğunu göstermiştir. Maruziyetin ise, vücut ağırlığı düştükçe pediyatrik hastalarda arttığı gözlenmiştir. Bu değişiklikler, atezolizumab konsantrasyonunun terapötik hedef maruziyetinin altına düşmesi ile bağlantılı değildir. 2 yaş altındaki çocuklar için veriler sınırlıdır, bu nedenle kesin sonuçlara varılamamaktadır.
Geriyatrik popülasyon:
Yaşlı hastalarda TECENTRIQ için özel bir çalışma yapılmamıştır. Yaşın atezolizumabın farmakokinetiği üzerindeki etkisi bir popülasyon farmakokinetik analizinde değerlendirilmiştir. Yaş, 21-89 yaş aralığındaki (n=472) ve medyan 62 yaşındaki hastalar temel alındığında, atezolizumabın farmakokinetiğini etkileyen önemli bir kovaryant olarak tanımlanmamıştır.
<65 yaşındaki (n=274), 65-75 yaşındaki (n=152) ve >75 yaşındaki (n=46) hastalarda atezolizumabın farmakokinetiğinde klinik açıdan anlamlı bir fark gözlenmemiştir (bkz. Bölüm 4.2).
Böbrek yetmezliği:
Böbrek yetmezliği olan hastalarda TECENTRIQ için özel bir çalışma yapılmamıştır. Popülasyon farmakokinetik analizinde, böbrek fonksiyonu normal (90 mL/dk/1,73 m veya üzeri tahmini glomerüler filtrasyon hızı [eGFR]; n=140) olan hastalarla karşılaştırıldığında, hafif (60 - 89 mL/dk/1,73 m eGFR; n=208) veya orta şiddetli (30 - 59 mL/dk/1,73 m eGFR; n=116) böbrek yetmezliği olan hastalarda atezolizumabın klerensinde klinik açıdan önemli farklar bulunmamıştır. Yalnızca birkaç hastada şiddetli böbrek yetmezliği vardır (eGFR 15 - 29 mL/dk/1,73 m; n=8) (bkz. Bölüm 4.2). Şiddetli böbrek yetmezliğinin atezolizumabın farmakokinetiği üzerindeki etkisi bilinmemektedir.
Karaciğer yetmezliği:
Karaciğer bozukluğu olan hastalarda TECENTRIQ için özel bir çalışma yapılmamıştır. Popülasyon farmakokinetik analizinde, hafif (bilirubin ≤NÜS ve AST>NÜS veya bilirubin >1 – 1,5 x NÜS ve herhangi bir AST) veya orta (bilirubin > 1,5 – 3 x NÜS ve herhangi bir AST) karaciğer yetmezliği olan ve normal karaciğer fonksiyonu (bilirubin ≤ NÜS ve AST ≤NÜS) olan hastalar arasında atezolizumabın klerensi bakımından klinik olarak önemli farklar bulunmamıştır. Şiddetli (bilirubin > 3 x NÜS ve herhangi bir AST) karaciğer yetmezliği olan hastalara ilişkin veri mevcut değildir. Karaciğer yetmezliği, Ulusal Kanser Enstitüsü (NCI) karaciğer fonksiyon bozukluğu kriterlerine göre tanımlanmıştır (bkz. Bölüm 4.2). Şiddetli karaciğer yetmezliğinin (bilirubin > 3 x NÜS ve herhangi bir AST) atezolizumabın farmakokinetiği üzerindeki etkisi bilinmemektedir.
5.3. Klinik öncesi güvenlilik verileri
Karsinojenite:
TECENTRIQ'in karsinojenik potansiyelini belirlemek için karsinojenite çalışması yapılmamıştır.
Mutajenite:
TECENTRIQ'in mutajenik potansiyelini belirlemek için mutajenite çalışması yapılmamıştır. Bununla birlikte, monoklonal antikorların DNA veya kromozomları değiştirmesi beklenmemektedir.
Fertilite:
TECENTRIQ ile herhangi bir doğurganlık çalışması yapılmamıştır. Bununla birlikte, kronik toksisite çalışmasına sinomolgus maymunlarında erkek ve dişi üreme organlarının değerlendirilmesi dahil edilmiştir. Atezolizumabın dişi maymunlara tahmini EAA'da uygulanması (önerilen dozu alan hastalardaki EAA'nın yaklaşık 6 katı), geri dönüşümlü olarak düzensiz adet döngüsüne ve yumurtalıklarda yeni oluşturulmuş korpus lutea eksikliğine neden olmuştur. Erkek üreme organları üzerinde herhangi bir etkisi olmamıştır.
Teratojenite:
TECENTRIQ ile hayvanlarda üreme ve teratojenite çalışmaları yapılmamıştır. Hayvan çalışmaları, PD-L1/PD-1 yolağının inhibisyonunun, gelişen fetüsün bağışıklıkla ilişkili reddine yol açarak fetal ölümle sonuçlanabileceğini göstermiştir. TECENTRIQ uygulaması, embriyo-fetal ölüm dahil olmak üzere fetal zarara neden olabilir.
6. FARMASÖTİK ÖZELLİKLER
6.1. Yardımcı maddelerin listesi
L-histidin
Glasiyal asetik asit Sükroz
Polisorbat 20 Enjeksiyonluk su
6.2. Geçimsizlikler
Bu tıbbi ürün, Bölüm 6.6'da belirtilenlerin dışında diğer tıbbi ürünlerle karıştırılmamalıdır.
