VALAMOR 200 mg film kapl� tablet (63 tablet) K�sa �r�n Bilgisi
{ Ribosiklib }
1. BE�ER� TIBB� �R�N�N ADI
VALAMOR 200 mg film kapl� tablet
2. KAL�TAT�F VE KANT�TAT�F B�LE��M
Etkin madde
Ribosiklib s�ksinat 254,4 mg
(200 mg ribosiklibe kar��l�k gelmektedir)
Yard�mc� maddeler
Soya lesitin 0,344 mg
Yard�mc� maddelerin tam listesi i�in b�l�m 6.1'e bak�n�z.
3. FARMAS�T�K FORMU
Film kapl� tablet
A��k grimsi mor, �entiksiz, yuvarlak, e�imli kenarl�, bir taraf�nda “RIC” ve di�er taraf�nda “NVR” i�lemeli.
E�er QTcF ≥481 milisaniye tekrar g�r�l�rse, QTcF <481 milisaniyeye d�zelene kadar ara verilir ve sonra VALAMOR'a bir sonraki d���k doz d�zeyinden devam edilir.
4. KL�N�K �ZELL�KLER
4.1. Terap�tik endikasyonlar
VALAMOR, letrozol ile kombine tedavilerde, en az %10 �strojen resept�r� pozitif ve insan epidermal b�y�me fakt�r� resept�r� 2 (HER2) negatif postmenopozal metastatik meme kanseri olan, adjuvan tedavinin tamamlanmas�ndan 12 ay sonra relaps yapm�� ya da ileri evre meme kanseri i�in daha �nce hi�bir endokrin tedavi almam�� hastalarda endikedir.
VALAMOR, fulvestrant ile kombine tedavilerde, en az %10 �strojen resept�r� pozitif ve insan epidermal b�y�me fakt�r� resept�r� 2 (HER) negatif postmenopozal metastatik meme kanseri olan, daha �nce fulvestrant ile tedavi edilmemi� hastalarda endikedir,
Metastatik hastal���n tedavisi i�in en az 6 ay boyunca ve en az bir basamak aromataz inhibit�r� ald�ktan sonra klinik ve/veya radyolojik hastal�k progresyonu varl���nda fulvestrant ile kombinasyon halinde;
4.2. Pozoloji ve uygulama �ekli
VALAMOR ile tedavi, anti-kanser tedavilerin kullan�m�nda deneyimli bir hekim taraf�ndan ba�lat�lmal�d�r.
Pozoloji:
VALAMOR'un �nerilen dozu, 28 g�nl�k bir siklusu tamamlayacak �ekilde arka arkaya 21 g�n s�reyle g�nde bir kez oral yoldan al�nan 600 mg (�� adet 200 mg film kapl� tablet) ve bunu izleyen tedavisiz 7 g�n �eklindedir. Tedavi, hasta tedaviden klinik fayda g�rd��� s�rece veya kabul edilemez toksisite meydana gelene kadar s�rd�r�lmelidir.
VALAMOR, 2,5 mg letrozol ya da 500 mg fulvestrant ile birlikte kullan�lmal�d�r. VALAMOR letrozol ile birlikte kullan�ld���nda letrozol, 28 g�nl�k siklus boyunca g�nde bir
kez al�nmal�d�r. L�tfen ilave bilgiler i�in letrozol�n K�sa �r�n Bilgisine ba�vurunuz.
VALAMOR fulvestrant ile birlikte kullan�ld���nda fulvestrant, intram�sk�ler olarak 1., 15. ve
29. g�nlerde ve daha sonra ayda bir kere uygulan�r. L�tfen ilave bilgiler i�in fulvestrant�n K�sa �r�n Bilgisine ba�vurunuz.
VALAMOR a� veya tok karn�na al�nabilir (bkz. b�l�m 4.5). Hastalar dozlar�n� her g�n yakla��k olarak ayn� saatte, tercihen sabah saatlerinde almaya te�vik edilmelidir. Hasta dozu ald�ktan sonra kusarsa veya bir dozu atlarsa, o g�n ilave bir doz al�nmamal�d�r. Bir sonraki re�ete edilmi� doz normal zaman�nda al�nmal�d�r.
Doz modifikasyonlar�
A��r veya tolere edilemeyen advers ila� reaksiyonlar�n�n kontrol edilebilmesi i�in dozlara ge�ici olarak ara vermek, dozu azaltmak veya VALAMOR tedavisini kesmek gerekebilir. Dozda azaltma gerekiyorsa �nerilen doz azalt�m� k�lavuzlar� Tablo 1'de verilmektedir.
Tablo 1 �nerilen doz modifikasyonu k�lavuzlar�
| VALAMOR |
|
Doz | 200 mg'l�k tablet say�s� | |
Ba�lang�� dozu �lk doz azalt�m� �kinci doz azalt�m� | 600 mg/g�n 400 mg/g�n 200 mg*/g�n | 3 2 1 |
* E�er 200 mg/g�n�n de alt�nda doz azalt�m� gerekiyorsa tedavi bir daha ba�lanmamak �zere kesilmelidir. | ||
Tablo 2, 3, 4 ve 5'te belirli advers ila� reaksiyonlar�n�n kontrol� i�in dozlara ara verme, doz
azaltma ve tedavinin kesilmesi ile ilgili �neriler �zetlenmektedir. Her hastan�n tedavi plan�na, hasta baz�nda yap�lan fayda/risk de�erlendirmesine dayal� olarak tedaviden sorumlu hekimin klinik de�erlendirmesi y�n vermelidir (bkz. b�l�m 4.4).
VALAMOR ile tedaviye ba�lanmadan �nce tam kan say�m� yap�lmal�d�r. VALAMOR ile tedaviye ba�land�ktan sonra tam kan say�m� izlemi ilk 2 siklus s�resince iki haftada bir, sonraki her 4 siklusun ba��nda ve sonras�nda klinik durum gerektirdik�e yap�lmal�d�r.
Tablo 2 Doz modifikasyonu ve y�netimi – N�tropeni
| Derece 1 veya 2* (MNS 1000/mm - ≤NAS) | Derece 3* (MNS 500 - <1000/mm) | Derece 3* febril n�tropeni ** | Derece 4* (MNS <500/mm) |
N�tropeni | Herhangi bir doz ayarlamas� gerekmez | Derece ≤2'ye d�zelme olana kadar dozlara ara verilir.
VALAMOR'a ayn� doz d�zeyinden devam edilir.
E�er toksisite 3. derecede tekrar g�r�l�rse: Derece ≤2'ye d�zelme olana kadar dozlara ara verilir, ard�ndan VALAMOR'a devam edilir ve doz 1 seviye d���r�l�r. | Derece ≤2'ye d�zelme olana kadar dozlara ara verilir.
VALAMOR'a devam edilir ve doz 1 seviye d���r�l�r. | Derece ≤2'ye d�zelme olana kadar dozlara ara verilir.
VALAMOR'a devam edilir ve doz 1 seviye d���r�l�r. |
* Derecelendirme AOOTK Versiyon 4.03'e g�redir (AOOTK= Advers Olaylar i�in Ortak Terminoloji Kriteri). ** >38,3°C'lik tek bir ate�le derece 3 n�tropeni (ya da bir saatten fazla >38°C ve/veya e� zamanl� enfeksiyon) MNS: Mutlak n�trofil say�s�, NAS: Normalin alt s�n�r� | ||||
VALAMOR ile tedaviye ba�lanmadan karaci�er fonksiyon testleri yap�lmal�d�r. VALAMOR ile tedaviye ba�land�ktan sonra karaci�er fonksiyon testleri izlemi ilk 2 d�ng� s�resince iki haftada bir, sonraki her 4 siklusun ba��nda ve sonras�nda klinik durum gerektirdik�e yap�lmal�d�r. E�er derece ≥2 anomaliler g�r�l�rse daha s�k izlem yap�lmas� �nerilir.
Tablo 3 Doz modifikasyonu ve y�netimi – Hepatobiliyer toksisite
| Derece 1* (>N�S – 3 x N�S) | Derece 2* (>3 ila 5 x N�S) | Derece 3* (>5 ila 20 x N�S) | Derece 4* (>20 x N�S) |
2 x N�S | Herhangi | Ba�lang�� derece < 2: | Ba�lang��taki | VALAMOR |
�zerinde total bilirubin art���n�n e�lik etmedi�i ba�lang�ca g�re AST ve/veya ALT y�kselmeleri**, | bir doz ayarlamas� gerekmez | ba�lang��taki dereceye e�it veya bu derece alt�na d���� olana kadar dozlara ara verilir, ard�ndan VALAMOR'a ayn� doz d�zeyinden devam edilir. E�er derecede 2 tekrar g�r�l�rse, VALAMOR'a bir sonraki d���k doz d�zeyinden devam edilir. | dereceye e�it veya bu derece alt�na d���� olana kadar VALAMOR dozuna ara verilir, ard�ndan bir sonraki d���k doz d�zeyinden devam edilir. E�er derecede 3 tekrar g�r�l�rse, VALAMOR tedavisi kesilir. | tedavisi kesilir. |
Ba�lang�� derece 2: Dozlara herhangi bir ara verilmez. | ||||
Kolestaz yoklu�unda AST ve/veya ALT'ta total bilirubin art���n�n e�lik etti�i kombine y�kselmeler | E�er hastalar ba�lang��taki derece fark etmeksizin total bilirubin >2x N�S ile birlikte ALT ve/veya AST > 3x N�S geli�tirirse, VALAMOR tedavisi kesilir. | |||
* Derecelendirme AOOTK Versiyon 4.03'e g�redir (AOOTK= Advers Olaylar i�in Ortak Terminoloji Kriteri) ** Ba�lang�� = tedavi ba�lang�c� �ncesinde N�S=Normalin �st s�n�r� | ||||
Tedaviye ba�lanmadan �nce EKG de�erlendirmesi yap�lmal�d�r. VALAMOR ile tedaviye ba�land�ktan sonra EKG, ilk siklusun yakla��k 14. g�n�nde ve ikinci siklusun ba��nda, sonras�nda klinik durum gerektirdik�e tekrarlanmal�d�r. Tedavi s�ras�nda QTcF uzamas� olursa daha s�k EKG izlemi �nerilir.
Tablo 4 Doz modifikasyonu ve y�netimi – QT uzamas�
QTcF >480 milisaniye g�steren EKG'ler | |
QTcF >500 milisaniye g�steren EKG'ler | E�er QTcF de�eri EKG'de >500 milisaniye ise, QTcF <481 milisaniye olana kadar VALAMOR'a ara verilir, ard�ndan VALAMOR'a bir sonraki d���k doz d�zeyinden devam edilir. |
Doz kesilmelidir.
