TRUXIMA 100 mg/10 ml IV infüzyonluk çözelti hazırlamak için konsantre (2 flakon) Kısa Ürün Bilgisi
{ Rituksimab }
1. BEŞERİ TIBBİ ÜRÜNÜN ADI
TRUXIMA 100 mg/10 mL iv infüzyonluk çözelti hazırlamak için konsantre Steril
2. KALİTATİF VE KANTİTATİF BİLEŞİM
Etkin madde
Her bir flakon 10 mL'lik çözelti içinde 100 mg rituximab içerir. Çözeltinin her mL'sinde 10 mg rituximab bulunur.
Rituximab insan IgGl sabit bölgeleri ve sırasıyla değişken mürin hafif zincir ve ağır zincir içeren bir glikozile immünoglobulin sunan, genetik mühendisliği ile üretilen kimerik fare/insan monoklonal antikorudur. Antikor, memelilerin (Çin hamster over hücresi) hücre süspansiyon kültüründe üretilir ve spesifik viral inaktivasyon ve çıkarma prosedürlerini içerecek şekilde afinite kromotografisi ve iyon değiştirme ile saflaştırılan bir biyobenzer ilaçtır.
Yardımcı maddeler
Tri-sodyum sitrat dihidrat : 7,36 mg/mL
3. FARMASÖTİK FORMU
İnfüzyon için konsantre çözelti içeren flakon. Çözelti berrak, renksiz bir sıvıdır.
4. KLİNİK ÖZELLİKLER
4.1. Terapötik endikasyonlar
TRUXIMA yetişkinlerde aşağıdaki endikasyonlar için endikedir:
Hodgkin-dışı lenfoma (NHL)
TRUXIMA'nın, daha önce tedavi edilmemiş evre III-IVfoliküler lenfoması olan yetişkin hastaların tedavisinde kemoterapi ile birlikte kullanılması endikedir. TRUXIMA idame tedavisi, indüksiyon tedavisine yanıt veren yetişkin foliküler lenfoma hastalarının tedavisinde endikedir.
TRUXIMA monoterapisi, kemoresistan veya kemoterapiden sonra ikinci ya da daha sonraki relapslarında olan evre III-IV foliküler lenfomalı yetişkin hastaların tedavisinde endikedir.
TRUXIMA'nın, CD20 pozitif diffüz büyük B hücreli hodgkin-dışı lenfoması olan yetişkin
hastaların tedavisinde CHOP (siklofosfamid, doksorubisin, vinkristin, prednizolon) kemoterapisi ile birlikte kullanılması endikedir. TRUXIMA'nın yaş aralığı ≥6 ay ila <18 yaş olan pediyatrik hastalarda, daha önceden tedavi edilmemiş ileri evre CD20 pozitif diffüz büyük B hücreli lenfoma (DLBCL), burkitt lenfoma (BL)/burkitt lösemi (olgun B-hücreli akut lösemi) (BAL) veya burkitt-benzeri lenfoma (BLL) tedavisinde kemoterapi ile birlikte kullanılması endikedir.
Kronik lenfositik lösemi (KLL)
TRUXIMA, daha önce tedavi edilmemiş ve relaps/refrakter (nükseden/dirençli) KLL hastalarının tedavisinde kemoterapi ile kombinasyon halinde endikedir. TRUXIMA dahil monoklonal antikorlar ile daha önce tedavi edilmiş ya da daha önceki kemoterapi ile kombine kullanılan TRUXIMA tedavisine refrakter hastalarda etkililik ve güvenlilik ile ilgili sınırlı veri mevcuttur.
Daha fazla bilgi için Bölüm 5.1'e bakınız.
Granülomatöz polianjiitis (Wegener) (GPA) ve mikroskobik polianjiitis (MPA)
Siklofosfamide dirençli veya siklofosfamid tedavisi verilemeyen ciddi, aktif granülomatöz polianjiitis (GPA, Wegener granülomatözü olarak da bilinir) ve mikroskobik polianjiitis (MPA) hastalarının tedavisinde glukokortikoidlerle kombine olarak TRUXIMA kullanılır.
Pemfigus vulgaris (PV)
TRUXIMA, orta ila şiddetli pemphigus vulgarisi olan hastaların tedavisinde endikedir.
4.2. Pozoloji ve uygulama şekli
TRUXIMA, acil uygulama için tüm resüsitasyon olanaklarının bulunduğu bir ortamda deneyimli bir sağlık mesleği mensubunun yakın gözetimi altında uygulanmalıdır (Bkz. Bölüm 4.4).
Premedikasyon ve profilaktik ilaçlar
Her TRUXIMA infüzyonundan önce, analjezik/antipiretik (örn. parasetamol) ve antihistaminik ilaçtan (örn. difenhidramin) oluşan bir premedikasyon her zaman yapılmalıdır.
Hodgkin-dışı lenfoma ve kronik lenfositik lösemili yetişkin hastalarda, TRUXIMA glukortikoid içeren kemoterapiyle kombinasyon halinde verilmiyorsa, glukokortikoidlerle premedikasyon göz önünde bulundurulmalıdır.
Hodgkin-dışı lenfomalı pediyatrik hastalara TRUXIMA infüzyonunun başlangıcından 30 ila 60 dakika öncesinde parasetamol ve H1 antihistamin (=difenhidramin veya eşdeğeri) ile premedikasyon uygulanmalıdır. Ek olarak, prednizon Tablo 1'de belirtildiği şekilde uygulanmalıdır.
KLL hastaları için, tümör lizis sendromu (TLS) riskini azaltmak amacıyla tedavi başlangıcından 48 saat öncesinde yeterli hidrasyon ve ürikostatik uygulanmaya başlanması ile profilaksi önerilmektedir. Lenfosit sayıları > 25x10/L olan tüm KLL hastalarında akut infüzyon reaksiyonları ve/veya sitokin salıverilme sendromunun oranını ve ciddiyetini azaltmak amacıyla, TRUXIMA infüzyonundan kısa bir süre önce 100 mg i.v. prednizon/prednizolon uygulanması önerilmektedir.
Granülamatöz polianjiitis (Wegener) veya mikroskobik polianjiitis veya pemfigus vulgaris hastalarında infüzyonla ilişkili reaksiyonların (IRR) sıklığını ve ciddiyetini azaltmak için her TRUXIMA infüzyonundan 30 dakika önce tamamlanacak şekilde 100 mg i.v. metilprednizolon
uygulanmalıdır.
Granülomatöz polianjiitis (Wegener) ve mikroskobik polianjiitis hastalarında TRUXIMA'nın ilk infüzyonundan önce 1 ila 3 günlük 1.000 mg i.v./gün metilprednizolon önerilmektedir (metilprednizolonun son dozu TRUXIMA'nın ilk infüzyonu ile aynı günde verilebilir). Ardından TRUXIMA tedavisinin 4 haftalık indüksiyon kürü sırasında ve sonrasında oral prednizon 1 mg/kg/gün (en fazla 80 mg/gün ve daha sonra klinik duruma göre mümkün olduğunca hızlı bir şekilde azaltılır) uygulanmalıdır.
GPA ve MPA veya PV hastalarına TRUXIMA tedavisi sırasında ve sonrasında yerel klinik uygulama kılavuzlarına uygun şekilde Pnömosistis jiroveci pnömoni (PCP) profilaksisi önerilmektedir.
Pozoloji/uygulama sıklığı ve süresi:
Hodgkin-dışı lenfoma
Foliküler Hodgkin-dışı lenfoma Kombinasyon tedavisi
Önceden tedavi edilmemiş veya relaps/refrakter (nükseden/dirençli) foliküler lenfoma hastalarının indüksiyon tedavisinde, kemoterapi ile kombinasyon halinde önerilen TRUXIMA dozu, her siklusta 375 mg/m vücut yüzey alanı (BSA) olacak şekilde en fazla 8 siklustur.
TRUXIMA, kemoterapinin glukokortikoid bileşeninin i.v. yolla verilmesinden sonra her bir kemoterapi siklusunun ilk gününde uygulanmalıdır.
İdame tedavisi
Önceden tedavi edilmemiş foliküler lenfoma
İndüksiyon tedavisine yanıt vermiş, önceden tedavi edilmemiş, foliküler lenfomalı hastalar için idame tedavisi olarak kullanılan TRUXIMA'nın önerilen dozu hastalık progresyonuna kadar ya da en fazla 2 yıllık süre (toplam 12 infüzyon) boyunca (indüksiyon tedavisinin son dozundan 2 ay sonra başlayarak) 2 ayda bir 375 mg/m vücut yüzey alanıdır.
Relaps/refrakter (nükseden/dirençli) foliküler lenfoma
İndüksiyon tedavisine yanıt vermiş relaps/refrakter foliküler lenfomalı hastalar için idame tedavisi olarak kullanılan TRUXIMA'nın önerilen dozu, hastalık progresyonuna kadar veya en fazla 2 yıllık süre (toplam 8 infüzyon) boyunca (indüksiyon tedavisinin son dozundan 3 ay sonra başlayarak) 3 ayda bir 375 mg/m vücut yüzey alanıdır.
Monoterapi
Relaps/Refrakter (Nükseden/Dirençli) foliküler lenfoma
Evre III-IV foliküler lenfoma olan, kemoterapiye dirençli veya kemoterapiden sonra ikinci kez veya daha fazla nüks oluşan yetişkin hastalar için indüksiyon tedavisi olarak kullanılan TRUXIMA monoterapisinin önerilen dozu: 4 hafta süreyle haftada bir kere i.v. infüzyon yoluyla verilen 375 mg/m vücut yüzey alanıdır.
Nüks eden/refrakter foliküler lenfoma için TRUXIMA monoterapisi ile geçmiş tedaviye yanıt veren hastalarda TRUXIMA monoterapisiyle yeniden tedavi için önerilen doz: 4 hafta süreyle haftada bir kere i.v. infüzyon yoluyla verilen 375 mg/m vücut yüzey alanıdır (Bkz. Bölüm 5.1).
Yetişkinlerde diffüz büyük B hücreli Hodgkin-dışı lenfoma
TRUXIMA, CHOP (siklofosfamid, doksorubisin, prednizolon ve vinkristin) kemoterapisi ile kombinasyon şeklinde kullanılmalıdır. Önerilen TRUXIMA dozu, her kemoterapi siklusunun
1. gününde, 8 siklus için, CHOP rejiminin glukokortikoid bileşeni i.v. yoldan uygulandıktan sonra verilmek üzere, 375 mg/m vücut yüzey alanıdır. CHOP rejiminin öteki bileşenleri, TRUXIMA uygulandıktan sonra verilmelidir. Diffüz büyük B hücreli Hodgkin-dışı lenfomada diğer kemoterapilerle kombinasyon halinde TRUXIMA'nın güvenliliği ve etkililiği belirlenmemiştir.
Tedavi sırasında doz ayarlamaları
TRUXIMA dozunda herhangi bir azaltma önerilmemektedir. TRUXIMA, kemoterapi ile kombine halde uygulandığında, kemoterapötik ilaçlar için geçerli standart doz azaltmaları yapılmalıdır.
Kronik lenfositik lösemi (KLL)
Daha önce tedavi edilmemiş ve relaps/refrakter hastalar için kemoterapiyle kombinasyon halinde önerilen TRUXIMA dozu, toplam 6 siklus olmak üzere, ilk tedavi siklusunun 0. gününde uygulanan 375 mg/m vücut yüzey alanı ve sonrasındaki her siklusun 1. gününde uygulanan 500 mg/m vücut yüzey alanıdır. Kemoterapi, TRUXIMA infüzyonundan sonra verilmelidir.
Granülomatöz polianjiitis (Wegener) (GPA) ve mikroskobik polianjiitis (MPA)
GPA ve MPA remisyon tedavisinin indüksiyonu için önerilen TRUXIMA dozu, 4 hafta süreyle haftada bir kere i.v. infüzyon yoluyla verilen 375 mg/m vücut yüzey alanıdır.
Pemfigus vulgaris
TRUXIMA'nın pemfigus vulgaris tedavisinde önerilen dozu, azaltılarak kesilen glukokortikoid kürü ile birlikte, i.v. infüzyon olarak uygulanan 1.000 mg ve bunu takiben iki hafta sonra uygulanan ikinci bir 1.000 mg i.v. infüzyondur.
İdame tedavisi
500 mg'lık idame i.v. infüzyon 12. ve 18. aylarda ve sonrasında klinik değerlendirme sonucunda gerek duyulması halinde her 6 ayda bir verilmelidir.
Relaps tedavisi
Relaps gelişmesi halinde, hastalar 1.000 mg i.v. alabilir. Hekim klinik değerlendirme sonucunda hastanın glukokortikoide tekrar başlamasını veya dozunun arttırılmasını düşünmelidir.
Devam infüzyonları, bir önceki infüzyondan en az 16 hafta sonra yapılabilir.
Uygulama şekli:
TRUXIMA sadece ona ayrılmış damar yoluyla, tek başına intravenöz infüzyon olarak uygulanmalıdır. TRUXIMA tüm resüsitasyon olanaklarının eksiksiz olarak hazır bulunduğu bir sağlık kuruluşunda ve uzman bir hekimin yakın gözetimi altında uygulanmalıdır.
Hazırlanmış infüzyon çözeltilerini i.v. puşe veya bolus yoluyla uygulamayınız (Bkz. Bölüm 6.6).
Hastalar, sitokin salıverilme sendromu başlaması açısından yakından izlenmelidir (Bkz. Bölüm 4.4). Özellikle şiddetli dispne, bronkospazm veya hipoksi şeklinde şiddetli reaksiyon belirtileri gelişen hastalarda infüzyon derhal kesilmelidir. Hodgkin-dışı lenfomalı hastalar daha sonra, uygun laboratuvar testleri ile tümör lizis sendromu belirtileri açısından ve göğüs röntgeni ile pulmoner infiltrasyon açısından değerlendirilmelidir. Semptomlar tamamen düzelene kadar ve laboratuvar değerleriyle göğüs röntgeni bulguları normalleşene dek tüm hastalarda infüzyona tekrar başlanmamalıdır. Ayrıca infüzyona başlangıç olarak daha önce uygulananın en çok yarısı kadar bir hızda yeniden başlanmalıdır. Aynı şiddetli advers reaksiyonlar ikinci kez meydana gelirse tedavinin durdurulması yönünde bir karar vaka bazında ciddi olarak düşünülmelidir.
Hafif veya orta dereceli infüzyonla ilişkili reaksiyonlar (IRR) (Bkz. Bölüm 4.8), genelde infüzyon hızının azaltılmasına yanıt vermektedir. Semptomlar düzeldikten sonra infüzyon hızı arttırılabilir.
İlk infüzyon
Önerilen ilk infüzyon hızı 50 mg/saattir; ilk 30 dakikadan sonra 30 dakikada bir 50 mg/saatlik artışlarla hız maksimum 400 mg/saate çıkarılabilir.
İzleyen İnfüzyonlar Tüm endikasyonlar
Sonraki TRUXIMA infüzyonlarına 100 mg/saat hızıyla başlanabilir ve daha sonra her 30 dakikada bir 100 mg/saatlik artışlarla hız maksimum 400 mg/saate çıkarılabilir.
Pediyatrik hastalar – Hodgkin-dışı Lenfoma İlk İnfüzyon
Önerilen ilk infüzyon hızı 0,5 mg/kg/saattir (en fazla 50 mg/saat); aşırı duyarlılık veya infüzyon ile bağlantılı reaksiyonlar görülmezse 30 dakikada bir 0,5 mg/kg/saatlik artışlarla hız maksimum 400 mg/saate çıkarılabilir.
İzleyen İnfüzyonlar
Sonraki TRUXIMA infüzyonlarına 1 mg/kg/saat hızıyla başlanabilir (en fazla 50 mg/saat) ve daha sonra her 30 dakikada bir 1 mg/kg/saatlik artışlarla hız maksimum 400 mg/saate çıkarılabilir.
Özel popülasyonlara ilişkin ek bilgiler:
Böbrek/ Karaciğer yetmezliği:
Böbrek/ karaciğer yetmezliği olan hastalarda özel bir kullanım söz konusu değildir.