6.3. Raf ömrü
Açılmamış flakon: 36 ay
Seyreltilmiş çözelti: Hazırlama zamanından sonra 2-8°C'de 24 saat ve ortam sıcaklığında (≤30°C) 24 saat içinde kullanımdaki kimyasal ve fiziksel stabilite gösterilmiştir.
Mikrobiyolojik açıdan, hazırlanan infüzyonluk çözelti hemen kullanılmalıdır. Hemen kullanılmazsa, kullanım sırasındaki saklama süreleri ve kullanım öncesi koşullar kullanıcının sorumluluğundadır ve normalde 2-8°C'de 24 saatten fazla veya ortam sıcaklığında (≤25°C) 8 saatten fazla olmamalıdır.
6.4. Saklamaya yönelik özel tedbirler
Buzdolabında saklayınız (2°C-8°C).
Işıktan korumak için flakonu karton kutusunda saklayınız. Dondurmayınız. Çalkalamayınız.
Tıbbi ürünün seyreltme sonrasında saklama koşulları için bkz. Bölüm 6.3.
6.5. Ambalajın niteliği ve içeriği
14 mL çözelti içeren tapalı (butil kauçuk) flakon (Tip I cam). Bir flakonluk paket.
6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler
TECENTRIQ herhangi bir antimikrobiyal koruyucu içermez ve bir sağlık meslek mensubu tarafından aseptik teknik kullanılarak hazırlanmalıdır.
Seyreltme talimatları:
14 mL TECENTRIQ konsantresi flakondan çekilmeli ve içinde sodyum klorür 9 mg/mL (% 0,9) enjeksiyonluk çözelti bulunan 250 mL'lik bir PVC, polietilen (PE) veya poliolefin infüzyon torbası içine seyreltilmelidir. Seyreltmeden sonra çözeltinin bir mL'si yaklaşık 3,2 mg TECENTRIQ (840 mg/264 mL) içermelidir. Torba, köpük oluşumuna izin vermeden çözeltiyi karıştırmak için yavaşça alt üst edilmelidir. İnfüzyon hazırlandığında hemen uygulanmalıdır (bkz. Bölüm 6.3).
Parenteral tıbbi ürünler uygulanmadan önce partiküller ve renk değişimi açısından çıplak gözle incelenmelidir. Partiküller veya renk değişimi gözlenirse çözelti kullanılmamalıdır.
TECENTRIQ ile ürüne temas eden polivinil klorür (PVC), polietilen (PE) veya poliolefin (PO) yüzeyleri olan intravenöz torbalar arasında geçimsizlik gözlenmemiştir. İlave olarak,
polietersülfon veya polisülfon içeren düz eksenli filtre membranları ve infüzyon setleri ile PVC, PE, polibutadien veya polieterüretan içeren diğer infüzyon yardımcıları ile de geçimsizlik gözlenmemiştir. Düz eksenli filtre membranlarının kullanılması seçime bağlıdır.
Kullanılmamış/son kullanma tarihi geçmiş ilaçların imhası:
Tıbbi ürünlerinin çevreye salınması en aza indirilmelidir. İlaçlar, atık suyla birlikte atılmamalıdır ve evsel atıklarla imhasından kaçınılmalıdır.
Kullanılmamış olan ürünler ya da atık materyaller, “Tıbbi Atıkların Kontrolü Yönetmeliği'' ve “Ambalaj Atıklarının Kontrolü Yönetmeliğiâ€ne uygun olarak imha edilmelidir.
 Grip, Soğuk Algınlığı ve Öksürük
Grip ve soğuk algınlığı (nezle) semptomları arasındaki farkı bilmek önemlidir. Soğuk algınlığı gripten daha hafif belirtiler gösteren bir solunum yolu hastalığıdır.
Grip, Soğuk Algınlığı ve Öksürük
Grip ve soğuk algınlığı (nezle) semptomları arasındaki farkı bilmek önemlidir. Soğuk algınlığı gripten daha hafif belirtiler gösteren bir solunum yolu hastalığıdır. |
 Doğum Sonrası Depresyonu
Doğum sonrası depresyonu, doğumdan sonra her on kadından biri tarafından
tecrübe edilen stresli bir durumdur.
Doğum Sonrası Depresyonu
Doğum sonrası depresyonu, doğumdan sonra her on kadından biri tarafından
tecrübe edilen stresli bir durumdur. |
 |
Astım Astımlı kişilerin akciğerlerindeki hava boruları (bronşlar) hassastır. Bu kişiler belirli tetikleyici faktörlere maruz kaldıklarında, hava boruları nefes almalarını güçleştirecek şekilde daralır. |
 |
Pankreas Kanseri Pankreas karnın alt kısmında yatay şekilde bulunan bir organdır. Sindirime yardımcı olan enzimleri ve kan şekerini yönetmeye yardımcı olan hormonları vücuda dağıtmakla görevlidir. |
 |
Deri Kanseri Deri kanseri çok rastlanan bir hastalıktır. Üç ana türü bulunur ;genelde kemirici ülser olarak bilinen bazal hücreli karsinom, yassı hücreli karsinom ve kötü huylu tümör. |
İLAÇ GENEL BİLGİLERİ
Roche Müstahzarları Sanayi A.Ş.
| Satış Fiyatı | 73857.88 TL [ 8 Aug 2025 ] |
| Önceki Satış Fiyatı | 73857.88 TL [ 1 Aug 2025 ] |
| Original / Jenerik | Original İlaç |
| Reçete Durumu | Kırmızı Reçeteli bir ilaçdır. |
| Barkodu | 8699505763323 |
| Etkin Madde | Atezolizumab |
| İthal ( ref. ülke : Litvanya ) ve Beşeri bir ilaçdır. |