4.3. Kontrendikasyonlar
Etkin madde
4.4. �zel kullan�m uyar�lar� ve �nlemleri
Kritik visseral hastal�k
Ribosiklibin etkilili�i ve g�venlili�i kritik visseral hastal��� olan hastalarda ara�t�r�lmam��t�r. N�tropeni
N�tropeninin �iddetine ba�l� olarak Tablo 2'de tarif edildi�i gibi VALAMOR ile tedaviye ara
verilmesi, dozun azalt�lmas� veya tedavinin kesilmesi gerekebilir (bkz. B�l�m 4.2 ve 4.8). Hepatobiliyer toksisite
VALAMOR ile tedaviye ba�lanmadan karaci�er fonksiyon testleri yap�lmal�d�r. VALAMOR ile tedaviye ba�land�ktan sonra karaci�er fonksiyon testleri izlenmelidir (bkz. B�l�m 4.2 ve 4.8).
Transaminaz y�kselmelerinin �iddetine ba�l� olarak Tablo 3'te tarif edildi�i gibi VALAMOR ile tedaviye ara verilmesi, dozun azalt�lmas� veya tedavinin kesilmesi gerekebilir (bkz. B�l�m 4.2 ve 4.8). Ba�lang��ta AST/ALT derece ≥ 3 y�kselmeleri olan hastalar i�in �neriler belirlenmemi�tir.
QT aral���nda uzama
�al��ma E2301'de (MONALEESA-7) ribosiklib art� tamoksifen alan hastalar�n 14/87'sinde (%16,1) ve ribosiklib art� bir nonsteroidal aromataz inhibit�r� (NSAI) alan hastalar�n 18/245'inde (%7,3), �al��ma ba�lang�c�na k�yasla > 60 milisaniyelik bir QTcF aral��� art��� g�zlenmi�tir. VALAMOR'un tamoksifen ile kombinasyon halinde kullan�m� �nerilmemektedir (bkz. b�l�m 4.8 ve 5.1).
Tedaviye ba�lanmadan �nce EKG de�erlendirmesi yap�lmal�d�r. VALAMOR ile tedavi sadece QTcF de�erleri <450 milisaniye olan hastalarda ba�lat�lmal�d�r. EKG, ilk siklusun yakla��k 14. g�n�nde ve ikinci siklusun ba��nda, sonras�nda klinik durum gerektirdik�e tekrarlanmal�d�r (bkz. B�l�m 4.2 ve 4.8).
Tedaviye ba�lanmadan �nce, ilk alt� siklusun ba��nda ve sonras�nda klinik durum gerektirdik�e serum elektrolitlerinin (potasyum, kalsiyum, fosfor ve magnezyum dahil) izlemi yap�lmal�d�r. Anormallik varsa VALAMOR ile tedaviye ba�lanmadan �nce ve VALAMOR ile tedavi s�ras�nda d�zeltilmelidir.
Halihaz�rda QTc uzamas� olan ya da QTc uzamas� a��s�ndan �nemli riske sahip hastalarda VALAMOR kullan�m�ndan ka��n�lmal�d�r. Bu hastalar:
Uzun QT sendromu olan hastalar.
Yak�n zamanda ge�irilmi� miyokard enfarkt�s�, konjestif kalp yetmezli�i, stabil olmayan anjina ve bradiaritmiler dahil kontrol edilmemi� veya �nemli kalp hastal��� olan hastalar.
Elektrolit anormallikleri olan hastalard�r.
QTcF aral���nda klinik olarak anlaml� uzamaya yol a�abilece�inden VALAMOR'un QTc aral���n� uzatt��� bilinen ila�lar ve/veya g��l� CYP3A4 inhibit�rleri ile birlikte kullan�m�ndan ka��n�lmal�d�r (bkz. B�l�m 4.2, 4.5 ve 5.1). G��l� bir CYP3A4 inhibit�r� ile tedavinden ka��n�lamazsa, doz g�nde bir kez 400 mg'a d���r�lmelidir (bkz. B�l�m 4.2 ve 4.5).
Tedavi s�ras�nda g�zlenen QT uzamas�na ba�l� olarak Tablo 4'te tarif edildi�i gibi VALAMOR ile tedaviye ara verilmesi, dozun azalt�lmas� veya tedavinin kesilmesi gerekebilir (bkz. B�l�m 4.2, 4.8 ve 5.2).
�iddetli kutan�z reaksiyonlar
VALAMOR tedavisi ile toksik epidermal nekroliz (TEN) bildirilmi�tir. �iddetli kutan�z reaksiyonlar� d���nd�ren i�aret ve semptomlar (�rn., s�kl�kla kabarc�klar veya mukozal
lezyonlar ile birlikte progresif geni� �apl� deri d�k�nt�s�) ortaya ��karsa, VALAMOR acilen b�rak�lmal�d�r.
�nterstisyel Akci�er Hastal���/Pn�monit
VALAMOR ve di�er siklin-ba��ml� kinaz 4/6 (CDK4/6) inhibit�rleriyle tedavi edilen hastalarda �iddetli, ya�am� tehdit eden veya �l�mc�l interstisiyel akci�er hastal��� (�AH) ve/veya pn�monit olu�abilir.
�� Faz-III klinik �al��mada (MONALEESA-2, MONALEESA-3, MONALEESA-7) ribosiklib tedavisi alan hastalar�n %1,1'inde herhangi bir dereceden �AH/pn�monit, %0,3'�nde Derece 3 ya da Derece 4, %0,1'inde �l�m rapor edilmi�tir.
Ruhsatland�rma sonras�nda ek �AH/Pn�monit olgular�na rastlanm�� olup, �l�ml� vakalar bildirilmi�tir. (bkz. B�l�m 4.8)
Hastalar, �AH/Pn�monit d���nd�ren akci�er semptomlar� (�rn., hipoksi, �ks�r�k, dispne) a��s�ndan takip edilmelidir. �AH/Pn�monit ku�kusu yaratacak yeni veya k�t�le�en solunum semptomlar� olan hastalarda hemen VALAMOR kullan�m� durdurularak hasta de�erlendirmeye tabi tutulmal�d�r. Tekrarlayan semptomatik veya �iddetli �AH veya pn�monit g�zlenen hastalarda VALAMOR kullan�m� kal�c� olarak b�rak�lmal�d�r. (bkz. B�l�m 4.2)
Kan kreatinin y�kselmesi
Ribosiklib, proksimal t�b�llerden kreatinin aktif salg�lanmas�nda rol oynayan renal ta��y�c�lar organik katyon ta��y�c� 2 (OCT2) ve �oklu ila� ve toksin ekstr�zyon proteini 1'in (MATE1) bir inhibit�r� olarak kan kreatinin art���na neden olabilir (bkz. B�k�m 4.5). Tedavi s�ras�nda kan kreatinin art��� durumunda, b�brek yetmezli�ini d��lamak i�in b�brek fonksiyonunun daha fazla de�erlendirilmesi �nerilir.
CYP3A4 substratlar�
Ribosiklib 600 mg dozda g��l� bir CYP3A4 inhibit�r� ve 400 mg dozda orta g��te bir CYP3A4 inhibit�r�d�r. Bu nedenle, ribosiklib CYP3A4 ile metabolize edilen t�bbi �r�nlerle etkile�ime girebilir ki bu da CYP3A4 substratlar�n�n serum konsantrasyonlar�nda art��a yol a�abilir (bkz. b�l�m 4.5). Dar terap�tik indekse sahip duyarl� CYP3A4 substratlar� ile e� zamanl� kullan�m durumunda dikkat �nerilmektedir ve CYP3A4 inhibit�rleri ile birlikte uygulamaya ili�kin �neriler i�in di�er �r�n�n K�B'�ne dan���lmal�d�r.
B�brek yetmezli�i
�iddetli b�brek yetmezli�i olan hastalar i�in �nerilen 200 mg'l�k ba�lang�� dozunun, normal b�brek fonksiyonu olan hastalardaki standart ba�lang�� dozuna k�yasla yakla��k %45 daha d���k maruziyet ile sonu�land��� tahmin edilmektedir. Bu ba�lang�� dozundaki etkinlik ara�t�r�lmam��t�r. �iddetli b�brek yetmezli�i olan hastalarda, toksisite belirtileri a��s�ndan yak�ndan izlenen hastalarda dikkatli olunmal�d�r (bkz. B�l�m 4.2 ve 5.2).
�ocuk do�urma potansiyeli olan kad�nlar
�ocuk do�urma potansiyeli olan kad�nlar�n, VALAMOR'u kullan�rken ve son dozdan en az 21 g�n sonras�na kadar etkili bir do�um kontrol y�ntemi kullanmalar� �nerilmelidir (bkz. B�l�m 4.6).
Soya lesitini
VALAMOR soya lesitini i�erir. Yer f�st���na veya soyaya a��r� duyarl�l��� olan hastalar VALAMOR almamal�d�r (bkz. b�l�m 4.3).
4.5. Di�er t�bbi �r�nler ile etkile�imler ve di�er etkile�im �ekilleri
Ribosiklibin plazma konsantrasyonlar�n� y�kseltebilen maddeler
Ribosiklib ba�l�ca CYP3A4 ile metabolize olur. Dolay�s�yla, CYP3A4 enzimi aktivitesini etkileyebilen t�bbi �r�nler, ribosiklibin farmakokineti�ini etkileyebilir. G��l� CYP3A4 inhibit�r� ritonavirin (14 g�n boyunca g�nde iki kez 100 mg) 400 mg'l�k tekli ribosiklib dozu ile birlikte uygulanmas� sa�l�kl� g�n�ll�lerde ribosiklib maruziyetini (EAA) ve pik konsantrasyonunu (C) tek ba��na verildi�inde 400 mg'l�k tekli ribosiklib dozuna k�yasla s�ras�yla 3,2 ve 1,7 kat art�rm��t�r. LEQ803 (ana maruziyetin %10'undan az�n� olu�turan bask�n ribosiklib metaboliti) i�in Cve EAAs�ras�yla %96 ve %98 azalm��t�r.
Bunlarla s�n�rl� olmamak �zere klaritromisin, indinavir, itrakonazol, ketokonazol, lopinavir, ritonavir, nefazodon, nelfinavir, posakonazol, sakuinavir, telaprevir, telitromisin verapamil ve vorikonazol dahil g��l� CYP3A4 inhibit�rlerinin e� zamanl� kullan�m�ndan ka��n�lmal�d�r (bkz. B�l�m 4.4). CYP3A4'y� inhibe etme potansiyeli daha d���k alternatif e� zamanl� t�bbi �r�nler g�z �n�nde bulundurulmal� ve hastalar ribosiklib ile ili�kili advers olaylar a��s�ndan izlenmelidir (bkz. B�l�m 4.2, 4.4 ve 5.2).