Pediyatrik popülasyon:
Hodgkin-dışı Lenfoma
Yaşı ≥ 6 ay ila 18 yaş arasında olan daha önceden tedavi edilmemiş, ileri evre CD20 pozitif DLBCL/BL/BAL/BLL'li pediyatrik hastalarda, TRUXIMA sistemik Lenfoma Malign B (LMB) kemoterapisi ile kombinasyon halinde kullanılmalıdır (Tablo 1 ve Tablo 2'ye bakınız). TRUXIMA'nın önerilen dozu i.v. infüzyon yoluyla verilen 375 mg/m vücut yüzey alanıdır. TRUXIMA için vücut yüzey alanı haricinde doz ayarlaması gerekli değildir.
TRUXIMA'nın güvenliliği ve etkililiği yaşı ≥ 6 ay ila 18 yaş arasında olan pediyatrik hastalarda daha önceden tedavi edilmemiş, ileri evre CD20 pozitif DLBCL/BL/BAL/BLL endikasyonu dışındaki endikasyonlarda ortaya konmamıştır. 3 yaşın altındaki hastalarda sadece sınırlı veri mevcuttur. Daha fazla bilgi için Bölüm 5.1'e bakınız.
TRUXIMA, CD20 pozitif diffüz büyük B hücreli lenfoması olan 0 ila 6 ay arasındaki pediyatrik hastalarda kullanılmamalıdır (Bkz. Bölüm 5.1).
Tablo 1. Pediyatrik Hastalarda Non-Hodgkin Lenfoma için TRUXIMA Uygulamasının Pozolojisi
Siklus | Tedavi Günü | Uygulama Detayları | |
Prefaz (COP) | TRUXIMA verilmez | - | |
İndüksiyon kürü 1 (COPDAM1) | -2. Gün (prefazın 6. Gününe tekabül eder) 1. TRUXIMA infüzyonu | uygulanmalıdır. | |
TRUXIMA, ilk TRUXIMA infüzyonundan 48 saat sonra verilecektir. | |||
İndüksiyon kürü 2 (COPDAM2) | -2. Gün 3. TRUXIMA infüzyonu | 2. indüksiyon prednizon uygulaması verilmez. | küründe, TRUXIMA zamanında |
1. Gün 4. TRUXIMA infüzyonu | TRUXIMA, üçüncü TRUXIMA infüzyonundan 48 saat sonra verilecektir. | ||
Konsolidasyon kürü 1 (CYM/CYVE) | 1. Gün 5. TRUXIMA infüzyonu | Prednizon uygulaması verilmez. | TRUXIMA zamanında |
Konsolidasyon kürü 2 (CYM/CYVE) | 1. Gün 6. TRUXIMA infüzyonu | Prednizon uygulaması verilmez. | TRUXIMA zamanında |
İdame kür 1 (M1) | Konsolidasyon kürü (CYVE) 25 ila 28. Günü TRUXIMA verilmez. | 2'nin | Konsolidasyon kürü 2'den (CYVE) sonra periferik sayılar düzeldiğinde (ANC>1,0 x 10/L ve trombositler 100 x 10/L) başlar |
İdame kür 2 (M2) | İdame kür 1'nün (M1) 28. Günü TRUXIMA verilmez | - | |
ANC = Mutlak Nötrofil Sayısı; COP = Siklofosfamid, Vinkristine, Prednizon; COPDAM = Siklofosfamid, Vinkristin, Prednizolon, Doksorubisin, Metotreksat; CYM = CYtarabin (Aracytine, Ara-C), Metotreksat; CYVE = CYtarabin (Aracytine, Ara-C), VEposid (VP16) | |||
indüksiyon kürü esnasında, prednizon kemoterapi kürünün bir parçası olarak verilir ve TRUXIMA'dan önce
4.3. Kontrendikasyonlar
TRUXIMA'nın Hodgkin-dışı lenfoma ve kronik lenfositik lösemide kullanım için kontrendike olduğu durumlar:
Etkin madde
4.4. Özel kullanım uyarıları ve önlemleri

infüzyon reaksiyonlarının yaklaşık %80'i ilk infüzyonla ilişkili olarak görülmüştür. İnfüzyon sırasında hastaları dikkatlice gözlemleyiniz. Evre 3 veya 4 infüzyon reaksiyonları gelişirse TRUXIMA infüzyonunu kesiniz ve tıbbi tedavi uygulayınız (Bkz. Bölüm 4.4, Bölüm 4.8).
Tümör Lizis Sendromu (TLS)
Hodgkin-dışı lenfoma (NHL) hastalarının TRUXIMA tedavisi sonrasında, TLS sonucu, diyaliz gerektiren ve ölümle sonuçlanan akut renal yetmezlik görülebilir.
Ciddi Mukokütanöz Reaksiyonlar
TRUXIMA kullanan hastalarda ölümcül olabilen, ciddi mukokütanöz reaksiyonlar meydana gelebilir (Bkz. Bölüm 4.4, Bölüm 4.8).
Progresif Multifokal Lökoensefalopati (PML)
TRUXIMA kullanan hastalarda PML ile sonuçlanan John Cunningham (JC) virüsü aktivasyonu ve ölüm meydana gelebilir.
Hepatit B Virüs (HBV) Reaktivasyonu
TRUXIMA ile tedavi edilen hastalarda fulminan hepatit, hepatik yetmezlik ve ölümle sonuçlanabilen hepatit B reaktivasyonu gerçekleşebilir. TRUXIMA tedavisine başlamadan önce bütün hastalar HBV enfeksiyonu açısından taranmalı ve tedavi süresince ve sonrasında hastalar izlenmelidir. HBV reaktivasyonu gelişen hastalarda acilen TRUXIMA ve birlikte kullanılan kemoterapi ilaçları kesilmelidir.
Progresif multifokal lökoensefalopati (PML)
Rituximabın kullanımı sonrasında çok seyrek ölümcül progresif multifokal lökoensefalopati (PML) vakaları bildirilmiştir. Hastalar, herhangi bir yeni veya kötüleşen nörolojik semptomlar açısından veya PML'yi düşündürebilecek belirtiler açısından düzenli aralıklarla izlenmelidir. PML'den şüphelenilmesi durumunda, PML dışlanana kadar doz uygulaması askıya alınmalıdır. Klinisyen, semptomların nörolojik disfonksiyonu gösterip göstermediğine ve nörolojik disfonksiyon varsa, bu semptomların PML'yi düşündürüp düşündürmediğine karar vermek için hastayı değerlendirmelidir. Klinik gereklilik nedeniyle, bir nörolog tarafından konsültasyon yapılması da dikkate alınmalıdır.
Herhangi bir şüphe olması durumunda tercihen kontrastlı MRG, JC Viral DNA için beyin- omurilik sıvısı (BOS) testi ve tekrarlı nörolojik değerlendirmeler dahil detaylı muayene dikkate alınmalıdır.
Hekim, özellikle hastanın fark etmeyebileceği PML semptomları (örn. bilişsel, nörolojik veya psikiyatrik semptomlar) açısından dikkatli olmalıdır. Hastanın farkında olmadığı semptomları fark edebilmeleri nedeniyle, hastaların ayrıca eşleri ve hastaya bakanlar tedavi konusunda bilgilendirilmelidirler.
Eğer bir hastada PML gelişirse, rituximabın kullanımı kalıcı şekilde durdurulmalıdır.
PML olan bağışıklığı zayıflamış hastalarda immün sistemin yeniden düzenlenmesi ardından, stabilizasyon veya sonuçlarda iyileşme olduğu görülmüştür. PML'nin erken saptanmasının ve rituximab tedavisinin askıya alınmasının benzer stabilizasyon veya sonuçlarda iyileşme sağlayıp sağlamayacağı bilinmemektedir.
Hodgkin-dışı lenfoma ve kronik lenfositik lösemi İnfüzyonla ilişkili reaksiyonlar
Rituximab sitokinlerin ve/veya diğer kimyasal aracıların salınması ile bağlantılı olabilen
infüzyon reaksiyonları ile ilişkilendirilmektedir. Sitokin salıverilme sendromu klinik olarak akut aşırı duyarlılık reaksiyonlarından ayırt edilemeyebilir.
Sitokin salıverilme sendromu, tümör lizis sendromu ve anafilaktik ve aşırı duyarlılık reaksiyonlarını içeren reaksiyonlar topluluğu aşağıda tarif edilmiştir.
Rituximabın intravenöz formülasyonunun pazarlama sonrası deneyimlerde kullanımı esnasında, ilk intravenöz rituximab infüzyonunun başlamasının ardından 30 dakika ila 2 saat içerisinde başlayan ve ölümle sonuçlanan ciddi infüzyon reaksiyonları rapor edilmiştir. Bunlar pulmoner olaylar ile karakterizedir ve bazı vakalarda ateş, titreme, kasılma, hipotansiyon, ürtiker, anjiyoödem ve diğer semptomlara ek olarak hızlı tümör lizis ve tümör lizis sendromu özelliklerini de içermiştir (Bkz. Bölüm 4.8).
Şiddetli sitokin salıverilme sendromu ateş, titreme, kasılma, ürtiker ve anjiyoödeme ek olarak sıklıkla bronkospazm ve hipoksinin eşlik ettiği şiddetli dispne ile karakterizedir. Bu sendrom tümör lizis sendromunun hiperürisemi, hiperkalemi, hipokalemi, hiperfosfatemi, akut böbrek yetmezliği, laktat dehidrojenaz (LDH) artışı gibi bazı özellikleriyle ilişkili olabilir ve akut solunum yetmezliğine ve ölüme yol açabilir. Akut solunum yetmezliğine, göğüs röntgeninde görünebilen pulmoner interstisyel infiltrasyon veya ödem gibi olaylar eşlik edebilir. Sendrom, genellikle ilk infüzyonun başlatılmasından sonraki bir ya da iki saat içinde kendini gösterir. Geçmişte pulmoner yetmezliği olan hastalar veya pulmoner tümör infiltrasyonu bulunan hastalar zayıf sonuçlar açısından daha fazla risk altında olabilirler ve bu hastalar daha dikkatli tedavi edilmelidirler. Şiddetli sitokin salıverilme sendromu gelişen hastaların infüzyonu derhal kesilmelidir (Bkz. Bölüm 4.2) ve bu hastalara agresif semptomatik tedavi uygulanmalıdır. Klinik semptomlarda başta görülen iyileşmenin ardından kötüleşme olabileceğinden, bu hastalar tümör lizis sendromu ve pulmoner infiltrasyon geçene kadar veya dışlanana kadar yakından izlenmelidir. Belirtilerin ve semptomların tamamen ortadan kalkması ardından hastalara uygulanan tedavi, nadiren şiddetli sitokin salıverilme sendromunun tekrarlamasıyla sonuçlanmıştır.
Özellikle şiddetli sitokin salıverilme sendromu açısından yüksek risk altında olabilecek, KLL'si olan hastalar gibi yüksek tümör yükü veya dolaşımda yüksek sayıda malign hücresi (≥ 25 x 10/L) olan hastalar aşırı dikkatle tedavi edilmelidir. Bu hastalar ilk infüzyonun başından sonuna kadar çok yakından gözlemlenmelidir. Bu hastalarda, ilk infüzyon sırasında düşük bir infüzyon hızının kullanılması veya ilk siklus sırasında ve lenfosit sayısının hala >25 x 10/L olması durumunda takip eden sikluslarda dozun iki güne bölünerek verilmesi göz önünde bulundurulmalıdır.
İnfüzyonla ilişkili tüm advers reaksiyon tipleri, tedavi uygulanan hastaların %77'sinde gözlenmiştir (hastaların %10'unda hipotansiyon ve bronkospazmın eşlik ettiği sitokin salıverilme sendromu dahil, Bkz. Bölüm 4.8). Bu semptomlar genellikle rituximab infüzyonunun kesilmesiyle ve bir antipiretik, bir antihistaminik ve bazı durumlarda oksijen, intravenöz serum fizyolojik veya bronkodilatörlerin ve gerektiğinde glukokortikoidlerin uygulanmasıyla geri döndürülebilir olmuştur. Şiddetli reaksiyonlar için yukarıda yer alan sitokin salıverilme sendromuna bakınız.
Hastalara intravenöz yolla protein verilmesinden sonra anafilaktik reaksiyonlar veya diğer aşırı duyarlılık reaksiyonları bildirilmiştir. Sitokin salıverilme sendromunun tersine, gerçek aşırı
duyarlılık reaksiyonları tipik şekilde infüzyona başlanmasından sonra dakikalar içinde oluşur. Rituximab uygulaması sırasındaki alerjik reaksiyon olgularında acil kullanım için, aşırı duyarlılık reaksiyonlarının tedavisine yönelik ilaçlar örn., epinefrin (adrenalin), antihistaminikler ve glukokortikoidler kullanıma hazır bulundurulmalıdır. Anafilaksinin klinik belirtileri, sitokin salıverilme sendromunun klinik belirtilerine (yukarıda tanımlanmıştır) benzer görünebilir. Aşırı duyarlılığa bağlı reaksiyonlar, sitokin salıverilmesine bağlı reaksiyonlardan daha az sıklıkta bildirilmiştir.
Bazı vakalarda bildirilen diğer reaksiyonlar miyokard enfarktüsü, atriyal fibrilasyon, pulmoner ödem ve akut geri döndürülebilir trombositopeni olmuştur.
Rituximab infüzyonu sırasında hipotansiyon oluşabileceğinden, rituximab infüzyonundan önceki 12 saatlik süre boyunca herhangi bir antihipertansif ilacın alınmamış olmasına dikkat edilmelidir.
Kardiyak hastalıklar
Rituximab ile tedavi edilen hastalarda anjina pektoris, atriyal flatter ve fibrilasyon gibi kardiyak aritmiler, kalp yetmezliği ve/veya miyokard enfarktüsü meydana gelmiştir. Bu nedenle kardiyak hastalık ve/veya kardiyotoksik kemoterapi öyküsü olan hastalar yakından izlenmelidir.
Hematolojik toksisiteler
Monoterapi şeklinde uygulanan rituximab miyelosupresif olmadığı halde, nötrofil sayısı < 1,5 x 10/L ve/veya trombosit sayısı < 75 x 10/L olan hastalar rituximab ile tedavi edilirken dikkatli olunmalıdır, çünkü bu tip hastalarla ilgili klinik deneyimler sınırlıdır. Rituximab, otolog kemik iliği transplantasyonu olan 21 hastada ve miyelotoksisite indüklenmediği halde kemik iliği fonksiyonlarında azalma olan diğer risk gruplarında kullanılmıştır.
Rituximab tedavisi sırasında düzenli olarak nötrofil ve trombosit sayımı dahil, kan hücrelerinin sayımı yapılmalıdır.
Enfeksiyonlar
Rituximab tedavisi sırasında ölümcül, ciddi enfeksiyonlar meydana gelebilir (Bkz. Bölüm 4.8). Rituximab aktif, şiddetli enfeksiyonu (örn. tüberküloz, sepsis ve fırsatçı enfeksiyonlar, Bkz. Bölüm 4.3) bulunan hastalara uygulanmamalıdır.
Doktorlar, tekrarlayan veya kronik enfeksiyon öyküsü bulunan veya hastayı ciddi enfeksiyonlara eğilimli hale getirecek şekilde altta yatan koşullara sahip hastalarda rituximab kullanırken dikkatli olmalıdırlar (Bkz. Bölüm 4.8).
Rituximab alan olgularda, ölümle sonuçlanan fulminan hepatit de dahil olmak üzere hepatit B reaktivasyonu vakaları bildirilmiştir. Bu vakaların büyük çoğunluğu ayrıca sitotoksik kemoterapiye maruz kalmıştır. Nükseden/refrakter KLL hastalarında yapılan bir çalışmadan sağlanan kısıtlı veriler, rituximab tedavisinin ayrıca primer hepatit B enfeksiyonlarının sonucunu kötüleştirebildiğini göstermektedir. Rituximab ile tedaviye başlanmadan önce bütün hastalar (sadece HBV enfeksiyon riski olanlar değil) Hepatit B virüsü (HBV) açısından taranmalıdır. Bu ölçümler en azından hepatit B yüzey antijeni (HBsAg)-durumu ve hepatit B çekirdek antikoru (HBcAb)-durumunu içermelidir. Bunlar yerel kılavuzlara göre diğer uygun markörler ile tamamlanmalıdır. Aktif hepatit B hastalığı olan hastalar rituximab ile tedavi edilmemelidir. Pozitif hepatit B serolojisi olan hastalar (HBsAg veya HBcAb) tedavi başlangıcından önce karaciğer hastalıkları uzmanına danışmalıdır ve bu hastalar hepatit B reaktivasyonunun önlenmesi için yerel medikal standartlara göre takip edilmeli ve yönetilmelidir.
NHL ve KLL'de rituximabın pazarlama sonrası kullanımı sırasında çok seyrek progresif multifokal lökoensefalopati (PML) vakaları bildirilmiştir (Bkz. Bölüm 4.8). Hastaların büyük çoğunluğu rituximabı kemoterapi ile birlikte veya hematopoetik kök hücre transplantasyonunun bir parçası olarak almışlardır.
İmmünizasyonlar
NHL ve KLL hastalarında, rituximab tedavisini takiben canlı viral aşılarla yapılan immünizasyonun güvenliliği üzerinde çalışma yapılmamıştır ve canlı virüs aşılarıyla aşılama yapılması önerilmemektedir. Rituximab ile tedavi edilen hastalar canlı olmayan aşılarla aşılanabilirler. Ancak canlı olmayan aşılara yanıt oranları düşebilir. Randomize olmayan bir çalışmada monoterapi olarak rituximab alan relaps, düşük evreli yetişkin NHL hastaları ile sağlıklı, tedavi görmemiş kontrol vakaları karşılaştırıldığında, tetanoz hatırlatıcı antijenine (%16'ya karşılık %81) ve Keyhole Limpet Haemocyanin (KLH) neoantijenine (antikor titrelerinde >2 katı artışa göre değerlendirildiğinde %4'e karşılık %76) daha düşük oranda aşı yanıtı gerçekleşmiştir. Her iki hastalık arasındaki benzerlikler dikkate alındığında, KLL olan hastalar için benzer bulgular öngörülebilir fakat klinik çalışmalarda incelenmemiştir.
Bir grup antijene karşı (Streptococcus pneumoniae, influenza A, kabakulak, kızamıkçık ve suçiçeği) ortalama tedavi öncesi antikor titreleri rituximab tedavisi sonrasında en az 6 ay süreyle korunmuştur.
Deri reaksiyonları
Toksik Epidermal Nekroliz (Lyell sendromu) ve Stevens-Johnson sendromu gibi bazılarının ölümcül sonuçları olabilen ciddi cilt reaksiyonları bildirilmiştir (Bkz. Bölüm 4.8). Bu gibi durumlarda olayın rituximab ile ilişkili olduğundan şüpheleniliyorsa tedavi kalıcı olarak durdurulmalıdır.
Pediyatrik popülasyon
3 yaşın altındaki hastalar için sınırlı veri mevcuttur. Daha fazla bilgi için Bölüm 5.1'e bakınız. Granülomatöz polianjiitis ve mikroskobik polianjiitis ve pemfigus vulgaris
İnfüzyonla ilişkili reaksiyonlar
Rituximab, sitokinlerin ve/veya diğer kimyasal mediyatörlerin salıverilmesine bağlı olabilen infüzyonla ilgili reaksiyonlarla (IRR) ilişkilendirilmiştir.
Pazarlama sonrası koşullarda, romatoid artritli hastalarda ölümcül sonuçlara neden olabilen infüzyonla ilgili şiddetli reaksiyonlar bildirilmiştir. Klinik çalışmalarda romatoid artrit hastalarında bildirilen infüzyona bağlı olguların çoğu hafif ve orta ciddiyette olmuştur. En yaygın semptomlar baş ağrısı, kaşıntı, boğazda tahriş, kızarıklık, döküntü, ürtiker, hipertansiyon ve pireksidir. Genel olarak, herhangi bir tedavi kürünün birinci infüzyonunun ardından herhangi bir infüzyon reaksiyonu yaşayan hastaların oranı, ikinci infüzyonun ardından görülene oranla daha yüksek olmuştur. IRR insidansı sonraki kürlerle azalmıştır (Bkz. Bölüm 4.8). Bildirilen reaksiyonlar rituximab infüzyonunun hızının azaltılması ya da kesilmesine ve bir antipiretik, antihistaminik ve seyrek olarak oksijen, intravenöz serum fizyolojik veya bronkodilatörün ve gerektiğinde glukokortikoidlerin uygulanmasına bağlı olarak genellikle geri dönüşümlü olmuştur. Önceden kalp sorunları bulunan ve önceden kardiyopulmoner advers reaksiyon yaşamış olan hastalar dikkatlice izlenmelidir. İnfüzyonla ilgili reaksiyonun ciddiyetine göre ve gereken müdahaleye göre, rituximab kullanımı geçici veya kalıcı olarak bırakılmalıdır. Olguların çoğunda, semptomlar tamamen giderildiğinde, infüzyon hızı %50 oranında (örn. 100 mg/saatten 50 mg/saat hızına) azaltılarak infüzyona devam edilebilir.
Rituximab uygulaması sırasındaki alerjik reaksiyon olgularında acil kullanım için, aşırı duyarlılık reaksiyonlarının tedavisine yönelik ilaçlar örn. epinefrin (adrenalin), antihistaminikler ve glukokortikoidler kullanıma hazır bulundurulmalıdır.
Orta şiddette kalp yetmezliği (NYHA sınıf III) veya ciddi, kontrol altına alınmamış kardiyovasküler hastalığı olan hastalarda rituximab kullanımıyla ilgili güvenlilik verisi bulunmamaktadır. Rituximab ile tedavi edilen hastalarda, atriyal fibrilasyon ve flatter ile anjina pektoris gibi önceden mevcut olan iskemik kardiyak koşulların belirti verdiği gözlemlenmiştir. Bu sebeple, rituximab tedavisinden önce, bilinen bir kardiyak öyküsü olan ve önceden kardiyopulmoner advers reaksiyon yaşamış olan hastalarda, infüzyon reaksiyonlarından kaynaklanan kardiyovasküler komplikasyonların riski dikkate alınmalı ve hastalar uygulama sırasında dikkatle gözlenmelidir. Rituximab infüzyonu sırasında hipotansiyon oluşabileceğinden, rituximab infüzyonu öncesindeki 12 saatlik süre boyunca herhangi bir antihipertansif ilacın alınmamış olmasına dikkat edilmelidir.
Granülomatöz polianjiitis ve mikroskobik polianjiitisi ve pemfigus vulgarisi bulunan hastalarda infüzyonla ilgili reaksiyonlar, klinik çalışmalarda romatoid artrit hastalarında görülenlere benzer olmuştur (Bkz. Bölüm 4.8).
Kardiyak hastalıklar
Rituximab ile tedavi edilen hastalarda anjina pektoris, atriyal flatter ve fibrilasyon gibi kardiyak aritmiler, kalp yetmezliği ve/veya miyokard enfarktüsü meydana gelmiştir. Bu nedenle kardiyak hastalık öyküsü olan hastalar yakından izlenmelidir (Bkz. yukarıda infüzyonla ilgili reaksiyonlar).
Enfeksiyonlar
TRUXIMA'nın etki mekanizması ve B hücrelerinin normal immün cevabın sürdürülmesinde önemli rol oynaması bilgisine dayanarak, rituximab tedavisi ardından hastalar artan enfeksiyon riski taşımaktadır (Bkz. Bölüm 5.1). Rituximab tedavisi sırasında ölümcül, ciddi enfeksiyonlar meydana gelebilir (Bkz. Bölüm 4.8). Rituximab aktif, şiddetli enfeksiyonu (örn. tüberküloz, sepsis ve fırsatçı enfeksiyonlar, Bkz. Bölüm 4.3) bulunan veya bağışıklığı ciddi düzeyde baskılanmış (örn. CD4 veya CD8 düzeyleri çok düşük olan) hastalara uygulanmamalıdır. Doktorlar, tekrarlayan veya kronik enfeksiyon öyküsü bulunan veya hastayı ciddi enfeksiyonlara eğilimli hale getirecek şekilde altta yatan koşullara (örn. hipogamaglobulinemi) sahip hastalarda kullanırken dikkatli olmalıdırlar (Bkz. Bölüm 4.8). Rituximab tedavisine başlanmadan önce immünoglobulin düzeylerinin saptanması önerilir.
Rituximab tedavisi ardından enfeksiyon belirti ve semptomları bildiren hastalar dikkatli şekilde değerlendirilmeli ve uygun şekilde tedavi edilmelidir. Sonraki rituximab kürü uygulanmadan önce, hastalar potansiyel enfeksiyon riski açısından tekrar değerlendirilmelidir.
Sistemik Lupus Eritematozus (SLE) ile vaskülit dahil otoimmün hastalıkların tedavisi için rituximab kullanımı ardından çok seyrek fatal progresif multifokal lökoensefalopati (PML) vakaları bildirilmiştir.
Hepatit B Enfeksiyonları
Rituximab alan granülomatöz polianjiitis ve mikroskobik polianjiitis hastalarında, ölümle sonuçlananlar da dahil olmak üzere hepatit B reaktivasyonu vakaları bildirilmiştir.
Rituximab ile tedaviye başlanmadan önce bütün hastalar (sadece HBV enfeksiyon riski olanlar değil) Hepatit B virüsü (HBV) açısından taranmalıdır. Bu ölçümler en azından hepatit B yüzey antijeni (HBsAg)-durumu ve hepatit B çekirdek antikoru (HBcAb)-durumunu içermelidir. Bunlar yerel kılavuzlara göre diğer uygun markörler ile tamamlanmalıdır. Aktif hepatit B hastalığı olan hastalar rituximab ile tedavi edilmemelidir. Pozitif hepatit B serolojisi olan hastalar (HBsAg veya HBcAb) tedavi başlangıcından önce karaciğer hastalıkları uzmanına danışmalıdır ve bu hastalar hepatit B reaktivasyonunun önlenmesi için yerel medikal standartlara göre takip edilmeli ve yönetilmelidir.
Geç nötropeni
Rituximab ile tedaviye başlanmadan önce ve tedavinin sonlandırılması ardından 6 aya kadar düzenli olarak ve enfeksiyon belirti veya semptomlarının görülmesi durumunda, kan nötrofil ölçümü yapılmalıdır (Bkz. Bölüm 4.8).
Deri reaksiyonları
Toksik Epidermal Nekroliz (Lyell sendromu) ve Stevens-Johnson sendromu gibi bazılarının ölümcül sonuçları olabilen ciddi cilt reaksiyonları bildirilmiştir (Bkz. Bölüm 4.8). Bu gibi durumlarda olayın rituximab ile ilişkili olduğundan şüpheleniliyorsa tedavi kalıcı olarak durdurulmalıdır.
İmmünizasyon
Hekimler, rituximab tedavisinden önce hastanın aşılanma durumunu değerlendirmeli ve mevcut immünizasyon kılavuzlarını izlemelidir. Aşılama, ilk rituximab uygulamasından en az 4 hafta önce tamamlanmış olmalıdır.
Rituximab tedavisini takiben canlı viral aşılarla yapılan immünizasyonun güvenliliği üzerinde çalışma yapılmamıştır. Bu nedenle rituximab tedavisi sırasında veya periferik B hücre deplesyonu varken, canlı virüs aşılarıyla aşılama yapılması önerilmemektedir.
Rituximab ile tedavi edilen hastalar canlı olmayan aşılarla aşılanabilirler. Ancak canlı olmayan aşılara yanıt oranları düşebilir. Randomize bir çalışmada, rituximab ve metotreksat ile tedavi edilen RA hastaları ile yalnızca metotreksat alan hastalar rituximab kullanımından en az 6 ay sonra aşılandıklarında, tetanoz hatırlatıcı antijenine karşı benzer yanıt oranı (%39'a karşılık
%42), pnömokokkal polisakkarid aşısına (en az 2 pnömokokkal antikor serotipine karşı %43'e karşılık %82) karşı ve KLH neoantijenine (%47'ye karşılık %93) karşı ise azalmış yanıt oranı göstermişlerdir. Rituximab tedavisi sırasında canlı olmayan aşılama gerekli olursa, bunlar sonraki rituximab kürüne başlanmadan en az 4 hafta önce tamamlanmalıdır.
Malignite
İmmünomodülatör ilaçlar malignite riskini arttırabilir. Mevcut veriler otoimmün endikasyonlarda rituximab kullanılması sebebiyle, halihazırda altta yatan otoimmün hastalıkla ilişkili malignite riskinden daha fazla bir malignite riski olduğunu düşündürmemektedir. Ancak solid tümör gelişimine ilişkin olası risk göz ardı edilemez.
Yardımcı maddeler
TRUXIMA, flakon başına yaklaşık 2,3 mmol (52,6 mg) sodyum ihtiva eder. Sodyum miktarı 1 mmol'den (23 mg) fazladır. Bu durum, kontrollü sodyum diyetinde olan hastalar için göz önünde bulundurulmalıdır.
Biyobenzer ürünlerin takip edilebilirliği
Biyobenzer tıbbi ürünlerin takip edilebilirliğinin sağlanması için uygulanan ürünün ticari ismi ve seri numarası mutlaka hasta dosyasına kaydedilmelidir.
4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri
Rituximab ile olası ilaç etkileşimleri konusunda sınırlı veri vardır.
KLL hastalarında, rituximab ile kombine kullanımın fludarabin veya siklofosfamidin farmakokinetiği üzerine bir etkisinin olmadığı, bununla birlikte fludarabin veya siklofosfamidin de rituximab farmakokinetiği üzerine görünür bir etkisinin olmadığı görülmüştür.
İnsan anti-mürin antikoru (HAMA) veya anti-ilaç antikoru (ADA) titrelerine sahip hastalar tanı veya tedavi amacıyla başka monoklonal antikorlarla tedavi edildiklerinde alerjik reaksiyonlar veya aşırı duyarlılık reaksiyonları geliştirebilirler.
Özel popülasyonlara ilişkin ek bilgiler
Herhangi bir etkileşim çalışması yapılmamıştır.
4.6. Gebelik ve laktasyon
Gebelik kategorisi: C
Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (kontrasepsiyon)
B hücre deplesyonu olan hastalarda rituximabın uzun retansiyon süresi nedeniyle, üreme çağındaki kadınlar rituximab tedavisi sırasında ve bu tedaviyi takip eden 12 ay boyunca etkili doğum kontrol yöntemleri kullanmalıdır.
Gebelik dönemi
Rituximabın gebe kadınlarda kullanımına ilişkin yeterli veri mevcut değildir. Hayvanlar üzerinde yapılan çalışmalar üreme toksisitesinin bulunduğunu göstermiştir (Bkz. Bölüm 5.3). İnsanlara yönelik potansiyel risk bilinmemektedir.
IgG immünoglobulinlerinin plasenta engelini geçtiği bilinmektedir.
Anne vasıtasıyla rituximaba maruz kalan insan yenidoğanındaki B hücre seviyeleri klinik çalışmalarla araştırılmamıştır. Gebe kadınlarda yapılmış çalışmalarda yeterli ve kontrollü veri elde edilememiştir, ancak gebelik süresince anneleri rituximaba maruz kalmış olan bazı yenidoğanlarda geçici B hücre deplesyonu ve lenfositopeni bildirilmiştir. Benzer etkiler hayvan çalışmalarında da gözlenmiştir (Bkz. Bölüm 5.3). Bu sebeplerle rituximab, muhtemel faydalar potansiyel riskten fazla olmadığı sürece gebe kadınlarda uygulanmamalıdır.
Laktasyon dönemi
Rituximabın anne sütüyle atılmasına dair sınırlı veriler, anne sütünde çok düşük seviyede (rölatif infant dozu %0,4'den az) olduğunu düşündürmektedir. Takip edilen anne sütüyle beslenen az sayıdaki bebek 1,5 yaşına kadar normal büyüme ve gelişim göstermiştir. Ancak, bu veriler sınırlı olduğu için ve bebeklerin emzirilmesinin uzun dönem sonuçları bilinmediği için,
annelerin rituximab tedavisi sırasında ve rituximab tedavisinden sonraki 12 ay boyunca bebeklerini emzirmemeleri tavsiye edilir.
Üreme yeteneği/ Fertilite
Hayvan çalışmaları, rituximabın üreme organları üzerinde zararlı etkileri olduğunu göstermemiştir.
4.7. Araç ve makine kullanımı üzerindeki etkiler
Rituximabın araç veya makine kullanma becerisine etkisini belirleyecek çalışmalar yapılmamıştır, ancak farmakolojik aktivite ve bugüne kadar bildirilen yan etkiler rituximabın araç ve makine kullanımı üzerinde hiçbir etkisinin olmadığını ya da göz ardı edilebilir etkilerinin olduğunu düşündürmektedir.