E�er VALAMOR'un g��l� bir CYP3A4 inhibit�r� ile bir arada kullan�m�ndan ka��n�lam�yorsa, VALAMOR'un dozu b�l�m 4.2'de tan�mland��� �ekilde azalt�lmal�d�r. �te yandan, bu doz ayarlamas� ile ilgili klinik veri bulunmamaktad�r. Hastalar aras�ndaki de�i�kenlik nedeniyle �nerilen doz ayarlamalar� t�m hastalarda optimal olmayabilir ve bu nedenle ribosiklib ile ili�kili advers olaylar i�in yak�n izlem �nerilir. VALAMOR ile ili�kili toksisite g�r�lmesi durumunda doz modifiye edilmeli veya toksisite d�zelene kadar tedaviye ara verilmelidir (bkz. B�l�m 4.2 ve 5.2). G��l� CYP3A4 inhibit�r� b�rak�l�rsa CYP3A4 inhibit�r�n�n en az 5 yar�lanma �mr�nden sonra (s�z konusu CYP3A4 inhibit�r�n�n K�B'�n� inceleyiniz), VALAMOR, g��l� CYP3A4 inhibit�r�n�n ba�lat�lmas�ndan �nce kullan�lanla ayn� dozda yeniden ba�lat�lmal�d�r.
Fizyoloji temelli farmakokinetik sim�lasyonlar�, 600 mg'l�k ribosiklib dozunda orta g��te bir CYP3A4 inhibit�r�n�n (eritromisin) ribosiklib kararl� durum Cve EAA de�erini s�ras�yla 1,2 kat ve 1,3 kat art�rabilece�ini d���nd�rm��t�r. Ribosiklib dozlar� g�nde bir kez 400 mg'a azalt�lm�� hastalar i�in kararl� durum Cve EAA de�erindeki art���n s�ras�yla tahminen 1,4 ve 2,1 kat oldu�u hesaplanm��t�r. 200 mg'l�k g�nl�k dozdaki etkinin s�ras�yla 1,7 ve 2,8 kat olaca�� �ng�r�lm��t�r. Hafif veya orta �iddette CYP3A4 inhibit�rleri ile tedavinin ba�lat�lmas�nda ribosiklib dozunda ayarlama gerekli de�ildir. Bununla birlikte, ribosiklib ile ili�kili advers olaylar�n izlenmesi �nerilmektedir.
Hastalara greyfurt veya greyfurt suyundan uzak durmalar� s�ylenmelidir. Bunlar�n sitokrom CYP3A4 enzimlerini inhibe etti�i bilinmektedir ve ribosiklibe maruziyeti art�rabilirler.
Ribosiklibin plazma konsantrasyonlar�n� d���rebilen maddeler
G��l� CYP3A4 ind�kleyici rifampisinin (14 g�n boyunca g�nde 600 mg) 600 mg'l�k tekli ribosiklib dozu ile birlikte uygulanmas� ribosiklib EAAve Cde�erlerini sa�l�kl� g�n�ll�lerde tek ba��na verilen tekli 600 mg ribosiklib dozuna k�yasla s�ras�yla %89 ve %81 azaltm��t�r. LEQ803 Cde�eri 1,7 kat artarken EAAde�eri %27 azalm��t�r. B�ylece g��l� CYP3A4 ind�kleyicilerinin e� zamanl� kullan�m� azalm�� maruziyete ve sonu�ta etkisizlik riskine yol a�abilir. G��l� CYP3A4 ind�kleyicilerinin e� zamanl� kullan�m�ndan ka��n�lmal�d�r; bunlar fenitoin, rifampisin, karbamazepin ve sar� kantaronu (Hypericum perforatum) i�ermekle beraber bunlarla s�n�rl� de�ildir. CYP3A4'� ind�kleme potansiyeli olmayan ya da minimum potansiyele sahip alternatif bir e� zamanl� t�bbi �r�n d���n�lmelidir.
Orta g��te CYP3A4 ind�kleyicisinin ribosiklib maruziyeti �zerindeki etkisi ara�t�r�lmam��t�r. Fizyoloji bazl� farmakokinetik sim�lasyonlar� orta g��te CYP3A4 ind�kleyicisinin (efavirenz) kararl� durum ribosiklib Cve EAA de�erini s�ras�yla %51 ve %70 azaltabilece�ini d���nd�rm��t�r. B�ylece orta g��te CYP3A4 ind�kleyicilerinin e� zamanl� kullan�m� azalm�� maruziyete ve sonu� olarak �zellikle g�nde bir kez 400 mg veya 200 mg'da ribosiklib ile tedavi edilen hastalarda yetersiz etkililik riskine yol a�abilir.
Plazma konsantrasyonlar� VALAMOR ile de�i�ebilen maddeler
Ribosiklib orta g�� ila g��l� bir CYP3A4 inhibit�r� olup, CYP3A4 ile metabolize edilen t�bbi substratlarla etkile�ime girebilir ki bu da e� zamanl� kullan�lan t�bbi �r�n�n serum konsantrasyonlar�nda art��a yol a�abilir.
Sa�l�kl� g�n�ll�lerde tek ba��na midazolam (CYP3A4 substrat�) uygulamas� ile kar��la�t�r�ld���nda midazolam�n �oklu ribosiklib (400 mg) dozlar� ile birlikte uygulanmas� midazolam maruziyetini %280 y�kseltmi�tir (3,8 kat). Fizyolojik esasl� farmakokineti�in (FEFK) kullan�ld��� sim�lasyonlar, klinik olarak anlaml� 600 mg dozunda verilen VALAMOR'un midazolam EAA de�erini 5,2 kat art�rmas�n�n beklendi�ini g�stermi�tir. Bu nedenle, genel olarak, VALAMOR ba�ka bir t�bbi �r�n ile birlikte uyguland���nda, CYP3A4 inhibit�rleri ile birlikte kullan�ma ili�kin �neriler i�in di�er �r�n�n K�B'�ne ba�vurulmal�d�r. VALAMOR, dar terap�tik endekse sahip CYP3A4 substratlar� ile birlikte uygulan�rken dikkatli olunmas� �nerilir (bkz. b�l�m 4.4). Bunlarla s�n�rl� olmamak �zere alfentanil, siklosporin, everolimus, fentanil, sirolimus ve takrolimus dahil dar terap�tik endekse sahip duyarl� CYP3A4 substratlar�n�n dozunun azalt�lmas� gerekebilir ��nk� ribosiklib bunlar�n maruziyetini art�rabilir.
600 mg dozundaki ribosiklibin �u CYP3A4 substratlar� ile e� zamanl� uygulanmas�ndan ka��n�lmal�d�r: alfuzosin, amiodaron, sisaprid, pimozid, kinidin, ergotamin, dihiroergotamin, ketiapin, lovastatin, simvastatin, sildenafil, midazolam, triazolam.
Sa�l�kl� g�n�ll�lerde tek ba��na kafein (CYP1A2 substrat�) al�nmas� ile kar��la�t�r�ld���nda kafeinin �oklu ribosiklib (400 mg) dozlar� ile birlikte al�nmas� kafein maruziyetini %20 (1,20 kat) art�rm��t�r. Klinik olarak anlaml� 600 mg dozunda FEFK'nin kullan�ld��� sim�lasyonlar, ribosiklibin CYP1A2 substratlar� �zerinde sadece �ok zay�f inhibit�r etkisini �ng�rm��t�r (EAA de�erinde <2 kat art��).
Ta��y�c�lar�n substratlar� olan maddeler
�n vitro de�erlendirmeler, ribosiklibin ila� ta��y�c�lar� P-gp, BCRP, OATP1B1/B3, OCT1, OCT2, MATE1 ve BSEP'in aktivitelerini inhibe etme y�n�nde potansiyele sahip oldu�una i�aret etmi�tir. Digoksin, pitavastatin, pravastatin, rosuvastatin ve metformini i�eren ancak bunlarla s�n�rl� olmayan dar terap�tik indeks sergileyen bu ta��y�c�lar�n duyarl� substratlar� ile e� zamanl� tedavisi s�ras�nda toksisite a��s�ndan dikkat ve takip tavsiye edilir.
�la�-besin etkile�imleri
VALAMOR a� veya tok karn�na uygulanabilir (bkz. B�l�m 4.2 ve 5.2). Gastrik pH de�erini y�kselten t�bbi �r�nler
Ribosiklib 4,5 pH de�erinde veya bu de�erin alt�nda ve biyo-uyumlu ortamlarda (pH 5 ve 6,5) y�ksek ��z�n�rl�k sergiler. Ribosiklibin gastrik pH'i y�kselten t�bbi �r�nler ile birlikte uygulanmas� klinik bir �al��mada de�erlendirilmemi�tir; ancak pop�lasyon farmakokineti�i ve kompart�manl� olmayan farmakokinetik analizlerinde de�i�mi� ribosiklib emilimi g�zlenmemi�tir.
Ribosiklib ve letrozol aras�ndaki ila�-ila� etkile�imi
Meme kanseri olan hastalarda y�r�t�len klinik �al��madan ve pop�lasyon farmakokineti�i analizinden veriler bu t�bbi �r�nlerin birlikte uygulanmas�ndan sonra ribosiklib ve letrozol aras�nda ila� etkile�imi olmad���n� g�stermi�tir.
Ribosiklib ile anastrozol aras�ndaki ila�-ila� etkile�imi
Meme kanseri olan hastalardaki bir klinik �al��maya ait veriler, ribosiklib ile anastrozol�n bir arada uygulanmas�ndan sonra bu t�bbi �r�nler aras�nda klinik olarak anlaml� bir ila� etkile�imine i�aret etmemi�tir.
Ribosiklib ve fulvestrant aras�ndaki ila�-ila� etkile�imi
Meme kanserli hastalarda y�r�t�len klinik �al��madan elde edilen veri, bu iki t�bbi �r�n�n birlikte uygulanmas�n� takiben fulvestrant�n ribosiklib maruziyeti �zerinde klinik olarak ili�kili etkisinin olmad���n� g�stermi�tir.
Ribosiklib ile tamoksifen aras�ndaki ila�-ila� etkile�imi
Meme kanseri olan hastalardaki bir klinik �al��maya ait veriler, ribosiklib ile tamoksifenin bir arada uygulanmas�ndan sonra tamoksifen maruziyetinin yakla��k 2 kat artt���n� g�stermi�tir.
Ribosiklib ile oral kontraseptifler aras�ndaki ila�-ila� etkile�imi
Ribosiklib ile oral kontraseptifler aras�nda ila�-ila� etkile�imi �al��malar� ger�ekle�tirilmemi�tir
(bkz. B�l�m 4.6). Beklenen etkile�imler
QT aral���n� uzatabilen anti-aritmik t�bbi �r�nler ve di�er t�bbi �r�nler:
VALAMOR'un anti-aritmik t�bbi �r�nler gibi QT aral���n� uzatma potansiyeli oldu�u bilinen t�bbi �r�nler (bunlarla s�n�rl� olmamak �zere amiodaron, disopiramid, prokainamid, kinidin ve sotalol) ve QT aral���n� uzatt��� bilinen di�er t�bbi �r�nler (bunlarla s�n�rl� olmamak �zere klorokuin, halofantrin, klaritromisin, siprofloksazin, levofloksasin, azitromisin, haloperidol, metadon, moksifloksasin, bepridil, pimozid ve intraven�z ondansetron) ile birlikte uygulanmas�ndan ka��n�lmal�d�r (bkz. B�l�m 4.4). Ayr�ca, VALAMOR'un tamoksifen ile kombinasyon halinde kullan�lmas� �nerilmemektedir (bkz. B�l�m 4.1, 4.4 ve 5.1).