4.8. İstenmeyen etkiler
Hodgkin-dışı lenfoma ve kronik lenfositik lösemi deneyimi Güvenlilik profilinin özeti
Hodgkin-dışı lenfoma ve kronik lenfositik lösemide rituximabın genel güvenlilik profili, klinik çalışmalarda ve pazarlama sonrası gözetimde yer alan hastalardan gelen verilere dayanmaktadır. Bu hastalar rituximab monoterapisiyle (indüksiyon tedavisi şeklinde veya indüksiyon tedavisini takiben idame tedavi şeklinde) veya kemoterapi ile kombinasyon halinde tedavi edilmiştir.
Rituximab alan hastalarda en sık gözlenen advers ilaç reaksiyonları (AİR'ler), hastaların çoğunluğunda ilk infüzyon sırasında oluşan infüzyonla ilişkili reaksiyonlar olmuştur. İnfüzyonla ilişkili semptomların insidansı, sonraki infüzyonlarla belirgin şekilde azalmıştır ve sekiz doz rituximabdan sonra %1'den düşük olmuştur.
Enfeksiyöz olaylar (ağırlıklı şekilde bakteriyel ve viral), yapılan klinik çalışmalar sırasında NHL olan hastalarda yaklaşık hastaların %30-55'inde ve KLL olan hastalarda hastaların %30- 50'sinde meydana gelmiştir.
En sık bildirilen veya gözlenen ciddi advers ilaç reaksiyonları:
İnfüzyonla ilişkili reaksiyonlar (IRR) (sitokin salıverilme sendromu, tümör lizis sendromu dahil), Bkz. Bölüm 4.4
4.9. Doz aşımı ve tedavisi
İntravenöz rituximab formülasyonunun onaylanmış dozundan daha yüksek dozlarla ilgili olarak insanlarda yapılan klinik çalışmalarda deneyim kısıtlıdır. Bugüne kadar insanlarda test edilen
en yüksek intravenöz rutiximab dozu kronik lenfositik lösemi (KLL) hastaları ile yürütülen doz artırma çalışmasında test edilen 5.000 mg'dır (2.250 mg/m). Hiçbir ek güvenlilik sinyali tespit edilmemiştir.
Doz aşımı görülen hastalarda infüzyon derhal kesilmelidir ve hastalar yakından izlenmelidir.
Pazarlama sonrası koşullarda rituximab doz aşımına ilişkin beş vaka bildirilmiştir. Üç vaka advers olay rapor etmemiştir. Bildirilen iki advers olay, 1,8 g'lık rituximab dozuyla raporlanan grip benzeri semptomlar ve 2 g'lık rituximab dozuyla raporlanan ölümcül solunum yetmezliğidir.
5. FARMAKOLOJİK ÖZELLİKLER
5.1. Farmakodinamik özellikler
Farmakoterapötik grubu: Antineoplastik ajanlar, monoklonal antikorlar ATC kodu: L01FA01
Bu ürün bir biyobenzer ilaçtır. Etki mekanizması
Rituximab spesifik olarak bir non-glikolize fosfoprotein olan CD20 adlı transmembran
antijenine bağlanan kimerik fare/insan monoklonal antikorudur. Bu antijen pre-B ve olgun B lenfositlerinde bulunur. Tüm B hücreli Hodgkin-dışı lenfomaların (NHL) >%95'inde bu antijen eksprese edilir.
CD20 hem normal hem de malignan B hücrelerinde bulunur ancak hematopoetik kök hücrelerde, pro-B hücrelerde, normal plazma hücrelerinde veya diğer normal dokuda bulunmaz. Antikora bağlandıktan sonra CD20 hücre içine alınmaz veya hücre membranından çevreye yayılmaz. CD20 plazmada serbest antijen olarak dolaşmaz ve dolayısıyla antikora bağlanmak için yarışmaz.
Rituximabın Fab alanı, B lenfositlerdeki CD20 antijenine bağlanır ve Fc alanı B hücresinin lizisine yol açan bağışıklığı etkileyen işlevleri başlatır. Etki-aracılı hücre yıkımındaki olası mekanizmalar C1q bağlanması sonucunda oluşan komplemana bağımlı sitotoksisite (CDC), granülositlerin, makrofajların ve NK hücrelerinin yüzeyindeki Fcγ reseptörlerinin bir veya daha fazlası aracılığıyla oluşan antikora bağımlı hücresel sitotoksisite (ADCC)'dir. B lenfositleri üzerindeki CD20 antijenine rituximab bağlanmasının apoptoz yoluyla hücre ölümünü indüklediği ortaya konmuştur.
Periferdeki B hücre sayısı ilk rituximab dozundan sonra normalin altına düşer. Hematolojik malignite tedavisi gören hastalarda B hücrelerinin normal düzeyine dönmesi tedavinin 6. Ayı içinde başlar, bazı hastalarda daha uzun sürse bile, genellikle tedavi tamamlandıktan sonraki 12 ay içinde de normal düzeylere döner (indüksiyon tedavisinden sonra medyan düzelme süresi 23 aya kadar uzayabilir). Romatoid artrit hastalarında, periferik kandaki B hücreleri sayısının hızla azalması 14 günlük arayla verilen 1.000 mg'lık iki rituximab infüzyonundan sonra gözlenmiştir. Rituximab monoterapi veya metotreksat ile beraber kombine olarak uygulandığında periferik kan B hücrelerinin sayısı 24. Haftadan sonra artmaya başlar ve hastaların büyük çoğunluğunda 40. Haftada eski sayıya döndüğünü gösteren işaretler gözlenmiştir. Hastaların küçük bir kısmında son rituximab dozundan sonraki 2 yıla kadar veya daha uzun süre devam eden periferik B hücre sayısı azalması olmuştur.
Granülomatöz polianjiitis ve mikroskobik polianjiitis hastalarında, iki haftalık 375 mg/m rituximab infüzyonu sonrasında periferik kan B hücreleri 10 hücre/µL'nin altına düşmüştür ve 6 ay boyunca hastaların çoğunda aynı seviyede kalmıştır. Hastaların büyük çoğunluğu (%81)
12. Ayda B hücrelerinin 10 hücre/µL seviyesinin üzerinde sayılarla tekrar artmaya başladığına dair işaretler göstermiştir, bu oran 18. Ay itibariyle hastaların %87'sine yükselmiştir.
Hodgkin-dışı lenfoma ve kronik lenfosittik lösemide klinik deneyim Foliküler lenfoma
Monoterapi
Başlangıç tedavisi, haftada bir uygulama, toplam 4 doz
Pivotal çalışmada, nüksetmiş veya kemorezistan düşük seviyeli ya da foliküler B hücreli NHL'ye sahip 166 hastaya haftada bir, toplam dört doz olarak i.v. infüzyon halinde 375 mg/m rituximab verilmiştir. Tedavi meyilli (ITT) popülasyonunda genel yanıt oranı (GYO) %48 (GA%41 - %56) olup tam yanıt (TY) oranı %6 ve kısmi yanıt (KY) oranı %42 olarak gerçekleşmiştir. Yanıt veren hastalarda hastalığın ilerlemesine kadar geçen medyan süre (TTP) 13 ay'dır. Yapılan bir alt-grup analizinde GYO, IWF B, C ve D histolojik alt-tiplerinde IWF A alt-tipine kıyasla daha yüksek (%58'e karşılık %12), en büyük lezyonu <5 cm olan hastalarda,
>7 cm olan hastalara kıyasla daha yüksek (%53'e karşılık %38) ve kemorezistan nüks grubu ile karşılaştırıldığında kemoduyarlı nüks hastalarında (yanıt süresi <3 ay olarak tanımlanır) daha yüksek (%22'ye karşılık %50) bulunmuştur. Önceden otolog kemik iliği transplantasyonu (OKİT) ile tedavi edilmiş hastalarda GYO %78 iken, OKİT tedavisi görmeyenlerde %43 olmuştur. Yaş, cinsiyet, lenfoma derecesi, başlangıçtaki tanı, büyük hacimli hastalık varlığı ya da yokluğu, normal ya da yüksek LDH düzeyleri veya ekstranodal hastalık varlığının rituximaba karşı verilen yanıt üzerinde istatistik olarak anlamlılık (Fischer'in exact testi) taşıyan bir etkisi bulunmamaktadır. Yanıt oranları ile kemik iliği tutulumu arasında istatistiksel olarak anlamlı bir bağıntı kaydedilmiştir. Kemik iliği tutulumu olan hastaların %40'ı yanıt verirken, kemik iliği tutulumu olmayan hastaların %59'u yanıt vermiştir (p=0,0186). Histolojik tip, başlangıçta bcl-2 pozitifliği, son kemoterapiye karşı direnç ve büyük hacimli hastalık faktörlerinin prognostik faktörler olarak tanımlandığı kademeli lojistik regresyon analizi bu bulguyu desteklememiştir.
Başlangıç tedavisi, haftada bir uygulama, toplam 8 doz
Çok merkezli, tek kollu bir çalışmada nüksetmiş veya kemorezistan, düşük dereceli veya foliküler B hücreli NHL'si olan 37 hastaya, toplam sekiz doz olacak şekilde, i.v. infüzyon yoluyla haftada bir kere 375 mg/m rituximab verilmiştir. GYO %57 (GA%41- %73; TY
%14, KY %43) ve medyan TTP 19,4 ay (5,3 ila 38,9 ay aralığında) bulunmuştur. Başlangıç tedavisi, büyük hacimli hastalık, haftada bir uygulama, toplam 4 doz
Üç çalışmanın bir araya getirilmiş verilerinde, nüksetmiş veya kemorezistan, büyük hacimli
hastalık (tek lezyon ≥ 10 cm çapında) özelliklerini taşıyan düşük dereceli veya foliküler B hücreli NHL'si olan 39 hastaya toplam dört doz, haftada bir kere 375 mg/m rituximab, i.v. infüzyon yoluyla verilmiştir. GYO %36 (GA%21-%51; TY %3, KY %33) ve yanıt veren hastalarda medyan TTP 9,6 ay olmuştur (4,5 ila 26,8 ay aralığında).
Tekrarlanan tedavi, haftada bir uygulama, toplam 4 doz
Çok merkezli, tek kollu bir çalışmada, önceki bir rituximab küründe objektif klinik yanıt alınan, nüksetmiş veya kemorezistan düşük evreli veya foliküler B hücreli NHL'si olan 58 hasta toplam dört doz, haftada bir kere, i.v. infüzyon yoluyla 375 mg/m rituximab ile yeniden tedavi edilmiştir. Hastaların üçü çalışmaya kaydolmadan önce iki seans rituximab tedavisi gördüğünden, bunlara çalışmada üçüncü bir seans uygulanmıştır. Çalışmada iki hasta, iki kez yeniden tedavi edilmiştir. Çalışmadaki 60 yeniden tedavi vakasında, yanıt veren hastalar için GYO %38 (GA%26-%51; TY %10, KY %28) ve yanıt veren hastalarda projekte edilen ortalama TTP, 17,8 ay (5,4 ila 26,6 ay aralığında) olmuştur. Bu değerler, önceki rituximab küründe elde edilen sonuçlar (12,4 ay) ile olumlu yönde karşılaştırılabilir niteliktedir.
Başlangıç tedavisi, kemoterapi ile kombinasyon halinde
Randomize, açık etiketli bir çalışmada, daha önce tedavi görmemiş foliküler lenfoması olan 322 hasta, 8 kür, her 3 haftada bir CVP kemoterapisi (siklofosfamid 750 mg/m, 1. Gün maksimum 2 mg doza kadar, vinkristin 1,4 mg/m ve 1-5. Gün arasında prednizolon 40 mg/m/gün) ya da CVP ile kombinasyon halinde rituximab 375 mg/m (R-CVP) alacak şekilde randomize edilmiştir. Rituximab her tedavi siklusunun ilk gününde uygulanmıştır. Toplam 321 hasta (162 R-CVP, 159 CVP) tedavi görmüş ve etkililik bakımından analiz edilmiştir. Hastaların medyan takip süresi 53 aydır. R-CVP, primer sonlanma noktası olan tedavi başarısızlığına kadar geçen süre açısından CVP'ye göre önemli bir üstünlük sağlamıştır (27 aya karşılık 6,6 ay, p < 0,0001, log-sıra testi). Tümör cevabı bulunan hastaların oranı (TY, TYo, KY), R-CVP grubunda (%80,9) CVP grubundan (%57,2) önemli oranda daha yüksek olmuştur (p< 0,0001, Ki-Kare testi). R-CVP ile yapılan tedavi, hastalık ilerlemesi veya ölüme kadar geçen süreyi belirgin bir şekilde artırmıştır (33,6 ay ve l4,7 ay) (p < 0,0001, log-sıra testi). R- CVP grubunda medyan yanıt süresi 37,7 ayken, CVP grubunda bu süre 13,5 ay olarak bulunmuştur (p < 0,0001, log-sıra testi).
Genel sağkalım açısından tedavi grupları arasındaki farklılık güçlü bir klinik yarar göstermiştir (p=0,029, katmanlı log-sıra testi): 53. Aydaki sağkalım oranları R-CVP grubunda %80,9 iken, CVP grubunda %71,1 'dir.
CVP dışındaki kemoterapi rejimleriyle (CHOP, MCP, CHVP/Interferon-α) yapılan diğer 3 randomize çalışmadan elde edilen sonuçlar yanıt oranlarında ve zamana bağlı parametrelerde olduğu gibi genel sağkalımda da belirgin iyileşmeler göstermiştir. Bu dört çalışmadan elde edilen anahtar sonuçlar aşağıda Tablo 6'da özetlenmektedir.
Tablo 6: Rituximabın foliküler lenfomada farklı kemoterapi rejimleriyle yararlarının değerlendirildiği dört faz III randomize çalışmadan elde edilen anahtar sonuçların özeti
Çalışma | Tedavi, N | Medyan Takip Süresi, ay |
GYO, % |
TY, % | Medyan TTF/PFS/ EFS ay | OS oranları, % |
M39021 |
CVP, 159 R-CVP, 162 |
53 |
57 81 |
10 41 | Medyan TTP: 14,7 33,6 p<0,0001 | 53 ay 71,1 80,9 p=0,029 |
GLSG'00 |
CHOP, 205 R-CHOP, 223 |
18
|
90 96 |
17 20 | Medyan TTF: 2,6 yıl Ulaşılamamıştır p< 0,001 | 18 ay 90 95 p = 0,016 |
OSHO-39 |
MCP, 96
R-MCP, 105 |
47 |
75
92 |
25 50 | Medyan PFS: 28,8 Ulaşılamamıştır p< 0,0001 | 48 ay 74 87 p = 0,0096 |
FL2000 | CHVP-IFN, 183
R-CHVP-IFN, 175 |
42 | 85
94 | 49 76 | Medyan EFS: 36 Ulaşılamamıştır p<0,0001 | 42 ay 84 91 p=0,029 |
GYO: Genel yanıt oranı TY: Tam yanıt
EFS: Olaysız sağkalım
TTP: Progresyona veya ölüme kadar geçen süre PFS: Progresyonsuz sağkalım
TTF: Tedavinin başarısızlığına kadar geçen süre
OS oranları: Analizler zamanında genel sağkalım oranları
İdame tedavisi
Daha önce tedavi uygulanmamış foliküler lenfoma
Prospektif, açık etiketli, uluslararası, çok merkezli bir faz III çalışmada daha önce tedavi uygulanmamış ileri seviye foliküler lenfoması olan 1.193 hastaya, araştırmacının tercihine göre R-CHOP (n=881), R-CVP (n=268) veya R-FCM (n=44) ile indüksiyon tedavisi uygulanmıştır. Toplam 1.078 hasta indüksiyon tedavisine yanıt vermiş, bu hastalardan 1.018'i rituximab idame tedavisine (n=505) veya gözlem grubuna (n=513) randomize edilmiştir. İki tedavi grubu, başlangıçtaki özellikler ve hastalık durumu açısından iyi dengelenmiştir. Rituximab idame tedavisi, hastalık progresyonuna kadar veya maksimum iki yıl boyunca 2 ayda bir 375 mg/m vücut yüzey alanı dozunda tek rituximab infüzyonundan oluşmuştur.
Randomizasyondan itibaren 25 aylık medyan gözlem süresinde yapılan, önceden belirlenmiş bir primer analiz sırasında rituximab idame tedavisi, önceden tedavi edilmemiş foliküler lenfoma hastalarında gözlem grubuna kıyasla, araştırmacı tarafından değerlendirilen progresyonsuz sağkalım (PFS) primer sonlanım noktasında klinik olarak tedaviyle alakalı ve istatistiksel olarak anlamlı iyileşme sağlamıştır (Tablo 7).
Primer analizde rituximab ile idame tedaviden elde edilen anlamlı yarar ayrıca sekonder
sonlanım noktaları olan olaysız sağkalım (EFS), bir sonraki anti-lenfoma tedavisine kadar geçen süre (TNLT), bir sonraki kemoterapiye kadar geçen süre (TNCT) ve genel yanıt oranında (GYO) da görülmüştür (Tablo 7).
Çalışmadaki hastaların uzun süreli takibinden (medyan takip 9 yıl) elde edilen veriler rituximabın idame tedavisinin PFS, EFS, TNLT ve TNCT açısından uzun süreli yararını teyit etmiştir (Tablo 7).
Tablo 7: Protokolde tanımlanmış primer analizlerde ve 9 yıllık medyan takip (sonuç analizi) sonrasında rituximabın idame tedavisinin etkililik sonuçlarının gözlem grubu ile karşılaştırılmasının genel özeti
| Primer analiz (medyan takip: 25 ay) | Son analiz (medyan takip: 9 yıl) | ||
| Gözlem N=513 | Rituximab N=505 | Gözlem N=513 | Rituximab N=505 |
Primer etkililik | ||||
Progresyonsuz sağkalım | NR NR | 4,06 yıl 10,49 yıl | ||
(medyan) |
|
| ||
log-sıra p değeri | <0,0001 | <0,0001 | ||
risk oranı (GA) | 0,50 (0,39, 0,64) | 0,61 (0,52, 0,73) | ||
risk azalması | %50 | %39 | ||
Sekonder etkililik | ||||
Genel sağkalım | NR NR | NR NR | ||
(medyan) |
|
| ||
log-sıra p değeri | 0,7246 | 0,7948 | ||
risk oranı (GA) | 0,89 (0,45, 1,74) | 1,04 (0,77, 1,40) | ||
risk azalması | %11 | %-6 | ||
Olaysız sağkalım | 38 ay NR | 4,04 yıl 9,25 yıl | ||
(medyan) |
|
| ||
log-sıra p değeri | <0,0001 | <0,0001 | ||
risk oranı (GA) | 0,54 (0,43, 0,69) | 0,64 (0,54, 0,76) | ||
risk azalması | %46 | %36 | ||
TNLT (medyan) | NR NR | 6,11 yıl NR | ||
log-sıra p değeri | 0,0003 | <0,0001 | ||
risk oranı (GA) | 0,61 (0,46, 0,80) | 0,66 (0,55, 0,78) | ||
risk azalması | %39 | %34 | ||
TNCT (medyan) | NR NR | 9,32 yıl NR | ||
log-sıra p değeri | 0,0011 | 0,0004 | ||
risk oranı (GA) | 0,60 (0,44, 0,82) | 0,71 (0,59, 0,86) | ||
risk azalması | %40 | %39 | ||
Genel Yanıt Oranı* | %55 %74 | %61 %79 | ||
Ki-kare testi p değeri | <0,0001 | <0,0001 | ||
Olasılık oranı (GA) | 2,33 (1,73, 3,15) | 2,43 (1,84, 3,22) | ||
Tam yanıt (TY/TYo) | %48 %67 | %53 %67 | ||
oranı* |
|
| ||
Ki-kare testi p değeri | <0,0001 | <0,0001 | ||
Olasılık oranı (GA) | 2,21 (1,65, 2,94) | 2,34 (1,80, 3,03) | ||
*İdame/gözlem sonunda; son analiz sonuçları 73 aylık medyan takip süresine dayanmaktadır. GA: güven aralığı; NR: klinik kesme tarihinde elde edilmemiş; TNCT: sonraki kemoterapiye kadar geçen süre; TNLT: sonraki anti-lenfoma tedavisine kadar geçen süre.
Rituximab idame tedavisi, önceden tanımlanmış tüm test edilen alt gruplarda tutarlı yarar sağlamıştır: cinsiyet (erkek, kadın), yaş (<60, ≥60), FLIPI skoru (≤1, 2 veya ≥3), indüksiyon tedavisi (R-CHOP, R-CVP veya R-FCM) ve indüksiyon tedavisine verilen yanıtın niteliğinden (TY/TYo veya KY) bağımsızdır. İdame tedavinin yararına ilişkin eksplatuar analizler, yaşlı hastalarda (>70 yaş) etkinin daha az belirgin olduğunu göstermiştir ancak örneklem sayısı azdır.
Relaps/refrakter foliküler lenfoma
Prospektif, açık etiketli, uluslararası, çok merkezli bir faz III çalışmada 465 relaps/refrakter foliküler lenfoma hastası, CHOP (siklofosfamid, doksorubisin, vinkristin, prednizolon; n=231) veya rituximab + CHOP (R-CHOP, n=234) ile yapılan indüksiyon tedavisine ilk basamakta randomize edilmiştir. İki tedavi grubu, başlangıç karakteristiklerine ve hastalık durumuna göre iyi dengelenmiştir. İndüksiyon tedavisinden sonra tam ya da kısmi remisyon sağlanan toplam 334 hasta, ikinci aşamada rituximab idame tedavisi (n=167) veya gözlem koluna (n= 167) randomize edilmiştir. Rituximab idame tedavisi, maksimum iki sene süresince ya da hastalık ilerleyene kadar, üç ayda bir 375 mg/m vücut yüzey alanı dozunda verilen tek rituximab uygulamasından ibarettir.
Son etkililik analizi, çalışmanın her iki bölümüne randomize edilen tüm hastaları içerir. İndüksiyon fazına randomize edilen hastaların 31 aylık medyan gözlem süresi sonunda, R- CHOP'un, CHOP ile kıyaslandığında relaps/refrakter foliküler lenfoma hastalarının klinik sonuçlarını belirgin olarak iyileştirdiği görülmüştür (Bkz. Tablo 8).
Tablo 8: İndüksiyon fazı: CHOP ile R-CHOP'un karşılaştırmalı etkililik sonuçlarına genel bakış (31 aylık medyan gözlem süresi)
| CHOP | R-CHOP | p değeri | Risk Azaltımı |
Primer Etkililik GYO TY KY |
%74 %16 %58 |
%87 %29 %58 |
0,0003 0,0005 0,9449 |
Yok Yok Yok |
Kısaltmalar: GYO: genel yanıt oranı; TY: tam yanıt; KY: kısmi yanıt
Çalışmanın idame fazına randomize edilen hastalar için medyan gözlem süresi, idame randomizasyonundan itibaren 28 aydır. Rituximab ile idame tedavisi, sadece gözlem koluna kıyasla, primer sonlanma noktası olan PFS'de (idame randomizasyonundan nükse, hastalık ilerlemesine ya da ölüme kadar olan süre) klinik olarak anlamlı ve istatistiksel olarak belirgin düzelme ile sonuçlanmıştır (p<0,000l, log-sıra testi). Medyan PFS, rituximab idame kolunda 42,2 ayken gözlem kolunda 14,3 aydır. Cox regresyon analizi kullanıldığında, hastalık ilerlemesi ya da ölüm riski, rituximab idame tedavisi ile gözleme göre %61 oranında azalmıştır (GA; %45-%72). 12 ayda Kaplan-Meier yöntemiyle hesaplanan progresyonsuz oranlar, rituximab idame grubunda %78 iken gözlem grubunda %57'dir. Genel sağkalım analizi, rituximab idamesinin, gözleme göre belirgin fayda sağladığını kanıtlamıştır (p=0,0039 log-sıra testi). Rituximab idame tedavisi, ölüm riskini %56 azaltmıştır (GA; %22-%75).
Tablo 9: İdame fazı: Rituximab ile gözlem gruplarının karşılaştırmalı etkililik sonuçlarına genel bakış (28 aylık medyan gözlem süresi)
Etkililik Parametresi | Olaya Kadar Medyan Sürenin (ay) Kaplan-Meier Yöntemiyle Hesaplanması |
Risk Azaltımı | ||
Gözlem (N = 167) | Rituximab (N=167) | Log-sıra p değeri | ||
Progresyonsuz sağkalım (PFS) | 14,3 | 42,2 | <0,0001 | %61 |
Genel sağkalım (OS) | NR | NR | 0,0039 | %56 |
Yeni lenfoma tedavisine kadar geçen süre | 20,1 | 38,8 | <0,0001 | %50 |
Hastalıksız sağkalım | 16,5 | 53,7 | 0,0003 | %67 |
Alt Grup Analizi |
|
|
|
|
PFS |
|
|
|
|
CHOP | 11,6 | 37,5 | <0,0001 | %71 |
R-CHOP | 22,1 | 51,9 | 0,0071 | %46 |
TY | 14,3 | 52,8 | 0,0008 | %64 |
KY | 14,3 | 37,8 | <0,0001 | %54 |
OS |
|
|
|
|
CHOP | NR | NR | 0,0348 | %55 |
R-CHOP | NR | NR | 0,0482 | %56 |
NR: ulaşılamamıştır; : sadece TY'ye ulaşan hastalar için
Rituximab idame tedavisinin faydası, indüksiyon rejimi (CHOP ya da R-CHOP) ya da indüksiyon tedavisine verilen yanıtların niteliğiyle (TY ya da KY) ilgili olmaksızın tüm alt gruplarda analiz edilmiştir (Tablo 9). Rituximab idame tedavisi, CHOP indüksiyon tedavisine yanıt veren hastalarda (medyan PFS 37,5 aya karşılık 11,6 ay, p<0,0001) olduğu kadar R-CHOP indüksiyon tedavisine yanıt veren hastalarda da (medyan PFS 51,9 aya karşılık 22,1 ay, p=0,0071) medyan PFS'yi önemli ölçüde uzatmıştır. Alt gruplar küçük olsa da, rituximab idame tedavisi genel sağkalım açısından hem CHOP'a yanıt veren hastalarda hem de R-CHOP'a yanıt veren hastalarda klinik açıdan anlamlı fayda sağlamıştır, bu gözlemi doğrulamak için daha uzun süreli takip gereklidir.
Yetişkinlerde diffüz büyük B hücreli Hodgkin-dışı lenfoma (DBBHL)
Randomize, açık etiketli bir çalışmada, diffüz büyük B hücreli lenfoması olan önceden tedavi görmemiş, yaşları 60 ile 80 arası değişen 399 yaşlı hastaya, sekiz siklus boyunca her üç haftada bir standart CHOP kemoterapisi (1. Günde siklofosfamid 750 mg/m, doksorubisin 50 mg/m,
1. Gün maksimum 2 mg'a kadar, vinkristin 1,4 mg/m ve 1-5. Günlerde prednizolon 40 mg/m/gün) veya 375 mg/m rituximab + CHOP (R-CHOP) verilmiştir. Rituximab tedavi siklusunun birinci gününde uygulanmıştır.
Nihai etkililik analizi randomize edilen tüm hastaları (197 CHOP, 202 R-CHOP) kapsamıştır ve ortalama izleme süresi yaklaşık 31 aydır. İki tedavi grubu, başlangıç düzeyi özellikleri ve hastalık durumu bakımından iyi dengelenmiştir. Nihai analiz, R-CHOP tedavisinin olaysız geçen sağkalım süresini (primer etkililik parametresi, buradaki olaylar ölüm, nüks veya lenfoma
ilerlemesi ya da yeni bir anti-lenfoma tedavisinin tesis edilmesidir) önemli oranda uzattığını doğrulamıştır (p=0,0001). Medyan olaysız sağkalım süresine ilişkin Kaplan-Meier tahminlerine göre, CHOP kolundaki 13 ay ile, R-CHOP kolunda 35 ayın karşılaştırılması riskin %41 azaldığını göstermektedir. 24. Ayda, genel sağkalıma ilişkin tahminler CHOP kolundaki
%57,4'lük orana kıyasla R-CHOP kolunda %68,2 olarak bulunmuştur. Medyan 60 aylık izleme süresi ile gerçekleştirilen daha sonraki bir genel sağkalım süresi analizi, R-CHOP tedavisinin CHOP tedavisinden daha yararlı olduğunu doğrulamış (p=0,0071) ve riskin %32 azaldığını göstermiştir.
Tüm sekonder parametrelerin analizi (yanıt oranları, progresyonsuz sağkalım, hastalıksız sağkalım, yanıt süresi), CHOP ile karşılaştırıldığında R-CHOP tedavisinin etkisini doğrulamıştır. 8. Siklustan sonra tam yanıt oranı, R-CHOP grubunda %76,2 ve CHOP grubunda
%62,4 bulunmuştur (p=0,0028). Hastalığın ilerleme riski %46 ve nüks riski %51 oranında azaltılmıştır.
Tüm hasta alt gruplarında (cinsiyet, yaş, yaşa göre ayarlanmış IPI, Ann Arbor evresi, ECOG, β2 Mikroglobulin, LDH, albümin, B semptomları, büyük hacimli hastalık, ekstranodal bölgeler, kemik iliği tutulumu), olaysız sağkalım ve genel sağkalıma ilişkin risk oranları (R-CHOP'a karşılık CHOP) sırasıyla 0,83 ve 0,95'den daha az bulunmuştur. Yaşa göre ayarlanmış IPI'ye göre R-CHOP hem yüksek hem de düşük risk taşıyan hastalarda, sonuçta ulaşılan iyileşme düzeyiyle ilişkili bulunmuştur.
Klinik laboratuvar bulguları
İnsan anti-fare antikoru (HAMA) açısından değerlendirilen 67 hastanın hiçbiri için yanıt bildirilmemiştir. İlaca karşı antikor (ADA) açısından değerlendirilen 356 hastanın %1,1'i (4 hasta) pozitif çıkmıştır.
Kronik lenfositik lösemi
Açık etiketli randomize iki çalışmada, daha önce tedavi görmemiş toplam 817 KLL hastası ve 552 relaps/refrakter KLL hastası, 6 siklus için 4 haftada bir FC kemoterapi (fludarabin 25 mg/m, siklofosfamid 250 mg/m, 1-3. Günler) veya FC ile kombinasyon halinde rituximab (R- FC) alacak şekilde randomize edilmiştir. Rituximab, ilk siklus sırasında kemoterapiden bir gün önce 375 mg/m dozunda ve sonraki her tedavi siklusunun 1. Gününde 500 mg/m dozunda uygulanmıştır. Relaps/refrakter KLL'de önceden monoklonal antikorlar ile tedavi edilmiş veya fludarabin ya da herhangi bir nükleozid analoğuna refrakter olan hastalar (en az 6 ay için kısmi remisyon gösterememe başarısızlığı olarak tanımlanmıştır) çalışmaya dahil edilmemiştir. Etkililik için birinci basamak çalışmasında (Tablo 10a) ve (Tablo 10b) toplam 810 hasta (403 R-FC, 407 FC), relaps/refrakter çalışmasında da (Tablo 11) 552 hasta (276 R-FC, 276 FC) analiz edilmiştir.
Birinci basamak çalışmasında 48,1 aylık medyan gözlem süresinden sonra medyan PFS, R- FC grubunda 55 ay ve FC grubunda 33 ay olmuştur (p<0,0001, log-sıra testi). Genel sağkalım analizi, yalnızca FC kemoterapisi kullanılan kola göre, R-FC kolu için anlamlı bir fayda göstermiştir (p=0,0319, log-sıra testi) (Tablo 10a). PFS açısından fayda, başlangıçtaki hastalık riskine göre (yani Binet A-C evreleri) (Tablo 10b) analiz edilen hasta alt gruplarının çoğunda tutarlı olarak gözlenmiştir.