�zel pop�lasyonlara ili�kin ek bilgiler:
Etkile�im a��s�ndan �zel pop�lasyonlara ili�kin veri bulunmamaktad�r.
Pediyatrik pop�lasyon:
Etkile�im a��s�ndan �zel pop�lasyonlara ili�kin veri bulunmamaktad�r.
4.6. Gebelik ve laktasyon
Gebelik kategorisi: C
�ocuk do�urma potansiyeli bulunan kad�nlar/Do�um kontrol� (Kontrasepsiyon)
VALAMOR ile tedaviye ba�lanmadan �nce gebelik durumu teyit edilmelidir.
VALAMOR kullanmakta olan �ocuk do�urma potansiyeline sahip kad�nlar�n tedavi s�ras�nda ve VALAMOR ile tedaviyi b�rakt�ktan sonra en az 21 g�n s�reyle etkili do�um kontrol� (�rn. �ift bariyer kontrasepsiyon) uygulamalar� gerekmektedir.
Gebelik d�nemi
Gebe kad�nlarda yeterli ve gerekli kontrol gruplar�na yer veren �al��malar bulunmamaktad�r. Hayvanlar �zerinde yap�lan ara�t�rmalar�n sonu�lar�na g�re, ribosiklib hamile kad�nlara uyguland���nda fetal zarara neden olabilir (bkz. B�l�m 5.3). VALAMOR'un hamilelik esnas�nda ve �ocuk do�urma potansiyeli olup kontrasepsiyon y�ntemi uygulamayan kad�nlarda uygulanmas� �nerilmemektedir.
Ribosiklibin gebe kad�nlarda kullan�m�na ili�kin yeterli veri mevcut de�ildir. Hayvanlar �zerinde yap�lan ara�t�rmalar �reme toksisitesinin bulundu�unu g�stermi�tir (bkz. b�l�m 5.3). �nsanlara y�nelik potansiyel risk bilinmemektedir.
VALAMOR, yarar-risk de�erlendirmesine g�re gerekli olmad�k�a gebelik d�neminde kullan�lmamal�d�r.
Laktasyon d�nemi
Ribosiklibin insan s�t�yle at�l�p at�lmad��� bilinmemektedir. Ribosiklibin emen bebek ve s�t �retimi �zerine etkisini g�steren veri bulunmamaktad�r. Ribosiklib ve metabolitleri s��an s�t�ne kolayl�kla ge�mi�tir. Hayvanlar �zerinde yap�lan �al��malar, ribosiklibin s�tle at�ld���n� g�stermektedir. Emzirmenin durdurulup durdurulmayaca��na ya da VALAMOR tedavisinin durdurulup durdurulmayaca��na/ tedaviden ka��n�l�p ka��n�lmayaca��na ili�kin karar verilirken, emzirmenin �ocuk a��s�ndan faydas� ve VALAMOR tedavisinin emziren anne a��s�ndan faydas� dikkate al�nmal�d�r. VALAMOR kullanan kad�nlar�n son VALAMOR dozundan en az 21 g�n sonras�na kadar emzirmemeleri �nerilir.
�reme yetene�i/Fertilite
Ribosiklibin fertilite �zerine etkilerini g�steren klinik veri bulunmamaktad�r. Hayvanlarda yap�lan �al��malara g�re, ribosiklib �reme potansiyeli bulunan erkeklerde fertiliteyi zay�flatabilir (bkz. B�l�m 5.3).
4.7. Ara� ve makine kullan�m� �zerindeki etkiler
VALAMOR, ara� ve makine kullanma becerisi �zerinde min�r etkiye sahip olabilir. Hastalara, VALAMOR ile tedavileri s�ras�nda yorgunluk, ba� d�nmesi veya vertigo deneyimlemeleri halinde ara� ve makine kullan�rken dikkatli olmalar� s�ylenmelidir (bkz. B�l�m 4.8).
4.8. �stenmeyen etkiler
G�venlilik profilinin �zeti
Birle�tirilmi� veri k�mesinde ribosiklib art� herhangi bir kombinasyon i�in s�kl���n plasebo art� herhangi bir kombinasyon i�in s�kl�ktan fazla oldu�u en yayg�n advers reaksiyonlar (≥ %20 s�kl�kla bildirilen) n�tropeni, enfeksiyonlar, bulant�, yorgunluk, diyare, l�kopeni, kusma, ba� a�r�s�, konstipasyon, alopesi, �ks�r�k, d�k�nt�, s�rt a�r�s�, anemi ve anormal karaci�er fonksiyonu testleridir.
Birle�tirilmi� veri k�mesinde ribosiklib art� herhangi bir kombinasyon i�in s�kl���n plasebo art� herhangi bir kombinasyon i�in s�kl�ktan fazla oldu�u en yayg�n derece 3/4 advers reaksiyonlar (≥2 s�kl�kla bildirilen) n�tropeni, l�kopeni, anormal karaci�er fonksiyonu testleri, lenfopeni, enfeksiyonlar, s�rt a�r�s�, anemi, yorgunluk, hipofosfatemi ve kusmad�r.
Nedensel ili�kiye bak�lmaks�z�n, faz III �al��malar�nda kombinasyona bakmaks�z�n ribosiklib alan hastalarda advers olaylara ba�l� doz azalt�m� hastalar�n % 39,5'inde; faz III �al��malar�nda herhangi bir kombinasyon ve ribosiklib alan hastalarda tedavinin kal�c� olarak durdurulmas� hastalar�n % 8,7'sinde bildirilmi�tir.
Advers reaksiyonlar�n tablo halinde �zeti
Ribosiklibin genel g�venlilik profili, ribosiklibi endokrin tedavisi (N=582, bir aromataz inhibit�r� ile kombinasyon halinde ve N=483, fulvestrant ile kombinasyon halinde) ile kombinasyon halinde alan ve HR-pozitif, HER2-negatif ileri veya metastatik meme kanseri �zerine randomize, �ift k�r, plasebo kontroll� faz III klinik �al��malara (MONALEESA-2, MONALEESA-7 NSAI alt grubu ve MONALEESA-3) kat�lan 1065 hastan�n birle�tirilmi� veri k�mesine dayanmaktad�r. Pazarlama sonras�nda, ilave advers reaksiyonlar g�r�lm��t�r.
Birle�tirilmi� faz III �al��malar�n veri k�mesi genelinde ribosiklib tedavisine medyan maruziyet s�resi 19,2 ay olup hastalar�n %61,7'sinde maruziyet ≥12 ayd�r.
Faz III �al��malardan bildirilen advers reaksiyonlar (Tablo 6) MedDRA sistem organ s�n�f�na g�re listelenmektedir. Her bir sistem organ s�n�f� i�inde advers reaksiyonlar s�kl��a g�re, en s�k reaksiyonlar ilk belirtilerek s�ralanmaktad�r. Her bir s�kl�k grupland�rmas� i�inde advers reaksiyonlar azalan ciddilik derecelerine g�re sunulmaktad�r. Ayr�ca, her advers reaksiyon i�in kar��l�k gelen s�kl�k kategorisi �u sisteme dayanmaktad�r (CIOMS III):
�ok yayg�n (≥1/10); yayg�n (≥1/100 ila <1/10); yayg�n olmayan (≥1/1.000 ila <1/100); seyrek (≥1/10.000 ila <1/1.000); �ok seyrek (<1/10.000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor).
Tablo 7 �� Faz III klinik �al��mada ve pazarlama sonras� deneyim s�ras�nda g�zlenen advers reaksiyonlar
Advers reaksiyon | S�kl�k |
Enfeksiyonlar ve enfestasyonlar | |
Enfeksiyonlar | �ok yayg�n |
Kan ve lenf sistemi hastal�klar� | |
N�tropeni, l�kopeni, anemi, lenfopeni Trombositopeni, febril n�tropeni | �ok yayg�n Yayg�n |
Metabolizma ve beslenme hastal�klar� | |
��tah azalmas� Hipokalsemi, hipokalemi, hipofosfatemi | �ok yayg�n Yayg�n |
Sinir sistemi hastal�klar� | |
Ba� a�r�s�, ba� d�nmesi Vertigo | �ok yayg�n Yayg�n |
G�z hastal�klar� | |
G�zya�� salg�s�nda art��, g�z kurulu�u | Yayg�n |
Kardiyak hastal�klar� | |
Senkop | Yayg�n |
Solunum, g���s bozukluklar� ve mediastinal hastal�klar | |
Dispne, �ks�r�k | �ok yayg�n |
�nterstisyel akci�er hastal��� (�AH)/pn�monit | Yayg�n |
Gastrointestinal hastal�klar | |
Bulant�, diyare, kusma, konstipasyon, kar�n a�r�s� , stomatit, dispepsi Disguzi | �ok yayg�n Yayg�n |
Hepatobiliyer hastal�klar | |
Hepatotoksisite | Yayg�n |
Deri ve deri alt� doku hastal�klar� | |
Alopesi, d�k�nt� , prurit Eritem, cilt kurulu�u, vitiligo Toksik epidermal nekroliz (TEN)* | �ok yayg�n Yayg�n Bilinmiyor |
Kas-iskelet bozukluklar, ba� doku ve kemik hastal�klar� | |
S�rt a�r�s� | �ok yayg�n |
Genel bozukluklar ve uygulama b�lgesine ili�kin durumlar | |
Yorgunluk, periferik �dem, pireksi, asteni Orofarengeal a�r�, a��z kurulu�u | �ok yayg�n Yayg�n |
Ara�t�rmalar | |
Anormal karaci�er fonksiyonu testleri Kan kreatinin d�zeyinde art��, elektrokardiyogramda QT uzamas� | �ok yayg�n Yayg�n |
Se�ilen advers reaksiyonlar�n tan�m�
N�tropeni
N�tropeni en s�k bildirilen advers reaksiyon (%75,4) olup, faz III �al��malar�nda ribosiklib ve herhangi bir kombinasyon kullanan hastalar�n %62'sinde n�trofil say�mlar�nda derece 3 veya 4 azalma (laboratuvar bulgular� temelinde) bildirilmi�tir.
Derece 2, 3 veya 4 n�tropeni g�r�len hastalar aras�nda, bir olay ya�am�� hastalar i�in ba�lang�ca kadar ge�en medyan s�re 17 g�nd�r. Derece ≥3 d�zelmeye (normalizasyona veya derece <3'e) kadar ge�en medyan s�re ribosiklib art� herhangi bir kombinasyon tedavisi kollar�nda tedavide ara vermeyi ve/veya azaltmay� ve/veya tedavinin b�rak�lmas�n� takiben 12 g�nd�r. Febril n�tropeni faz III �al��malar�nda ribosiklibe maruz kalm�� hastalar�n yakla��k %1,7'sinde bildirilmi�tir. Hastalara herhangi bir ate� olay�n� derhal bildirmeleri s�ylenmelidir.