Tablo 10a: Kronik lenfositik löseminin birinci basamak tedavisi
Tek başına FC'ye kıyasla rituximab+FC için etkililik sonuçlarına genel bakış – 48,1 aylık medyan gözlem süresi
Etkililik parametresi | Olaya kadar geçen medyan süre için Kaplan-Meier tahmini (Ay) | Risk azaltımı | ||
FC (N = 409) | R-FC (N=408) | Log-sıra p değeri | ||
Progresyonsuz sağkalım (PFS) | 32,8 | 55,3 | <0,0001 | %45 |
Genel sağkalım | NR | NR | 0,0319 | %27 |
Olaysız sağkalım | 31,3 | 51,8 | <0,0001 | %44 |
Yanıt oranı (TY, nKY veya KY) TY oranları | %72,6
%16,9 | %85,8
%36 | <0,0001
<0,0001 | n.a.
n.a. |
Yanıt süresi* | 36,2 | 57,3 | <0,0001 | %44 |
Hastalıksız sağkalım (DFS)** |
48,9 |
60,3 |
0,0520 |
%31 |
Yeni tedaviye kadar geçen süre | 47,2 | 69,7 | <0,0001 | %42 |
Yanıt oranı ve TY oranları Ki-kare Testi kullanılarak analiz edilmiştir. NR: ulaşılmadı; n.a.: geçerli değildir.
*: Yalnızca TY, nKY veya KY elde edilen hastalar için geçerlidir.
**: Yalnızca TY elde edilen hastalar için geçerlidir.
Tablo 10b: Kronik lenfositik löseminin birinci basamak tedavisi
Binet evresine göre (ITT) progresyonsuz sağkalım tehlike oranı - medyan gözlem süresi 48,1 ay
Progresyonsuz sağkalım (PFS) | Hasta sayısı | Tehlike Oranı (%95 GA) | p-değeri (Wald testi, ayarlanmamış) | |
FC | R-FC | |||
Binet evre A | 22 | 18 | 0,39 (0,15; 0,98) | 0,0442 |
Binet evre B | 259 | 263 | 0,52 (0,41; 0,66) | <0,0001 |
Binet evre C | 126 | 126 | 0,68 (0,49; 0,95) | 0,0224 |
GA: Güven aralığı
Relaps/refrakter çalışmasında, R-FC grubunda medyan progresyonsuz sağkalım (primer sonlanım noktası) 30,6 ay iken FC grubunda 20,6 aydır (p=0,0002, log-sıra testi). PFS açısından fayda, başlangıçtaki hastalık riskine göre analiz edilen hasta alt gruplarının neredeyse hepsinde gözlenmiştir. R-FC kolunda FC koluna kıyasla, genel sağkalımda (OS) az fakat anlamlı olmayan bir artış bildirilmiştir.
Tablo 11: Relaps/refrakter kronik lenfositik löseminin tedavisi - Tek başına FC'ye kıyasla rituximab-FC için etkililik sonuçlarına genel bakış (medyan gözlem süresi 25,3 ay)
Etkililik Parametresi | Olaya Kadar Geçen Medyan Süre için Kaplan-Meier Tahmini (Ay) | Risk azaltımı | ||
FC (N = 276) | R-FC (N=276) | Log-sıra p değeri | ||
Progresyonsuz sağkalım (PFS) | 20,6 | 30,6 | 0,0002 | %35 |
Genel sağkalım | 51,9 | ulaşılamadı | 0,2874 | %17 |
Olaysız sağkalım | 19,3 | 28,7 | 0,0002 | %36 |
Yanıt oranı (TY, nKY veya KY) | %58,0 | %69,9 | 0,0034 | uygulanabilir değil |
TY oranları | %13,0 | %24,3 | 0,0007 | uygulanabilir değil |
Yanıt süresi* | 27,6 | 39,6 | 0,0252 | %31 |
Hastalıksız sağkalım (DFS)** | 42,2 | 39,6 | 0,8842 | %-6 |
Yeni KLL tedavisine kadar geçen süre | 34,2 | ulaşılamadı | 0,0024 | %35 |
Yanıt oranı ve TY oranları Ki-kare Testi kullanılarak analiz edilmiştir.
*: Yalnızca TY, nKY, KY elde edilen hastalar için geçerlidir.
**: Yalnızca TY elde edilen hastalar için geçerlidir.
Önceden tedavi edilmemiş ve/veya relaps/refrakter KLL hastalarının tedavisinde diğer kemoterapi rejimleriyle (CHOP, FCM, PC, PCM, bendamustin ve kladribin dahil) kombinasyon halinde rituximab kullanılan diğer destekleyici çalışmalardan elde edilen sonuçlar, hafif yükselmiş toksisiteye (özellikle miyelotoksisite) rağmen, PFS oranları açısından faydalı yüksek genel yanıt oranları ortaya koymuştur. Bu çalışmalar rituximabın herhangi bir kemoterapi ile kullanımını desteklemektedir.
Daha önce rituximab ile tedavi edilmiş yaklaşık 180 hastaya ait veriler klinik faydayı ortaya koymuştur (TY dahil) ve bu veriler rituximab ile yeniden tedaviyi destekler niteliktedir.
Pediyatrik popülasyon
Daha önce tedavi edilmemiş ileri evre CD20 pozitif DLBCL/BL/BAL/BLL'si olan pediyatrik hastalarda rituximab ile beraber veya rutiximab olmaksızın Lenfoma Malign B (LMB) kemoterapi (kortikosteroidler, vinkristin, siklofosfamid, yüksek-doz metotreksat, sitarabin, doksorubisin, etoposid ve üçlü ilaç [metotreksat/sitarabin/kortikosteroid] intratekal terapi) için çok merkezli, açık-etiketli, randomize bir çalışma yapılmıştır. İleri evre, yükselmiş LDH seviyesi (“B-yüksekâ€) [LDH > yetişkin normal değerlerinin geleneksel üst sınırının iki katı (> Nx2)] ile beraber Evre III ya da herhangi bir Evre IV veya BAL olarak tanımlanmıştır. Hastalar LMB kemoterapi veya LMB şemasına göre LMB kemoterapisi ile kombine altı i.v. rituximab infüzyonunu (iki indüksiyon kürünün her birinde iki tane ve iki konsolidasyon kürünün her
birinde bir tane) 375 mg/m BSA dozunda almak üzere randomize edilmiştir. Toplamda randomize edilmiş 328 hasta etkililik analizlerine dahil edilmiştir, bu hastalar içinde LMB kemoterapi ile kombine rituximab alan bir hasta 3 yaşın altındaydı.
İki tedavi kolu, LMB (LMB kemoterapi) ve R-LMB (rituximab ile LMB kemoterapi), başlangıç karakteristikleri açısından iyi dengelenmişti. Hastaların medyan yaşı LMB kolunda ve R-LMB kolunda sırasıyla 7 ve 8 yıldı. Hastaların yaklaşık yarısı (LMB kolunda %50,6 ve R-LMB kolunda %49,4) Grup B'deydi, her iki kolda %39,6'sı Grup C1'deydi ve LMB ve R-LMB kolları için sırasıyla %9,8'i ve %11'i Grup C3'deydi. Murphy evrelemesine göre, hastaların çoğu ya BL evre III (LMB kolunda %45,7 ve R-LMB kolunda %43,3) ya da BAL, SSS negatif (LMB kolunda %21,3 ve R-LMB kolunda %24,4)'ti. Hastaların yarısından azında (her iki kolda da %45,1) kemik iliği tutulumu vardı ve hastaların çoğunda (LMB kolunda %72,6 ve R-LMB kolunda %73,2) SSS tutulumu yoktu. Primer etkililik sonlanım noktası Olaysız Sağkalım'dı (EFS), burada olay, hangisi önce meydana gelirse gelsin, hastalık ilerlemesi, relaps, ikinci malignite, herhangi bir sebepten ölüm veya ikinci CYVE küründen sonra canlı hücre kaldığının saptanması ile doğrulanan tedaviye yanıt-olmaması durumlarının meydana gelmesi olarak tanımlanmıştı. Sekonder etkililik sonlanım noktaları ise Genel Sağkalım ve Tam Remisyon olmuştur.
Yaklaşık 1 yıllık medyan takip süresinde, önceden tanımlanmış ara analizlerde, EFS primer sonlanım noktasında klinik olarak alakalı düzelmeler görülmüştür; 1-yıllık oran tahminleri R- LMB kolunda %94,2 (%95 GA, %88,5 - %97,2) olurken LMB kolunda %81,5 (%95 GA, %73,0
- %87,8) ve ayarlanmış Cox Risk Regresyonu (HR) 0,33 (%95GA, 0,14 – 0,79) olmuştur. Bağımsız Veri İzleme Komisyonu'nun bu sonuca dayanan tavsiyesi üzerine, randomizasyon durdurulmuş ve LMB kolundaki hastaların rituximab almak üzere geçiş yapmalarına izin verilmiştir.
Randomize edilmiş 328 hastada primer etkililik analizleri yapılmıştır, medyan takip süresi 3,1 yıldır. Sonuçlar Tablo 12'de açıklanmaktadır.
Tablo 12: Primer Etkililik Sonuçlarına Genel Bakış (ITT Popülasyonu)
Analiz | LMB (N = 164) | R-LMB (N = 164) |
Olaysız Sağkalım (EFS) | 28 olay | 10 olay |
Tek-yönlü log-sıra test p-değeri 0,0006 | ||
Ayarlanmış Cox HR 0,32 (%90 GA: 0,17; 0,58) | ||
3-yıl EFS oranları | %82,3 (%95 GA: %75,7; %87,5) | %93,9 (%95 GA: %89,1; %96,7) |
Genel Sağkalım (OS) | 20 ölüm | 8 ölüm |
Tek-yönlü log-sıra test p-değeri 0,0061 | ||
Ayarlanmış Cox HR 0,36 (%95 GA: 0,16; 0,81) | ||
3-yıl OS oranları | %87,3 (%95 GA: %81,2; %91,6) | %95,1 (%95 GA: %90,5; %97,5) |
Tam Remisyon | %93,6 (%95 GA: %88,2; %97,0) | %94,0 (%95 GA: %88,8; %97,2) |
Primer etkililik analizi, rituximab ilave edilmiş LMB kemoterapinin tek başına LMB kemoterapiye göre EFS avantajını göstermiştir; ulusal grup, histoloji ve terapötik grup için ayarlanmış Cox regresyon analizinden elde edilen EFS HR 0,32 (%90 GA: 0,17 – 0,58) olmuştur. İki tedavi grubunda Tam Remisyon elde eden hastaların sayısında büyük bir fark olmasada, LMB kemoterapiye rituximab ilavesinin sekonder sonlanım noktası olan Genel Sağkalım üzerine faydası da gösterilmiştir; OS HR 0,36 (%95 GA: 0,16 – 0,81) olmuştur.
Pediyatrik kullanım bilgisi için Bkz. Bölüm 4.2.
Granülamatöz polianjiitis (Wegener's) (GPA) ve mikroskobik polianjiitis (MPA)'de klinik deneyim
Yetişkin remisyon indüksiyonu
GPA/MPA klinik çalışmasına (çalışma 1), ciddi aktif GPA (%75) ve MPA'sı (%24) olan 15 yaş ve üzeri toplam 197 hasta dahil edilerek aktif karşılaştırmalı, randomize, çift-kör, çok merkezli, non-inferior bir çalışmada tedavi edilmiştir.
Hastalar 3-6 ay boyunca her gün oral siklofosfamid (2 mg/kg/gün) veya 4 hafta boyunca haftada bir rituximab (375 mg/m) almak üzere 1:1 oranında randomize edilmişlerdi. Siklofosfamid kolundaki tüm hastalar takip süresince azatioprin idame tedavisi almıştı. Her iki koldaki hastalar 1 ila 3 gün boyunca günde 1.000 mg pulse intravenöz (i.v.) metilprednizolon (veya eşdeğer-dozda başka bir glukokortikoid) ve ardından oral prednizon (l mg/kg/gün, en fazla 80 mg/gün) almıştı. Prednizon azaltımı çalışma tedavisinin başlamasından itibaren 6 ayda tamamlanmalıydı.
Primer sonuç ölçütü 6. Ayda tam remisyon sağlanmasıydı ve bu da Wegener Granülomatozisi için Birmingham Vaskülit Aktivite Skoru'nun (BVAS/WG) “0†olması ve glukokortikoid kullanımının bırakılması olarak tanımlanmaktaydı. Tedavi farkı için önceden belirlenen non- inferiorite marjini % 20'ydi. Bu çalışma, 6. Ayda tam remisyon (CR) açısından siklofosfamide karşı rituximabın non-interferiotesini göstermişti (Tablo 13).
Etkililik hem GPA ve MPA teşhisi yeni konan hastalarda hem de nükseden hastalığı olan hastalarda gözlenmiştir (Tablo 14).
Tablo 13: 6. Ayda tam remisyona erişen hastaların yüzdesi (tedavi meyilli popülasyon*)
| Rituximab (n = 99) | Siklofosfamid (n = 98) | Tedavi farkı (Rituximab – Siklofosfamid) |
Oran |
%63,6 |
%53,1 | %10,6 %95,1 GA (%-3,2, % 24,3) |
GA = güven aralığı.
* En kötü durum modeli
| Rituximab | Siklofosfamid | Fark (%95 GA) |
Tüm hastalar | n=99 | n=98 |
|
Yeni teşhis koyulmuş hastalar |
n=48 |
n=48 |
|
Relaps görülen hastalar |
n=51 |
n=50 |
|
Tam remisyon | |||
Tüm hastalar | %63,6 | %53,1 | %10,6 (-3,2, 24,3) |
Yeni teşhis | %60,4 | %64,6 | %-4,2 |
koyulmuş |
|
| (-23,6, 15,3) |
Relaps görülen | %66,7 | %42,0 | %24,7 |
hastalar |
|
| (5,8, 43,6) |
Tablo 14: Hastalık durumuna göre 6 aydaki tam remisyon
![]()
Verileri eksik olan hastalar için en kötü durum modeli geçerlidir. 12 ve 18. Aylarda tam remisyon (CR)
Rituximab grubunda hastaların %48'i 12 ayda ve %39'u 18 ayda CR'ye ulaşmıştır. Siklofosfamid (ve ardından tam remisyonun idamesi için azatioprin) ile tedavi edilen hastalarda, hastaların %39'u 12 ayda ve %33'ü 18 ayda CR'ye ulaşmıştır. 12. Aydan 18. Aya kadar rituximab grubunda 8 relaps görülürken siklofosfamid grubunda dört relaps görülmüştür.
Laboratuvar değerlendirmeleri
Remisyon tedavisinin indüksiyonu çalışmasında rituximab ile tedavi edilen toplam 99 hastanın 23'ü (%23) 18 aya kadar ADA açısından pozitif bulunmuştur. Rituximab ile tedavi edilen 99 hastanın hiçbiri çalışma başlangıcındaki taramada ADA açısından pozitif değildir. Remisyon tedavisinin indüksiyonu çalışmasında ADA varlığının güvenlilik veya etkililik üzerinde belirgin bir eğilimi veya olumsuz bir etkisi olmamıştır.
Pemfigus vulgaris'te klinik deneyim PV Çalışması 1 (Çalışma ML22196)
Rituximabın kısa-süreli, düşük-dozlu glukokortikoid (prednizon) tedavisi ile kombine kullanımının etkililik ve güvenliliği, yeni tanı konmuş, orta ila şiddetli pemfigusu (74 pemfigus vulgaris [PV] ve 16 pemfigus foliaseus [PF]) olan hastalarda yapılan bu randomize, açık- etiketli, kontrollü, çok-merkezli çalışmada değerlendirilmiştir. Hastalar 19 ve 79 yaş aralığındaydı ve daha önce pemfigus için tedavi görmemişlerdi. PV popülasyonunda, Harman kriterleri tarafından tanımlanmış hastalık şiddetine göre rituximab grubunda 5 hastanın (%13) ve standart prednizon grubunda 3 hastanın (%8) orta şiddette hastalığı ve 33 hastanın (%87) ve standart prednizon grubunda 33 hastanın (%92) şiddetli hastalığı vardı.
Hastalar başlangıçta hastalık şiddetine (orta ila şiddetli) gruplandırılmışlardı ve rituximab ve düşük-doz prednizon ya da standart doz prednizon alacak şekilde 1:1 oranında randomize edilmişlerdi. Rituximab grubuna randomize edilen hastalar Çalışmanın 1. Günü 1.000 mg'lık ilk rituximab intravenöz infüzyonu ile birlikte orta şiddette hastalığı olanlar 3 ay içerisinde azaltılarak kesilecek olan 0,5 mg/kg/gün oral prednizon, ya da hastalığı şiddetli olanlar 6 ay içerisinde azaltılarak kesilecek olan 1 mg/kg/gün oral prednizon almışlardır. Çalışmanın 15.