N�tropeni, �iddet derecesine dayal� olarak laboratuvar izlemi, dozlara ara verme ve/veya doz modifikasyonu ile kontrol edilmi�tir. N�tropeniye ba�l� tedavinin b�rak�lmas� d���k oranda ger�ekle�mi�tir (%0,8) (bkz. B�l�m 4.2 ve 4.4).
Hepatobiliyer toksisite
Faz III klinik �al��malarda, hepatobiliyer toksisite olaylar� ribosiklib art� herhangi bir kombinasyon kollar�ndaki hastalarda, plasebo art� herhangi bir kombinasyon kollar�ndaki hastalara k�yasla daha y�ksek bir oranda g�r�lm�� (s�ras�yla %27,3'e kar��l�k %19,6), ribosiklib art� herhangi bir kombinasyon ile tedavi edilen hastalar aras�nda daha fazla derece 3/4 advers olay bildirilmi�tir (s�ras�yla %13,2'ye k�yasla %6,1). Transaminazlarda y�kselme g�zlemlenmi�tir. Ribosiklib ve plasebo kollar�nda s�ras�yla ALT (%11,2'ye kar��l�k %1,7) ve AST'de (%7,8'e kar��l�k %2,1) derece 3 veya 4 art��lar bildirilmi�tir. Kolestaz yoklu�unda alkalen fosfataz normal iken ALT veya AST de�erlerinde normalin �st s�n�r�n�n �� kat�ndan b�y�k ve toplam bilirubin de�erinde normalin �st s�n�r�n�n iki kat�ndan b�y�k e� zamanl� y�kselmeler, 6 hastada g�r�lm��t�r (�al��ma A2301'de [MONALEESA-2] 4 hasta; ribosiklib tedavisi b�rak�ld�ktan sonra d�zeyleri 154 g�n i�inde normale d�nm��t�r ve �al��ma F2301'de [MONALEESA-3] 2 hasta; ribosiklib tedavisi b�rak�ld�ktan sonra d�zeyleri s�ras�yla 121 ve 532 g�n i�inde normale d�nm��t�r). �al��ma E2301'de (MONALEESA-7) bu gibi vakalar raporlanmam��t�r.
Hepatobiliyer toksisite olaylar� nedeniyle dozlara ara verme ve/veya doz ayarlamalar�, ribosiklib art� herhangi bir kombinasyon ile tedavi edilen hastalar�n %12,3'�nde, a��rl�kl� olarak ALT d�zeyinde y�kselme (%7,9) ve/veya AST d�zeyinde y�kselme (%7,3) nedeniyle bildirilmi�tir. Ribosiklib art� herhangi bir kombinasyon tedavisinin anormal karaci�er fonksiyonu testleri veya hepatotoksisite nedeniyle kesilmesi hastalar�n s�ras�yla %2,4 ve
%0,3'�nde s�z konusu olmu�tur (bkz. B�l�m 4.2 ve 4.4).
Faz III �al��malarda, derece 3 veya 4 ALT veya AST art��� olaylar�n�n %70,9'u (90/127) tedavinin ilk 6 ay� i�inde meydana gelmi�tir. Derece 3 veya 4 ALT/AST art��� g�r�len hastalar aras�nda ba�lang�ca kadar ge�en medyan s�re ribosiklib art� herhangi bir kombinasyon kollar� i�in 92 g�nd�r. Ribosiklib art� herhangi bir kombinasyon kollar�nda d�zelmeye (normalizasyona veya derece ≤2'ye) kadar ge�en medyan s�re 21 g�nd�r.
QT uzamas�
�al��ma E2301'de (MONALEESA-7) �al��ma ba�lang�c�na g�re g�zlenen ortalama QTcF art���, NSAI art� plasebo alt grubu ile kar��la�t�r�ld���nda tamoksifen art� plasebo alt grubunda yakla��k 10 milisaniye daha y�ksek olarak tek ba��na tamoksifenin, ribosiklib art� tamoksifen grubunda g�zlenen QTcF de�erlerine katk�da bulunmu� olabilecek bir QTcF uzatma etkisinin oldu�unu d���nd�rm��t�r. Plasebo kolunda, �al��ma ba�lang�c�na g�re >60 milisaniyelik QTcF aral��� art��� tamoksifen alan 6/90 (%6,7) hastada g�r�l�rken, NSAI alan hi�bir hastada g�r�lmemi�tir (bkz. b�l�m 5.2).Ribosiklib art� tamoksifen alan hastalar�n 14/87'sinde (%16,1) ve ribosiklib art� bir NSAI alan hastalar�n 18/245'inde (%7,3), �al��ma ba�lang�c�na k�yasla > 60 milisaniyelik bir QTcF aral��� art��� g�zlenmi�tir. Ribosiklibin tamoksifen ile kombinasyon halinde kullan�m� �nerilmemektedir (bkz. B�l�m 5.1).
Faz III klinik �al��malarda, ribosiklib art� letrozol veya fulvestrant kollar�nda hastalar�n %9,3'� ve plasebo art� letrozol veya fulvestrant kollar�nda hastalar�n %3,5'i en az bir QT aral��� uzamas� olay� ya�am��t�r (EKG'de QT uzamas� ve senkop dahil). EKG verilerinde inceleme 15 hastan�n (%1,4) >500 milisaniye ba�lang�� sonras� QTcF de�erine sahip oldu�unu ve 61 hastan�n (%5,8) QTcF aral�klar�nda ba�lang�ca g�re >60 milisaniye art�� ya�ad���n� g�stermi�tir. Torsade de pointes vakalar� bildirilmemi�tir. Elektrokardiyogramda QT uzamas� ve senkop nedeniyle dozlara ara verme/doz ayarlamalar�, ribosiklib art� letrozol veya fulvestrant ile tedavi edilen hastalar�n %2,9'unda bildirilmi�tir.
EKG verilerinin analizi, ribosiklib art� letrozol veya fulvestrant kollar�nda ve plasebo art� letrozol veya fulvestrant kollar�nda s�ras�yla 55 hasta (%5,2) ve 12 hastada (%1,5) en az bir
>480 milisaniye ba�lang�� sonras� QTcF sonucu g�stermi�tir. >480 milisaniye QTcF uzamas� olan hastalar aras�nda olay�n ba�lang�c�na kadar ge�en medyan s�re kombinasyona bak�lmaks�z�n 15 g�n olmu� ve dozlara ara verildi�inde ve/veya doz azalt�ld���nda bu de�i�ikliklerin geri d�n��l� oldu�u g�r�lm��t�r (bkz. B�l�m 4.2, 4.4 ve 5.2).
B�brek yetmezli�i olan hastalar
�� �nemli �al��mada, hafif b�brek yetmezli�i olan 341 hasta ve orta derecede b�brek yetmezli�i olan 97 hasta ribosiklib ile tedavi edilmi�tir. �iddetli b�brek yetmezli�i olan hastalar �al��maya dahil edilmemi�tir (bkz. B�l�m 5.1). Tedavi s�ras�nda ba�lang��taki b�brek yetmezli�inin derecesi ile kan kreatinin de�erleri aras�nda bir korelasyon bulunmaktad�r. Hafif veya orta derecede b�brek yetmezli�i olan hastalarda QT uzamas� ve trombositopeni oranlar�nda hafif art�� g�zlenmi�tir. Bu toksisiteler durumunda izleme ve doz ayarlama �nerileri i�in b�l�m 4.2 ve 4.4 bak�n�z.
��pheli advers reaksiyonlar�n raporlanmas�
Ruhsatland�rma sonras� ��pheli ila� advers reaksiyonlar�n�n raporlanmas� b�y�k �nem ta��maktad�r. Raporlama yap�lmas�, ilac�n yarar / risk dengesinin s�rekli olarak izlenmesine olanak sa�lar. Sa�l�k mesle�i mensuplar�n�n herhangi bir ��pheli advers reaksiyonu T�rkiye Farmakovijilans Merkezi (T�FAM)'ne bildirmeleri gerekmektedir. (www.titck.gov.tr; e- posta: tufam@titck.gov.tr; tel: 0 800 314 00 08; faks: 0 312 218 35 99)
4.9. Doz a��m� ve tedavisi
Raporlanm�� VALAMOR ile doz a��m� vakalar�na ait s�n�rl� deneyim bulunmaktad�r. Bir doz a��m� olay�nda, bulant� ve kusma gibi semptomlar meydana gelebilir. Buna ilaveten, hematolojik (�rn., n�tropeni, trombositopeni) toksisite ve olas� QTc uzamas� meydana gelebilir. T�m doz a��m� durumlar�nda gerekli genel semptomatik ve destekleyici �nlemler ba�lat�lmal�d�r.
5. FARMAKOLOJ�K �ZELL�KLER
5.1. Farmakodinamik �zellikler
Farmakoterap�tik grubu: Antineoplastik ajanlar, protein kinaz inhibit�rleri, sikline ba��ml� kinaz (CDK) inhibit�rleri
ATC kodu: L01EF02
Etki mekanizmas�:
Ribosiklib se�ici sikline ba��ml� kinaz (CDK) 4 ve 6 inhibit�r� olup, biyokimyasal analizlerde s�ras�yla 0,01 (4,3 ng/ml) ve 0,039 µM'lik (16,9 ng/ml) %50 inhibisyon (IC) de�erleri ile sonu�lan�r. Bu kinazlar D-sikline ba�lanma ile birlikte aktive olurlar ve h�cre d�ng�s� progresyonu ve h�cresel proliferasyona yol alan sinyal yolaklar�nda �nemli bir rol oynarlar.
Siklin D-CDK4/6 kompleksi h�cre d�ng�s� progresyonunu, retinoblastoma proteininin (pRb) fosforilasyonu yoluyla reg�le eder.
�n vitro ortamda ribosiklib pRb fosforilasyonunu azaltarak h�cre d�ng�s�n�n G1 faz�nda durmaya ve meme kanseri h�cre dizilerinde h�cre proliferasyonunda azalmaya yol a�m��t�r. �n vivo ortamda tek ajan ribosiklib ile tedavi, iyi tolere edilen dozlarda pRb fosforilasyonunun inhibisyonu ile korelasyon g�steren t�m�r regresyonuna yol a�m��t�r.
Hastadan elde edilmi� �strojen resept�r-pozitif meme kanseri ksenogreft modelinin kullan�ld��� in vivo �al��malarda, ribosiklib ve anti�strojen ajanlar� (�rn. letrozol) kombinasyonu, ajanlar�n tek ba��na uyguland��� durumla kar��la�t�r�ld���nda, kal�c� t�m�r regresyonu ve doz uygulamas� b�rak�ld�ktan sonra yeni t�m�r b�y�mesinde gecikme ile birlikte daha iyi bir t�m�r b�y�mesi inhibisyonu sa�lam��t�r. Ek olarak, ZR751 ER+ insan meme kanseri ksenogreftlerini ta��yan ve ba����kl�k yetmezli�i bulunan farelerde fulvestrant ile kombinasyon halindeki ribosiklibin in vivo anti-t�m�r aktivitesi de�erlendirilmi�tir ve fulvestrant ile kombinasyon, tam t�m�r b�y�mesi inhibisyonu ile sonu�lanm��t�r.
Bilinen ER durumuna sahip meme kanseri h�cre hatlar� paneli test edildi�inde, ribosiklibin ER- olanlara k�yasla ER+ meme kanseri h�cre hatlar�nda daha etkili oldu�u g�sterilmi�tir. �imdiye kadar test edilen klinik �ncesi modellerde, ribosiklib aktivitesi i�in intakt pRb gerekmi�tir.
Kardiyak elektrofizyoloji:
�leri evre kanseri olan hastalarda ribosiklibin QTc aral��� �zerindeki etkisini de�erlendirmek i�in tek dozdan sonra ve kararl� durumda seri, ��l� EKG'ler kaydedilmi�tir. Bir farmakokinetik- farmakodinamik analiz, 50-1200 mg aral���nda dozlarla ribosiklib ile tedavi edilen 997 hastay� i�ermi�tir. Bu analiz, ribosiklibin QTc aral���nda konsantrasyona ba��ml� art��lara neden oldu�unu d���nd�rm��t�r. NSAI veya fulvestrant ile kombinasyon halindeki 600 mg ribosiklib i�in ba�lang��a g�re tahmini ortalama QTcF de�i�imi, tamoksifen ile kombinasyon halinde 34,7 milisaniye (%90 GA: 31,64, 37,78) ile kar��la�t�r�ld���nda, s�ras�yla kararl� durumdaki geometrik ortalamada C'ta s�ras�yla 22 milisaniye (%90 GA: 20,56, 23,44) ve 23,7
milisaniye (%90 GA: 22,31, 25,08) olmu�tur (bkz. B�l�m 4.4). Klinik etkililik ve g�venlilik:
�al��ma CLEE011A2301 (MONALEESA-2)
Hormon resept�r� pozitif, HER2 negatif ileri evre meme kanseri olan, ileri evre hastal�k i�in daha �nce bir tedavi almam�� postmenopozal kad�nlarda tek ba��na letrozole k�yasla ribosiklibin letrozol ile kombine tedavisi randomize, �ift k�r, plasebo kontroll�, �ok merkezli bir faz III klinik �al��mada de�erlendirilmi�tir.
Toplam 668 hasta, karaci�er ve/veya akci�er metastazlar�n�n varl���na (Var [n=292 (44%)]) k�yasla Yok [n=376 (56%)]) g�re stratifiye edilerek 1:1 oran�nda ribosiklib 600 mg ve letrozol (n=334) veya plasebo ve letrozol (n=334) almak �zere randomize edilmi�tir. Demografik �zellikler ve ba�lang�� hastal�k karakteristikleri �al��ma kollar� aras�nda dengeli ve kar��la�t�r�labilir nitelikte olmu�tur. Ribosiklib, 28 g�n s�reyle g�nde bir kez 2,5 mg letrozol ile kombinasyon halinde oral yolla g�nde bir kez 600 mg dozunda arka arkaya 21 g�n verilmi�, ard�ndan 7 g�n tedavisiz ara b�rak�lm��t�r. �al��ma s�resince veya hastal�k progresyonundan sonra hastalar�n plasebodan ribosiklibe ge�i� yapmalar�na izin verilmemi�tir.
Bu �al��maya al�nan hastalar�n medyan ya�� 62'dir (aral�k 23 - 91). Hastalar�n %44,2'si 65 ya��n �zerindedir ve bu hastalar�nda 69'u 75 ya��n �zerindedir. Dahil edilen hastalar beyaz (%82,2), Asyal� (%7,6) ve siyaht�r (%2,5). T�m hastalar�n ECOG performans durumu 0 veya 1'dir. Ribosiklib kolunda, hastalar�n %46,6's� �al��maya girmeden �nce neoadjuvan veya adjuvan
ko�ullarda kemoterapi g�rm��t�r ve %51,3'� neoadjuvan veya adjuvan ko�ullarda anti- hormonal tedavi alm��t�r. Hastalar�n %34,1'i de novo hastad�r. Hastalar�n %22'sinde sadece kemi�i tutan hastal�k ve %58,8'inde visseral hastal�k vard�r. Daha �nce anastrozol veya letrozol ile (neo)adjuvan tedavi g�rm�� hastalar �al��ma randomizasyonundan en az 12 ay �nce bu tedaviyi tamamlam�� olmal�d�r.
Birincil analiz
�al��ma i�in birincil sonlan�m noktas� t�m pop�lasyonda (t�m randomize hastalar) ara�t�r�c� de�erlendirmesi temelinde Solid T�m�rlerde Yan�t De�erlendirme Kriterleri (RECIST v1.1) kullan�larak hedeflenen progresyonsuz sa�kal�m (PFS) olaylar�n�n %80'i g�zlendikten sonra y�r�t�lm�� planlanan ara analizde kar��lanm�� ve k�rlenmi� ba��ms�z merkezi radyolojik de�erlendirme ile do�rulanm��t�r.
Tam analiz setinde etkililik sonu�lar�, plasebo art� letrozol alan hastalara k�yasla, ribosiklib art� letrozol kombinasyonu ile tedavi edilen hastalar i�in klinik olarak anlaml� tedavi etkisi ile PFS'de istatistiksel olarak anlaml� d�zelme g�stermi�tir (tehlike oran� [HR] = 0,556, %95 GA: 0,429, 0,720, tek y�nl� stratifiye edilmi� log s�ra testi p de�eri 0,00000329).
Global sa�l�k durumu/QoL verileri ribosiklib art� letrozol kolu ve plasebo art� letrozol kolu aras�nda ilgili bir fark olmad���n� g�stermi�tir.
Tablo 8 ve 9'da etkililik verilerine ili�kin daha olgun bir g�ncelleme (02 Ocak 2017 veri kesme) sunulmaktad�r.
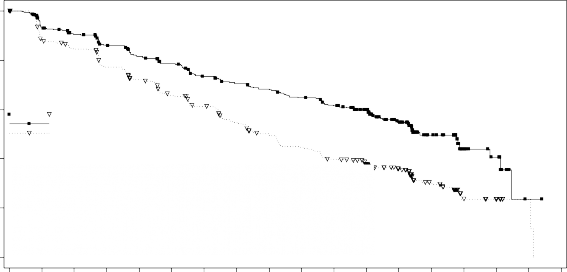
Medyan PFS ribosiklib art� letrozol ile tedavi edilen hastalar i�in 25,3 ay (%95 GA: 23, 30,3) iken, plasebo art� letrozol alan hastalar i�in 16 (%95 GA: 13,4, 18,2) ayd�r. Ribosiklib art� letrozol alan hastalar�n %54,7'sinin, plasebo art� letrozol alan hastalar�n ise %35,9'unun 24. ayda progresyonsuz oldu�u �ng�r�ld�.
Tablo 8 MONALEESA-2 - Ara�t�rmac�n�n radyolojik de�erlendirmesine dayal� etkililik sonu�lar� (PFS) (02 Ocak 2017 veri kesme)
| G�ncellenen analiz | |
| Ribosiklib art� letrozol N=334 | Plasebo art� letrozol N=334 |
Progresyonsuz sa�kal�m | ||
Medyan PFS [ay] (%95 GA) | 25,3 (23 – 30,3) | 16 (13,4 – 18,2) |
Tehlike oran� (%95 GA) | 0,568 (0,457 - 0,704) | |
p de�eri | 9,63x10 | |
GA=g�ven aral���; N=hasta say�s�; PFS = Progresyonsuz sa�kal�m | ||
�ekil 1 MONALEESA-2 - Ara�t�rmac�n�n de�erlendirmesine dayal� PFS'nin Kaplan- Meier grafi�i (02 Ocak 2017 veri kesme)
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
�nceden belirlenmi� alt grup PFS analizleri serisi prognostik fakt�rler ve ba�lang�� �zellikleri temelinde tedavi etkisinin tutarl�l���n� ara�t�rmak �zere y�r�t�lm��t�r. Ya�, �rk, �nceki adjuvan veya neo adjuvan kemoterapi veya hormonal tedaviler, karaci�er ve/veya akci�er tutulumu ve sadece kemi�e metastaz yapan hastal��� i�eren t�m bireysel hasta alt gruplar�nda hastal�k progresyonu veya �l�m riskinde ribosiklib art� letrozol kolu lehine bir azalma g�zlenmi�tir. Bu, karaci�er ve/veya akci�er metastazlar� olan hastalar i�in (HR 0,561 [%95 GA: 0,424, 0,743], medyan progresyonsuz sa�kal�m [mPFS] ribosiklib art� letrozol i�in 24,8 ay ve tek ba��na letrozol i�in 13,4 ay) ya da karaci�er ve/veya akci�er metastazlar� olmayan hastalar i�in (HR 0,597 [%95 GA: 0,426, 0,837], mPFS 27,6 aya kar��l�k 18,2 ay) belirgindir.
Genel yan�t ve klinik fayda oranlar� i�in g�ncellenmi� bulgular Tablo 9'da g�sterilmektedir.
Tablo 9 MONALEESA-2 - Ara�t�rmac� de�erlendirmesine dayal� etkililik sonu�lar� (ORR, CBR) (02 Ocak 2017 veri kesme)
Analiz | Ribosiklib art� letrozol (%, %95 GA) | Plasebo art� letrozol (%, %95 GA) | p de�eri |
Tam analiz seti | N=334 | N=334 |
|
Genel yan�t oran� | 42,5 (37,2; 47,8) | 28,7 (23,9; 33,6) | 9,18 × 10 |
Klinik fayda oran� | 79,9 (75,6; 84,2) | 73,1 (68,3; 77,8) | 0,018 |
�l��lebilir hastal��� olan hastalar | N=257 | N=245 |
|
Genel yan�t oran� | 54,5 (48,4; 60,6) | 38,8 (32,7;44,9) | 2,54 × 10 |
Klinik fayda oran� | 80,2 (75,3; 85) | 71,8 (66,2; 77,5) | 0,018 |
Nihai genel sa�kal�m analizi
Genel �al��ma pop�lasyonunda bu nihai genel sa�kal�m analizinden bulgular Tablo 10 ve �ekil 2'de sunulmaktad�r.