Günü ikinci bir 1.000 mg'lık intravenöz infüzyon uygulanmıştır. Rituximabın idame dozları 12. ve 18. Aylarda 500 mg dozunda uygulanmıştır. Standart-doz prednizon grubuna randomize edilen orta şiddette hastalığı olan hastalar başlangıçta 1 mg/kg/gün oral prednizonu 12 ay içerisinde azaltılarak kesilmek üzere ya da şiddetli hastalığı olan hastalar 1,5 mg/kg/gün oral prednizonu 18 ay içerisinde azaltılarak kesilmek üzere almışlardır. Rituximab grubunda relaps yaşayan hastalar ilave bir 1.000 mg rituximab infüzyonu ile beraber yeniden prednizolon almaya başlayabilmişlerdir ya da birlikte kullanılan prednizon dozu arttırılabilmiştir. İdame ya da relaps infüzyonları bir önceki infüzyonun ardından en az 16 hafta sonra uygulanmıştır.
Çalışmanın primer objektifi en az iki ay süresince prednizon tedavisi kullanmadan (CRoff ≥2 ay) 24. Ayda tam remisyondu (tam epitelizasyon ve yeni ve/veya önceden mevcut lezyonların olmaması).
PV Çalışma 1 sonuçları
Çalışma rituximab ve düşük-doz prednizonun standart doz prednizona göre, PV hastalarında
24. Ayda CRoff ≥2 ay hedefinin başarılmasında istatistiksel olarak anlamlı sonuçlar göstermiştir (Bkz. Tablo 14).
Tablo 14 24. Ayda kortikoid tedavisi olmadan en az 2 ay süresince tam remisyonu başarmış PV hastalarının yüzdesi (Tedavi Meyilli Popülasyon – PV)
| Rituximab + Prednizon N = 38 | Prednizon N = 36 | p-değeri | %95 GA |
Yanıt verenlerin sayısı | 34 (%89,5) | 10 (%27,8) | <0,0001 | %61,7 (38,4, 76,5) |
(yanıt oranı [%]) |
|
|
|
|
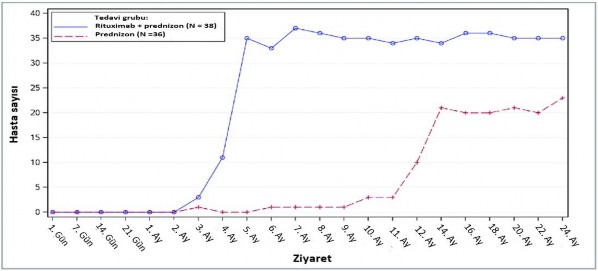
24 ay süren tedavi periyodu boyunca, rituximab artı düşük-doz prednizon hastalarında prednizon tedavisini bırakmış olan ya da minimal tedavi alan (günlük prednizon dozu 10 mg veya daha az) hastaların sayısının standart doz prednizon hastalarının sayısı ile kıyaslanması rituximabın steroid-azaltımı etkisini göstermiştir (Şekil 1).
Şekil 1: Zaman içerisinde kortikosteroidi bırakan ya da minimal kullanan (≤10 mg/gün) hastaların sayısı
Post-hoc geriye dönük laboratuvar değerlendirmesi
Rituximab ile tedavi edilen PV hastalarının toplam 19/37'si (%56) 18. aya kadar ADA antikorları açısından pozitif sonuç vermişlerdir. Rituximab ile tedavi edilen PV hastalarında ADA oluşumunun klinik ilişkisi net değildir.
PV Çalışması 2 (Çalışma WA29330)
Randomize, çift-kör, çift-sağır maskelenmiş, aktif- kontrollü, çok-merkezli bir çalışmada, orta ila şiddetli PV hastalığı olan ve çalışmaya katılırken 60-120 mg/gün dozunda oral prednizon veya eşdeğerini (1,0 – 1,5 mg/kg/gün) kullanan ve zaman içerisinde azaltılarak 1. Günde 60 veya 80 mg/gün dozuna ulaşan hastalarda, rituximabın etkililik ve güvenliliği mikofenolat mofetil (MMF) ile kıyaslanmıştır. Hastaların geçmiş 24 ay içerisinde PV teşhisi aldığı ve hastalıklarının orta ila şiddetli olduğuna dair kanıtlar (Pemfigus Hastalığı Alan Indeksi, PDAI, aktivite skoru ≥ 15) teyit edilmiştir.
135 hasta 24. Haftada prednizon dozunun 0 mg/güne doğru azaltılarak kesilmesi amacıyla, 60 veya 80 mg oral prednison ile kombine olarak 1. Gün, 15. Gün, 24. Hafta ve 26. Hafta'da uygulanacak 1.000 mg rituximab ile tedaviye ya da 52 hafta boyunca 2 g/gün dozunda oral MMF ile tedaviye randomize edilmiştir.
Bu çalışmanın primer etkililik amacı 52. Haftada 0 mg/gün dozunda prednizon veya eşdeğerini kullanırken hiç yeni aktif lezyon olmadan lezyonların iyileşmesinin sağlanması (örn. PDAI aktivite skoru 0) ve 52 haftalık tedavi periyodunda, bu yanıtın ardarda en az 16 hafta boyunca devam ettirilmesi olarak tarif edilen devamlı tam remisyonun elde edilmesinde rituximabın etkililiğinin MMF ile karşılaştırmasını değerlendirmekti.
PV Çalışması 2 sonuçları
Bu çalışma PV hastalarında 52. Haftada CRoff kortikosteroid ≥16 hafta hedefine ulaşılmasında kademeli olarak azaltılarak bırakılan oral kortikosteroid kürü ile beraber kullanılan rituximabın MMF'ye üstünlüğünü göstermiştir (Tablo 15). mITT popülasyonundaki hastaların çoğunluğu yeni teşhis konmuş hastalardı (%74) ve hastaların %26'sının hastalığı önceden mevcuttu (hastalığın süresi ≥6 ay ve PV için daha önce tedavi almışlardı).
Tablo 15 52. Haftada 16 Hafta veya Daha Uzun Süre Boyunca Kortikosteroid Tedavisi Almadan Devamlı Tam Remisyonda Kalan PV Hastalarının Yüzdesi
| Rituximab (N=62) | MMF (N=63) | Fark (%95 GA) | p-değeri |
Yanıt verenlerin sayısı (yanıt oranı [%]) Yeni teşhis konmuş hastalar Hastalığı önceden mevcut hastalar | 25 (%40,3) | 6 (%9,5) | %30,80 (%14,70, %45,15) | <0,0001 |
19 (%39,6) | 4 (%9,1) |
|
| |
6 (%42,9) | 2 (%10,5) |
|
| |
MMF = Mikofenolat mofetil GA = Güven aralığı Yeni teşhis konmuş hastalar = hastalık süresi <6 ay veya PV için daha önce tedavi görmemiş olmak Hastalığı önceden mevcut hastalar = hastalık süresi ≥6 ay ve PV için daha önce tedavi görmüş olmak p-değeri için Cochran-Mantel-Haenszel testi kullanılmıştır. | ||||
Sekonder parametrelerin (kümülatif oral kortikosteroid dozu, hastalık alevlenmelerinin toplam sayısı ve sağlıkla alakalı olarak Dermatoloji Hayat Kalite İndeksi ile ölçülen hayat kalitesinde değişim dahil) analizi rituximabın MMF'ye kıyasla istatistiksel olarak anlamlı sonuçlar ortaya koyduğunu doğrulamıştır.
Glukokortikoide maruziyet
Rituximab ile tedavi edilen hastalarda kümülatif oral kortikosteroid dozu anlamlı ölçüde daha düşük olmuştur. 52. Haftada medyan (min, maks) kümülatif prednizon dozu, rituximab grubunda 2.775 mg (450, 22.180) olurken MMF grubunda 4.005 mg (900, 19.920) olmuştur (p=0,0005).
Hastalık alevlenmeleri
Rituximab ile tedavi edilen hastalarda hastalık alevlenmelerinin toplam sayısı MMF ile tedavi edilen hastalardakine göre anlamlı ölçüde düşük olmuştur (6'ya karşılık 44, p<0,0001) ve en az bir hastalık alevlenmesi yaşayan hasta sayısı daha az olmuştur (%8,1'e karşılık %41,3).
Laboratuvar değerlendirmeleri
52. Haftaya kadar, rituximab ile tedavi edilen PV hastalarından toplamda 20/63'ü (%31,7) (19'u tedavi sebebiyle başlayan ve 1'i tedavi sebebiyle artan) ADA pozitif olarak test edilmiştir. ADA varlığının PV Çalışması 2'deki güvenlilik veya etkililik üzerine belirgin bir negatif etkisi olmamıştır.
5.2. Farmakokinetik özellikler
Genel özelliklerEmilim
Hodgkin-dışı lenfoma
Tekli ajan olarak ya da CHOP tedavisi ile kombinasyon halinde tek veya çok sayıda rituximab infüzyonu alan (uygulanan rituximab dozları 100 ile 500 mg/m arasında değişmiştir) 298 Hodgkin-dışı lenfoma hastasında yürütülen bir popülasyon farmakokinetiği analizi temelinde, spesifik olmayan klerens (CL), B hücreleri veya tümör yükünün katkıda bulunması olası spesifik klerens (CL) ve santral kompartıman dağılım hacmi (V) için tipik popülasyon tahminleri sırasıyla 0,14 L/gün, 0,59 L/gün ve 2,7 L'dir. Rituximabın hesaplanan medyan terminal eliminasyon yarılanma ömrü 22 gündür (6,1 ila 52 gün aralığında). 4 haftalık doz boyunca intravenöz infüzyon olarak 375 mg/m verilen 161 hastadan elde edilen verilerde başlangıç CD19-pozitif hücre sayımları ve ölçülebilir tümör lezyonlarının boyutu, rituximabın CL'sindeki değişkenliğin bir kısmına katkıda bulunmuştur. Daha yüksek CD19-pozitif hücre sayımlarına veya tümör lezyonlarına sahip hastalar daha yüksek CL'ye sahiptir. Bununla birlikte, CD19-pozitif hücre sayımları ve tümör lezyon boyutu için düzeltme sonrası CLiçin bireyler arası değişkenliğin büyük kısmı devam etmiştir. Vvücut yüzey alanı (BSA) ve CHOP tedavisine göre değişmiştir. Sırasıyla BSA'daki aralık (1,52 ila 2,32 m) ve eşzamanlı CHOP tedavisinin katkıda bulunduğu V'deki bu değişkenlik (%27,1 ve %19,0) nispeten küçüktür. Yaş, cinsiyet ve Dünya Sağlık Örgütü performans durumu rituximabın farmakokinetiği üzerinde bir etkiye sahip değildir. Bu analiz test edilen kovaryatlardan herhangi biri ile rituximab dozunun ayarlanmasının farmakokinetik değişkenliğinde anlamlı bir azalma ile sonuçlanmasının beklenmediğini düşündürmektedir.
Daha önce rituximab kullanmamış Hodgkin-dışı lenfomalı 203 hastaya 4 doz boyunca haftada bir aralıklarla 375 mg/m dozda intravenöz infüzyon olarak uygulanan rituximab, dördüncü infüzyonu takiben 486 mikrogram/mL'lik (77,5 ila 996,6 mikrogram/mL aralığında) ortalama
bir Cvermiştir. Rituximab son tedavinin tamamlanmasından 3-6 ay sonra hastaların serumunda tespit edilebilmiştir.
Rituximabın Hodgkin-dışı lenfomalı 37 hastaya 8 doz boyunca haftada bir aralarla intravenöz infüzyon olarak 375 mg/m'lik bir dozda uygulanmasından sonra ortalama Cher bir ardışık infüzyon ile artmış olup, ilk infüzyondan sonra ortalama 243 mikrogram/mL (16-582 mikrogram/mL aralığında) ve sekizinci infüzyondan sonra 550 mikrogram/mL (171-1.177 mikrogram/mL aralığında) arasında değişmiştir.
6 siklus CHOP kemoterapisi ile kombinasyon halinde 375 mg/m'lik 6 infüzyon olarak uygulandığında rituximabın farmakokinetik profili, tek başına rituximab ile görülene benzerdir.
Kronik lenfositik lösemi
Rituximab KLL hastalarında, fludarabin ve siklofosfamid ile kombinasyon halinde, i.v. infüzyon olarak ilk siklusta 375 mg/m, sonraki 5 siklusun her birinde 500 mg/m'ye artırılarak uygulanmıştır. Beşinci 500 mg/m'lik infüzyondan sonra ortalama C(n=15) 408 mikrogram/mL (97-764 mikrogram/mL aralığında), ortalama terminal yarılanma ömrü de 32 gündür (14 ila 62 gün aralığında).
Granülomatöz polianjiitis (Wegener) (GPA) ve mikroskobik polianjiitis (MPA)
Dört doz boyunca haftada bir kez 375 mg/m rituximab almış granülomatöz polianjiitis ve mikroskobik polianjiitis görülen 97 hastada popülasyon farmakokinetiği analizi temelinde hesaplanmış ortalama terminal eliminasyon yarılanma ömrü 23 gündür (9 ila 49 gün aralığında). Rituximab ortalama klerensi ve dağılım hacmi sırasıyla 0,313 L/gün (0,116 ila 0,726 L/gün aralığında) ve 4,50 L'dir (2,25 ila 7,39 L aralığında). İlk 180 gün boyunca maksimum konsantrasyon (C), 180. Günde minimum konsantrasyon (C180) ve 180 gün boyunca eğrinin altındaki kümülatif alan (EAA180) (medyan [aralık]) sırasıyla 372,6 (252,3-533,5) mikrogram/mL, 2,1 (0-29,3) mikrogram/mL ve 10.302 (3.653-21.874) mikrogram/mL/gün'dür. Bu hastalarda rituximabın farmakokinetik parametreleri romatoid artrit hastalarında gözlenene benzer görünmektedir.
Pemfigus vulgaris
1. Gün, 15. Gün, 168. Gün ve 182. Günde 1.000 mg dozda rituximab alan yetişkin PV hastalarındaki farmakokinetik parametreler Tablo 16'da özetlenmiştir.
Tablo 16 PV Çalışması 2'den Elde Edilen, Yetişkin PV Hastalarındaki Popülasyon Farmakokinetiği
Parametre | İnfüzyon Siklusu | |
| 1.000 mg'lık 1. Siklus 1. Gün ve 15. Gün N=67 | 1.000 mg'lık 2. Siklus 168. Gün ve 182. Gün N=67 |
Terminal yarılanma ömrü (gün) Medyan (Aralık) |
21,0 (9,3-36,2) |
26,5 (16,4-42,8) |
Klerens (L/gün) Ortalama (Range) |
391 (159-1.510) |
247 (128-454) |
Dağılımın Santral Hacmi (L) |
|
|
Ortalama | 3,52 | 3,52 |
(Aralık) | (2,48-5,22) | (2,48-5,22) |
İlk iki rituximab uygulamasını takiben (1. Siklusa denk gelen 1. Günde ve 15. Günde) PV'li hastalarda rituximabın farmakokinetik parametrelerin GPA/MPA ve romatoid artritli hastalarda görülenlere benzer olmuştur. Son iki uygulamanın ardından (2. Siklusa denk gelen 168. Günde ve 182. Günde), rituximab klerensi azalırken, dağılımın santral hacmi değişmeden kalmıştır.
Biyotransformasyon Hodgkin-dışı lenfoma Veri bulunmamaktadır.
Kronik lenfositik lösemi Veri bulunmamaktadır.
Granülomatöz polianjiitis (Wegener) (GPA) ve mikroskobik polianjiitis (MPA)
Veri bulunmamaktadır.
Pemfigus vulgaris
Veri bulunmamaktadır.
Dağılım
Hodgkin-dışı lenfoma
Tek ajan olarak ya da CHOP tedavisi ile kombinasyon halinde tek veya çok sayıda rituximab infüzyonu alan (uygulanan rituximab dozları 100 ila 500 mg/m arasında değişmiştir) 298 Hodgkin-dışı lenfoma hastasında yürütülen bir popülasyon farmakokineği analizine dayanarak, spesifik olmayan klerens (CL), muhtemelen B hücreleri veya tümör yükünün katkıda bulunduğu spesifik klerens (CL) ve santral kompartman dağılım hacmi (V) için tipik popülasyon tahminleri sırasıyla 0,14 L/gün, 0,59 L/ gün ve 2,7 L'dir.