Tablo 10 MONALEESA-2- Etkililik bulgular� (OS) (10 Haziran 2021 veri kesme)
Genel sa�kal�m, genel �al��ma pop�lasyonu | Ribosiklib art� letrozol N=334 | Plasebo art� letrozol N=334 |
Olay say�s� – n [%] | 181 (54,2) | 219 (65,6) |
Medyan OS [ay] (%95 GA) | 63,9 (52,4; 71) | 51,4 (47,2; 59,7) |
Tehlike oran� (%95 GA) | 0,765 (0,628; 0,932) | |
p-de�eri | 0,004 | |
OS olays�z oran, (%) (%95 GA) |
| |
24 ay | 86,6 (82,3; 89,9) | 85 (80,5; 88,4) |
60 ay | 52,3 (46,5; 57,7) | 43,9 (38,3; 49,4) |
72 ay | 44,2 (38,5; 49,8) | 32 (26,8; 37,3) |
GA = g�ven aral��� IRT'ye g�re akci�er ve/veya karaci�er metastazlar� durumuna g�re y�r�t�len s�n�fland�rma | ||
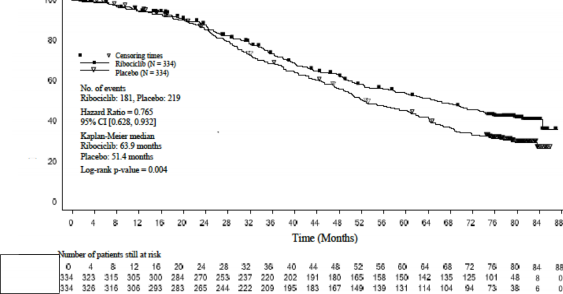
�ekil 2 MONALEESA-2- Genel pop�lasyonda OS'nin Kaplan-Meier grafi�i (10 Haziran 2021 veri kesme)
Log-s�ra testi ve Cox PH modeli IRT'ye g�re karaci�er ve/veya akci�er metastazlar� durumuna g�re s�n�fland�r�l�r.
Tek tarafl� P-de�eri s�n�fland�r�lm�� log-s�ra testinden elde edilir.
�al��ma CLEE011E2301 (MONALEESA-7)
Ribosiklib, randomize �ift k�r, plasebo kontroll�, �ok merkezli bir faz III klinik �al��mada, hormon resept�r� pozitif, HER2-negatif ilerlemi� meme kanseri olan premenopozal ve perimenopozal kad�nlar�n tedavisinde, NSAI veya tamoksifen art� goserelin ile kombinasyon halinde, bir NSAI veya tamoksifen art� goserelin ile kombinasyon halinde plasebo kar��s�nda de�erlendirilmi�tir. MONALEESA-7'deki hastalar ilerlemi� meme kanseri i�in �nceden endokrin tedavisi g�rmemi�lerdir.
Toplam 672 hasta, karaci�er ve/veya akci�er metastazlar�n�n varl���na (Var [n=344 (%51,2)] kar��s�nda Yok [n=328 (%48,8)]), ilerlemi� hastal�k i�in �nceki kemoterapi (Var [n=120 (%17,9)] kar��s�nda Yok [n=552 (%82,1)]) ve endokrin kombinasyon partneri (NSAI ve goserelin [n=493 (%73,4)] kar��s�nda tamoksifen ve goserelin [n=179 (%26,6)]) g�re katmanland�r�larak 1:1 oran�nda ya ribosiklib 600 mg art� NSAI/tamoksifen art� goserelin (n=335) ya da plasebo art� NSAI/tamoksifen art� goserelin (n=337) almak �zere randomize edilmi�tir. Demografik �zellikler ve �al��ma ba�lang�c� hastal�k karakteristikleri �al��ma kollar� aras�nda dengeli ve birbirine yak�n olmu�tur. Ribosiklib, 28 g�n s�reyle g�nde bir kez oral yolla NSAI (letrozol 2,5 mg veya anastrozol 1 mg) veya tamoksifen (20 mg) ve 28 g�nde bir subkutan yolla goserelin (3,6 mg) ile kombinasyon halinde, hastal�k progresyonuna veya kabul edilemez toksisiteye kadar oral yolla g�nde bir kez 600 mg dozunda arka arkaya 21 g�n verilmi�, ard�ndan 7 g�n tedavisiz ara b�rak�lm��t�r. �al��ma s�ras�nda veya hastal�k progresyonundan sonra hastalar�n plasebodan ribosiklibe ge�i� yapmalar�na izin verilmemi�tir. Endokrin kombinasyon partnerlerinin de�i�tirilmesine de izin verilmemi�tir.
Bu �al��maya kaydedilen hastalar�n medyan ya�� 44 y�ld�r (aral�k: 25 ila 58) ve hastalar�n
%27,7'si 40 ya��n alt�ndad�r. Dahil edilen hastalar�n �o�u Beyaz (%57,7), Asyal� (%29,5) veya Siyaht�r (%2,8) ve neredeyse t�m hastalar�n (%99) ba�lang�� ECOG performans durumu 0 veya 1'dir. �al��maya giri� �ncesinde bu 672 hastan�n %14'� �nceden metastatik hastal�k i�in
kemoterapi g�rm��, %32,6's� adjuvan ve %18'i neoadjuvan ko�ullarda kemoterapi alm��, ve
%39,6's� adjuvan ve %0,7'si neoadjuvan ko�ullarda endokrin tedavisi alm��t�r. �al��ma E2301'de hastalar�n %40,2'sinde de novo metastatik hastal�k, %23,7'sinde sadece kemi�i tutan hastal�k ve %56,7'sinde visseral hastal�k s�z konusudur.
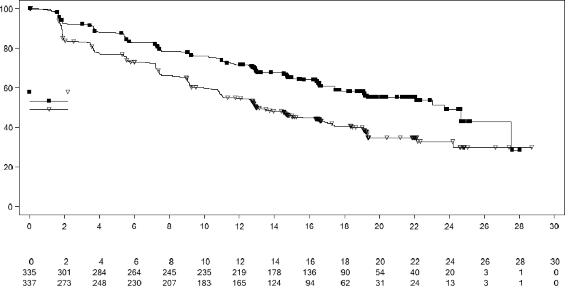
�al��ma, tam analiz setinde (randomize edilen t�m hastalar) ara�t�rmac� de�erlendirmesine dayanan ve RECIST v1.1 kriterleri kullan�larak 318 progresyonsuz sa�kal�m (PFS) olay�ndan sonra ger�ekle�tirilen birincil analizde birincil sonlan�m noktas�na ula�m��t�r. Birincil etkililik sonu�lar�, k�rlenmi� ba��ms�z merkezi radyolojik de�erlendirmeye dayal� PFS sonu�lar�yla desteklenmi�tir. Birincil PFS analizi tarihinde medyan takip s�resi 19,2 ay olmu�tur.
Genel �al��ma pop�lasyonunda etkililik sonu�lar�, plasebo art� NSAI/tamoksifen art� goserelin kullanan hastalar ile kar��la�t�r�ld���nda ribosiklib art� NSAI/tamoksifen art� goserelin kullanan hastalarda, klinik olarak anlaml� bir tedavi etkisiyle birlikte PFS'de istatistiksel olarak anlaml� bir d�zelme g�stermi�tir (Tehlike Oran�: 0,553, %95 GA: 0,441, 0,694, tek yanl� katmanland�r�lm�� log s�ra testi p de�eri 9,83x10). Medyan PFS, ribosiklib art� NSAI/tamoksifen art� goserelin ile tedavi edilen hastalarda 23,8 ay (%95 GA: 19,2, NE) ve plasebo art� NSAI/tamoksifen art� goserelin verilen hastalarda 13 ay (%95 GA: 11, 16,4) olmu�tur.
PFS i�in da��l�m�, �ekil 3'te, PFS i�in Kaplan-Meier e�risinde �zetlenmektedir.
�ekil 3 MONALEESA-7 – Genel pop�lasyonda PFS'nin ara�t�rmac� de�erlendirmesine dayal� Kaplan-Meier grafi�i
Randomize edilen hastalar�n yakla��k %40'�ndan rastgele se�ilmi� bir alt grubun k�rlenmi� ba��ms�z merkezi radyolojik de�erlendirmesine dayal� PFS sonu�lar�, ara�t�rmac� de�erlendirmesine dayal� birincil etkililik sonu�lar�n� destekler nitelikte olmu�tur (Tehlike Oran�: 0,427; %95 GA: 0,288, 0,633).
Birincil PFS analizi tarihinde genel sa�kal�m verileri haz�r olmay�p 89 (%13) �l�m (Tehlike Oran�: 0,916 [%95 GA: 0,601, 1,396]) s�z konusu olmu�tur.
RECIST v1.1'e g�re ara�t�rmac� de�erlendirmesine dayal� genel yan�t oran� (ORR) ribosiklib kolunda (%40,9; %95 GA: 35,6, 46,2), plasebo kolundan daha y�ksek bulunmu�tur (%29,7;
%95 GA: 24,8, 34,6, p=0,00098). G�zlenen klinik fayda oran� (CBR), plasebo kolu (%69,7;
%95 GA: 64,8:74,6, p=0,002) ile kar��la�t�r�ld���nda ribosiklib kolunda (%79,1; %95 GA:
74,8:83,5) daha y�ksektir.
NSAI art� goserelin ile kombinasyon halinde ribosiklib veya plasebo alan 495 hastan�n �nceden tan�mlanm�� alt grup analizinde, medyan PFS ribosiklib art� NSAI alt grubunda 27,5 ay (%95 GA: 19,1, NE) ve plasebo art� NSAI alt grubunda 13,8 ay (%95 GA: 12,6, 17,4) bulunmu�tur
[Tehlike Oran�: 0,569; %95 GA: 0,436, 0,743]. Etkililik sonu�lar� Tablo 11'de �zetlenmekte ve PFS i�in Kaplan-Meier e�rileri �ekil 4'te verilmektedir.
Tablo 11 MONALEESA-7 – NSAI alan hastalarda etkililik sonu�lar� (PFS)
| Ribosiklib + NSAI + goserelin N=248 | Plasebo + NSAI + goserelin N=247 |
Progresyonsuz sa�kal�m |
|
|
Medyan PFS [ay] (%95 GA) | 27,5 (19,1; NE) | 13,8 (12,6 – 17,4) |
Tehlike oran� (%95 GA) | 0,569 (0,436; 0,743) | |
GA=g�ven aral���; N=hasta say�s�; NE = Hesaplanabilir de�il. | ||
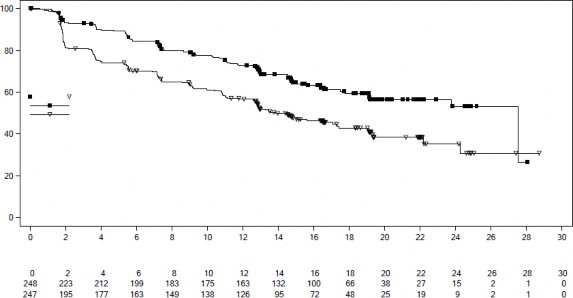
�ekil 4 MONALEESA-7 – NSAI alan hastalarda PFS'nin ara�t�rmac� de�erlendirmesine dayal� Kaplan-Meier grafi�i
Genel yan�t oran� (ORR) etkililik sonu�lar� ve RECIST v1.1'e dayal� ara�t�rmac� de�erlendirmesine g�re klinik fayda oran� (CBR) Tablo 12'de verilmektedir.