Kronik lenfositik lösemi Veri bulunmamaktadır.
Granülomatöz polianjiitis (Wegener) (GPA) ve mikroskobik polianjiitis (MPA)
Rituximab ortalama klerensi ve dağılım hacmi sırasıyla 0,313 L/gün (aralık, 0,116 ila 0,726 L/gün) ve 4,50 L'dir (aralık, 2,25 ila 7,39 L).
Pemfigus vulgaris
Dağılımın santral hacmi ortalama 3,52 L (2,48-5,22)'dir. İlk iki rituximab uygulamasını takiben (1. Siklusa denk gelen 1. Günde ve 15. Günde) ve son iki uygulamanın ardından (2. Siklusa denk gelen 168. Günde ve 182. Günde), rituximab dağılımının santral hacmi değişmeden kalmıştır.
Eliminasyon Hodgkin-dışı lenfoma
Rituximabın tahmin edilen medyan terminal eliminasyon yarılanma ömrü 22 gündür (6,1 ila 52 gün aralığında). Rituximabın, 4 haftalık dozlar halinde 375 mg/m i.v. infüzyon olarak verildiği 161 hastadan elde edilen verilerde CL'sindeki değişkenliğin bir kısmına başlangıç CD19- pozitif hücre sayısı ve ölçülebilir tümör lezyonlarının boyutu katkıda bulunmuştur. Daha yüksek CD19-pozitif hücre sayısı ya da tümör lezyonları olan hastaların daha yüksek CL'si olmuştur. Bununla birlikte, CD19-pozitif hücre sayısı ve tümör lezyonu boyutu için düzeltme yapıldıktan sonra, CLiçin bireyler arası değişkenliğin büyük kısmı aynen kalmıştır. V1, vücut yüzey alanı (BSA) ve CHOP tedavisi ile değişmiştir. Sırasıyla, BSA'daki aralık (1,53 ila 2,32 m) ve eşzamanlı CHOP tedavisinin katkıda bulunduğu V1'deki (%27,1 ve %19,0) bu değişkenlik, göreceli olarak küçüktür.
Kronik lenfositik lösemi
KLL hastalarında, fludarabin ve siklofosfamid ile kombinasyon halinde verilen beşinci 500 mg/m'lik i.v. infüzyondan sonra ortalama terminal yarılanma ömrü 32 gündür (14 ila 62 gün).
Granülamatöz polianjiitis (Wegener) (GPA) ve mikroskobik polianjiitis (MPA)
Dört doz boyunca haftada bir kere 375 mg/m rituximab alan 97 GPA ve MPA hastasındaki verilerin popülasyon farmakokinetik analizine dayanılarak, tahmini medyan terminal eliminasyon yarı ömrü 23 gündür (9 ila 49 gün aralığında). Rituximab ortalama klerensi 0,313 L/gün (0,116 ila 0,726 L/gün aralığında) olarak bulunmuştur.
Pemfigus vulgaris
İlk iki rituximab uygulamasını takiben (1. Siklusa denk gelen 1. Günde ve 15. Günde) medyan terminal yarılanma ömrü 21.0 (9,3-36,2) gündür. Son iki uygulamanın ardından (2. Siklusa denk gelen 168. Günde ve 182. Günde) medyan terminal yarılanma ömrü 26,5 (16,4-42,8)
gündür. 1. Siklusun ardından ortalama klerens 391 L/gün (159-1.510) olurken, 2. Siklus ardından ortalama klerens azalarak 247 L/gün (128-454) olmuştur.
Doğrusallık/doğrusal olmayan durum
Dört çalışmada, iki hafta aralıklarla 1. gün ve 15. günde uygulanan 500 mg ve 1.000 mg dozlardaki iki intravenöz rituximab infüzyonunu takiben, rituximab farmakokinetiği araştırılmıştır. Bu çalışmaların hepsinde, rituximab farmakokinetiği araştırılan limitli doz aralığı için doz ile orantılı olmuştur.
Pediyatrik popülasyon DLBCL/BL/BAL/BLL
Emilim
Pediyatrik DLBCL/BL/BAL/BLL'nin çalışıldığı klinik çalışmada, farmakokinetik, 3 yaşından büyük 35 hastalık bir alt grupta çalışılmıştır. İki yaş grubu (≥ 3 ila < 12 yaş ve ≥ 12 yaş ila < 18 yaş) arasında PK karşılaştırılabilir olmuştur. Her iki indüksiyon siklusunda (siklus 1 ve 2) 375 mg/m dozunda uygulanan iki rituximab infüzyonunu takip eden konsolidayon sikluslarının (siklus 3 ve 4) herbirinde 375 mg/m dozunda uygulanan bir rituximab infüzyonu sonrasında maksimum konsantrasyon dördüncü infüzyondan (siklus 2) sonra en yüksek olmuştur; geometrik ortalama 347 mikrogram/mL olmuştur, bunu maksimum konsantrasyonun daha düşük geometrik ortalamaları takip etmiştir (siklus 4: 247 mikrogram/mL). Bu doz rejimi ile,
vadi düzeyleri sürdürülmüştür (geometrik ortalama 41,8 mikrogram/mL (predoz Siklus 2; 1 siklusun arkasından), 67,7 mikrogram/mL (predoz Siklus 3, 2 siklusun ardından) ve 58,5 mikrogram/mL (predoz Siklus 4, 3 siklusun ardından).
Rituximabın DLBCL/BL/BAL/BLL'li pediyatrik hastalardaki PK karateristikleri yetişkin NHL hastalarında gözlenene benzerdir.
≥ 6 ay'dan < 3 yaşına kadar olan grup için PK verisi mevcut değildir ancak, popülasyon PK tahminleri ≥ 3 yaş grubu ile kıyaslandığında bu yaş grubunda karşılaştırılabilir sistemik maruziyeti (EAA, Cvadi) destekler (Tablo 17). Başlangıçtaki daha küçük tümör büyüklüğü, daha düşük zamana bağımlı klerens sebebiyle daha yüksek maruziyet ile ilişkilidir ancak farklı tümör büyüklüklerinden etkilenen sistemik maruziyet, etkili ve güvenlilik profili açısından kabul edilebilir maruziyet aralığı içerisinde kalmıştır.
Tablo 17: Pediyatrik DLBCL/BL/BAL/BLL'de Rituximab Doz Rejiminin Ardından Tahmini PK Parametreleri
Yaş Grubu | ≥ 6 ay ila < 3 yaş | ≥ 3 yaş ila < 12 yaş | ≥ 12 yaş ila < 18 yaş |
C(mikrogram/mL) | 47,5 (0,01 – 179) | 51,4 (0,00 – 182) | 44,1 (0,00 – 149) |
EAA (mikrogram*gün/mL) | 13.501 (278 – 31.070) | 11.609 (135 – 31.157) | 11.467 (110 – 27.066) |
Sonuçlar medyan olarak sunulmuştur (min – maks); Cpredoz Siklus 4'tür. Atılım
3 yaşından büyük pediyatrik hastalarda ortalama eliminasyon yarılanma ömrü 26 gün olmuştur. Hastalardaki karakteristik özellikler
Böbrek veya karaciğer yetmezliği olan hastalara ait farmakokinetik veri bulunmamaktadır.
Rituximabın farmakokinetiği üzerine yaş, cinsiyet, ırk ve Dünya Sağlık Örgütü performans statüsünün herhangi bir etkisi olmamıştır. Bu analiz, test edilen eşdeğişken faktörlerin herhangi biriyle rituximabın doz ayarlamasının, farmakokinetik değişkenlikte anlamlı bir azalmayla sonuçlanmasının beklenmediğini belirtmektedir.
5.3. Klinik öncesi güvenlilik verileri
Rituximabın B hücrelerindeki CD20 antijenine yüksek düzeyde spesifik olduğu gösterilmiştir. Sinomolgus maymunlarında yapılan toksisite çalışmaları, periferik kanda ve lenfoid dokuda B hücrelerinin beklenen farmakolojik tüketiminden başka bir etki göstermemiştir.
100 mg/kg'a kadarki dozlarda (gestasyonun 20-50. günleri arasında tedavi) sinomolgus maymunları üzerinde gelişimsel toksisite çalışmaları yapılmış ve fetüs için rituximabdan kaynaklanan herhangi bir toksisite kanıtının olmadığı gösterilmiştir. Ancak fetüsün lenfoid organlarında B hücrelerinin doza bağlı farmakolojik tüketimi gözlenmiş olup bu doğumdan sonra da devam etmiş ve buna etkilenen yeni doğan hayvanlarda IgG düzeylerinde bir azalma da eşlik etmiştir. B hücre sayısı, bu hayvanlarda doğumu takip eden 6 ay içerisinde normale dönmüş ve immünizasyon reaksiyonunu riske atmamıştır.
Mutajeniteyi araştıran standart testler bu molekül için uygun olmadığından yapılmamıştır. Rituximabın karsinojenik potansiyelini belirlemek için uzun dönem hayvan çalışmaları yapılmamıştır.
Rituximabın fertilite üzerine etkilerini tespit etmek için spesifik çalışmalar yapılmamıştır. Sinomolgus maymunlarındaki genel toksisite çalışmalarında, erkek ya da dişilerin üreme organları üzerine zararlı hiçbir etki gözlenmemiştir.
6. FARMASÖTİK ÖZELLİKLER
6.1. Yardımcı maddelerin listesi
Tri-sodyum sitrat dihidrat
6.2. Geçimsizlikler
Rituximab ile polivinilklorür veya polietilen torbalar veya infüzyon seti arasında geçimsizlik gözlenmemiştir.
6.3. Raf ömrü
Açılmamış flakonlar:
36 ay.
Seyreltilmiş ürünler:
Mikrobiyolojik açıdan, seyreltilmiş ürünlerin hemen kullanılması gerekir. Bu ürünler hemen kullanılmazsa, seyreltme işlemi kontrollü ve valide edilmiş aseptik koşullar altında gerçekleştirilmediği takdirde kullanım sırasındaki saklama süreleri ve kullanım öncesindeki koşullar kullanıcının sorumluluğundadır ve normal olarak 2°C-8°C'de 24 saati aşmamalıdır.
Ayrıca PE/PVC infüzyon torbasında seyreltilen ürün (%0,9 sodyum klorür veya %5'lik dekstrozun sudaki çözeltisi içerisinde) fiziksel ve kimyasal olarak 24 saat süresince 2°C-8°C'de ve sonra da 12 saat süresince 30°C altındaki oda sıcaklığında stabildir.
6.4. Saklamaya yönelik özel tedbirler
Flakonları 2°-8°C'de (buzdolabında) saklayınız. Dondurulmamalıdır. Flakonları doğrudan güneş ışığından korumak için ambalajında saklayınız.
Ürünün seyretmeden sonraki saklama koşulları için Bkz. Bölüm 6.3.
6.5. Ambalajın niteliği ve içeriği
10 mL'de 100 mg rituximab (10 mg/mL) içeren, klorobutil kauçuk tıpalı şeffaf Tip 1 cam flakon.
2 flakon içeren ambalajlarda.
6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler
TRUXIMA infüzyonları, acil uygulama için tüm resüsitasyon olanaklarının bulunduğu merkezlerde ve deneyimli bir onkolog/hematoloğun gözetimi altında uygulanmalıdır.
TRUXIMA steril, koruyucu maddeler içermeyen, tek dozluk flakonlarda sunulur.
TRUXIMA'yı hazırlamak için steril iğne ve şırınga kullanın. Gerekli miktarda TRUXIMA'yı aseptik koşullarda çekiniz ve içinde steril, pirojen bulundurmayan, %0,9'luk sodyum klorürün veya %5'lik dekstrozun sudaki çözeltisinden bulunan bir infüzyon torbasında (PE/PVC torba), hesaplanmış olan 1 - 4 mg/mL'lik rituximab konsantrasyonuna ulaşıncaya dek seyreltiniz. Çözeltiyi karıştırmak için, torbayı köpük oluşumunu önleyecek şekilde nazikçe ters çeviriniz. Hazırlanan çözeltinin steril olduğundan emin olunmalıdır. Bu ilaç herhangi bir antimikrobiyal koruyucu veya bakteriyostatik ajan içermediği için aseptik teknikler uygulanmalıdır.
Parenteral ilaçlar uygulanmadan önce, partiküllü maddeler ve renk değişikliğine dikkat edilmelidir.
Mikrobiyolojik açıdan, seyreltilmiş ürünlerin hemen kullanılması gerekir. Bu ürünler hemen kullanılmazsa, seyreltme işlemi kontrollü ve valide edilmiş aseptik koşullar altında gerçekleştirilmediği takdirde kullanım sırasındaki saklama süreleri ve kullanım öncesindeki koşullar kullanıcının sorumluluğundadır ve normal olarak 2°C-8°C'de 24 saati aşmamalıdır.
Ayrıca PE/PVC infüzyon torbasında seyreltilen ürün (%0,9 sodyum klorür veya %5'lik dekstrozun sudaki çözeltisi içerisinde) fiziksel ve kimyasal olarak 24 saat süresince 2°C-8°C'de ve sonra da 12 saat süresince 30°C altındaki oda sıcaklığında stabildir.
Kullanılmamış olan ürünler ya da atık materyaller “Tıbbi Atıkların Kontrolü Yönetmeliği†ve “Ambalaj ve Ambalaj Atıklarının Kontrolü Yönetmeliği'ne†uygun olarak imha edilmelidir.
 Mesane Kanseri
Mesane kanseri her zaman mukozada başlar. Erken safhalarda bu tabakada sınırlı kalır ve
hücre içindeki karsinom olarak nitelendirilir.
Mesane Kanseri
Mesane kanseri her zaman mukozada başlar. Erken safhalarda bu tabakada sınırlı kalır ve
hücre içindeki karsinom olarak nitelendirilir. |
 Mide Kanseri
Mide kanseri genellikle mideyi tümüyle kaplayan ve mukus üretmekle görevli hücrelerde başlar. Bu kanser tipine adenokarsinom denir.
Mide Kanseri
Mide kanseri genellikle mideyi tümüyle kaplayan ve mukus üretmekle görevli hücrelerde başlar. Bu kanser tipine adenokarsinom denir. |
İLAÇ EŞDEĞERLERİ
| Eşdeğer İlaç Adı | Barkodu | İlaç Fiyatı |
|---|---|---|
| NOVEX | 8699514760047 | 8,801.34TL |
| REDDITUX | 8680885576016 | 3,871.32TL |
| RIXATHON | 8681428761449 | 8,801.34TL |
| RUXIENCE | 8681308771032 | 2,793.08TL |
| TRUXIMA | 8680614140143 | 8,801.34TL |
| Diğer Eşdeğer İlaçlar |
 |
Artrit Artrit, oldukça yaygın bir hastalıktır ancak iyi anlaşılamamıştır. Aslında “artrit” tek bir hastalığın adı değildir; eklem ağrısı veya eklem hastalıklarını adlandırmanın gayri resmi yoludur. |
 |
Şizofrenlik Şizofrenliğin psikiatrik teşhisi hakkında çok fazla anlaşmazlık vardır. Bu sayfadaki bilgiler, şizofrenliğin teşhisi, nedenleri ve tedavisi hakkındaki faklı teoriler hakkında bilgi verecektir. |
 |
Pankreas Kanseri Pankreas karnın alt kısmında yatay şekilde bulunan bir organdır. Sindirime yardımcı olan enzimleri ve kan şekerini yönetmeye yardımcı olan hormonları vücuda dağıtmakla görevlidir. |
İLAÇ GENEL BİLGİLERİ
CELLTRION Healthcare İlaç San. ve Tic. Ltd.Şti
| Satış Fiyatı | 8801.34 TL [ 14 Apr 2025 ] |
| Önceki Satış Fiyatı | 8801.34 TL [ 7 Apr 2025 ] |
| Original / Jenerik | Original İlaç |
| Reçete Durumu | Normal Reçeteli bir ilaçdır. |
| Barkodu | 8680614140143 |
| Etkin Madde | Rituksimab |
| ATC Kodu | L01XC02 |
| Birim Miktar | 100 |
| Birim Cinsi | MG |
| Ambalaj Miktarı | 2 |
| Antineoplastik ve İmmünomodülatör Ajanlar > Diğer Kanser İlaçları > Rituksimab |
| İthal ( ref. ülke : Isvicre ) ve Beşeri bir ilaçdır. |