Tablo 12 MONALEESA-7 – NSAI alan hastalarda ara�t�rmac� de�erlendirmesine dayal� etkililik sonu�lar� (ORR, CBR)
Analiz | Ribosiklib art� NSAI art� goserelin (%, %95 GA) | Plasebo art� NSAI art� goserelin (%, %95 GA) |
Tam analiz seti | N=248 | N=247 |
Genel yan�t oran� (ORR) | 39,1 (33; 45,2) | 29,1 (23,5; 34,8) |
Klinik fayda oran� (CBR) | 80,2 (75,3; 85,2) | 67,2 (61,4; 73,1) |
�l��lebilir hastal��� olan hastalar | N=192 | N=199 |
Genel yan�t oran� | 50,5 (43,4; 57,6) | 36,2 (29,5; 42,9) |
Klinik fayda oran� | 81,8 (76,3; 87,2) | 63,8 (57,1; 70,5) |
Ribosiklib art� NSAI alt grubundaki sonu�lar ya�, �rk, �nceki adjuvan/ neoadjuvan kemoterapi veya hormonal tedaviler, karaci�er ve/veya akci�er tutulumu ve sadece kemi�i tutan metastatik hastal��� i�eren farkl� alt gruplar genelinde tutarl� olmu�tur.
Genel sa�kal�m verilerinin daha olgun bir g�ncellemesi (veri kesme tarihi 30 Kas�m 2018) Tablo 13 ve �ekil 5 ve 6'da verilmektedir.
�kinci OS analizinde �al��ma, OS'de istatistiksel olarak anlaml� bir iyile�me g�stererek anahtar ikincil sonlan�m noktas�n� kar��lam��t�r.
Tablo 13 MONALEESA-7 – Etkililik sonu�lar� (OS) (veri kesme tarihi 30 Kas�m 2018)
| G�ncellenmi� analiz | |
Genel sa�kal�m, genel �al��ma pop�lasyonu | Ribosiklib 600 mg N= 335 | Plasebo N=337 |
Olay say�s� – n [%] | 83 (24,8) | 109 (32,3) |
Medyan OS [ay] (%95 GA) | NE (NE;NE) | 40,9 (37,8; NE) |
Tehlike oran� ( %95 GA) | 0,712 (0,535;0,948) | |
p-de�eri | 0,00973 | |
Genel sa�kal�m, NSAI alt grubu | Ribosiklib 600 mg N=248 | Plasebo N=247 |
Olay say�s� - n [%] | 61 (24,6) | 80 (32,4) |
Medyan OS [ay] (%95 GA) | NE (NE; NE) | 40,7 (37,4; NE) |
Tehlike oran� (%95 GA) | 0,699 (0,501;0,976) | |
GA= g�ven aral���, NE = tahmin edilemez, N = hasta say�s�; | ||
�ekil 5 MONALEESA-7: Son OS analizinin Kaplan Meier grafi�i (veri kesme tarihi 30 Kas�m 2018)
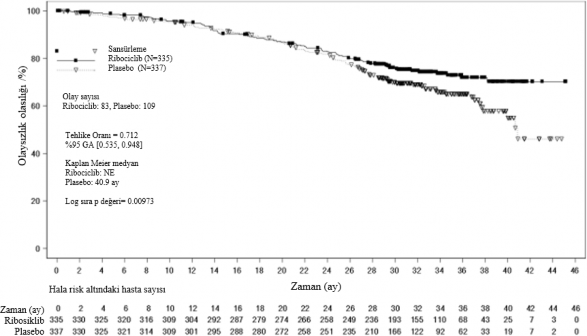
Log-s�ra testi ve Cox modeli akci�er ve/veya karaci�er metastaz�, ileri evre hastal�k i�in �nceki kemoterapi ve IRT ba��na endokrin kombinasyon partneri ile stratifiye edilmi�tir.
�ekil 6 MONALEESA-7: NSAI alan hastalarda son OS analizinin Kaplan Meier grafi�i
(veri kesme tarihi 30 Kas�m 2018)
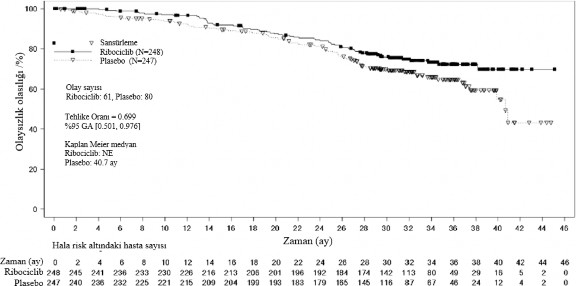
Tehlike oran� stratifiye edilmemi� Cox modeline dayanmaktad�r.
Ek olarak, genel �al��ma pop�lasyonundaki 0,692 HR de�eri (%95 GA: 0,548, 0,875) ile bu �al��mada daha �nce ribosiklib alan hastalarda sonraki basamak tedavide progresyon veya �l�m (PFS2) olas�l���, plasebo kolundaki hastalara g�re daha d���k olmu�tur. Medyan PFS2, plasebo kolunda 32,3 ay bulunmu�tur (%95 GA: 27,6, 38,3) ve ribosiklib kolunda ula��lmam��t�r (%95 GA: 39,4, NE). NSAI alt grubu i�in benzer sonu�lar g�zlenmi�, HR de�eri 0,660 (%95 GA: 0,503, 0,868) ile medyan PFS2 plasebo kolunda 32,3 ay olurken (%95 GA: 26,9, 38,3), ribosiklib kolunda ula��lmam��t�r (%95 GA: 39,4, NE).
�al��ma CLEE011F2301 (MONALEESA-3)
Ribosiklib, 2:1 randomize �ift k�r, plasebo kontroll�, �ok merkezli bir faz III klinik �al��mada, �nceden endokrin tedavi almam�� ya da sadece bir basamak endokrin tedavi alm�� hormon resept�r� pozitif, HER2-negatif ileri evre meme kanseri olan 726 postmenopozal kad�n�n tedavisinde, tek ba��na fulvestrant kar��s�nda fulvestrant ile kombinasyon halinde de�erlendirilmi�tir.
Bu �al��maya kaydedilen hastalar�n medyan ya�� 63't�r (aral�k 31 - 89). Hastalar�n %46,7'si 65 ya� ve �zeri olup bunlar aras�nda 75 ya� ve �zeri hasta oran� %13,8'dir. Dahil edilen hastalar beyaz (%85,3), Asyal� (%8,7) veya siyaht�r (%0,7) ve neredeyse t�m hastalar�n (%99,7) ECOG performans durumu 0 veya 1'dir. Bu �al��maya birinci ve ikinci basamak hastalar kaydedilmi�tir (hastalar�n %19,1'inde de novo metastatik hastal�k s�z konusudur). �al��maya giri� �ncesinde hastalar�n %42,7'si adjuvan ve %13,1'i neoadjuvan ko�ullarda kemoterapi alm��, %58,5'i adjuvan ve %1,4'� neoadjuvan ko�ullarda endokrin tedavisi alm��, %21'i ise ilerlemi� meme kanseri ko�ullar�nda �nceden endokrin tedavisi g�rm��t�r. �al��ma F2301'de %21,2'sinde sadece kemi�i tutan hastal�k ve %60,5'inde visseral hastal�k s�z konusudur.
Birincil analiz
�al��ma, tam analiz setinde (randomize edilen t�m hastalar, veri kesme tarihi: 3 Kas�m 2017) ara�t�rmac� de�erlendirmesine dayanan ve RECIST v1.1 kriterleri kullan�larak 361 progresyonsuz sa�kal�m (PFS) olay�ndan sonra ger�ekle�tirilen birincil analizde birincil sonlan�m noktas�na ula�m��t�r. Birincil PFS analizi tarihinde medyan takip s�resi 20,4 ay olmu�tur.
Birincil etkililik sonu�lar�, tam analiz setinde plasebo art� fulvestrant alan hastalar ile kar��la�t�r�ld���nda ribosiklib art� fulvestrant alan hastalarda, progresyon veya �l�m ba��l riskinde ribosiklib art� fulvestrant kolu lehine tahmini %41'lik azalmayla, PFS'de istatistiksel olarak anlaml� bir d�zelme g�stermi�tir (tehlike oran�: 0,593, %95 GA: 0,480, 0,732, tek yanl� stratifiye edilmi� log s�ra testi p de�eri 4,1x10).
Birincil etkililik bulgular� k�rlenmi� bir ba��ms�z merkezi radyolojik de�erlendirme ile %40 g�r�nt�leme alt setinin rastgele merkezi denetimi ile desteklenmi�tir (tehlike oran� 0,492; %95 GA: 0,345, 0,703).
�kinci OS ara analizi zaman�nda PFS i�in tan�mlay�c� bir g�ncelleme y�r�t�lm�� olup, genel pop�lasyon ve �nceki endokrin tedaviye g�re alt gruplarda g�ncellenmi� PFS bulgular� Tablo 14'te �zetlenmektedir ve Kaplan-Meier e�risi �ekil 7'de sunulmaktad�r.
Tablo 14 MONALEESA-3(F2301) – Ara�t�rmac� de�erlendirmesine dayal� g�ncellenmi� etkililik PFS sonu�lar� (veri kesme tarihi 3 Haziran 2019)
| Ribosiklib + fulvestrant N=484 | Plasebo + fulvestrant N=242 |
Progresyonsuz sa�kal�m genel �al��ma pop�lasyonu | ||
Olay say�s� – n [%] | 283 (58,5) | 193 (79,8) |
Medyan PFS [ay] (%95 GA) | 20,6 (18,6; 24) | 12,8 (10,9 – 16,3) |
Tehlike oran� (%95 GA) | 0,587 (0,488; 0,705) | |
Birinci basamak alt grubu | Ribosiklib + fulvestrant N=237 | Plasebo + fulvestrant N=128 |
Olay say�s� – n [%] | 112 (47,3) | 95 (74,2) |
Medyan PFS [ay] (%95 GA) | 33,6 (27,1; 41,3) | 19,2 (14,9; 23,6) |
Tehlike oran� (%95 GA) | 0,546 (0,415; 0,718) | |
�kinci basamak veya erken relaps alt grubu | Ribosiklib + fulvestrant N=237 | Plasebo + fulvestrant N=109 |
Olay say�s� – n [%] | 167 (70,5) | 95 (87,2) |
Medyan PFS [ay] (%95 GA) | 14,6 (12,5; 18,6) | 9,1 (5,8;11) |
Tehlike oran� (%95 GA) | 0,571 (0,443;0,737) | |
GA=g�ven aral���; daha �nce endokrin tedavi almam�� de novo ileri evre meme kanserli hastalar ve (neo)adjuvan endokrin tedavisinin tamamlanmas�ndan 12 ay sonra relaps olmu� hastalar hastal��� adjuvan tedavi s�ras�nda veya (neo)adjuvan endokrin tedavisinin tamamlanmas�ndan sonraki 12 ay i�inde relaps olmu� hastalar ve ileri hastal�k i�in bir basamak endokrin tedaviden sonra progresyon g�stermi� hastalar | ||
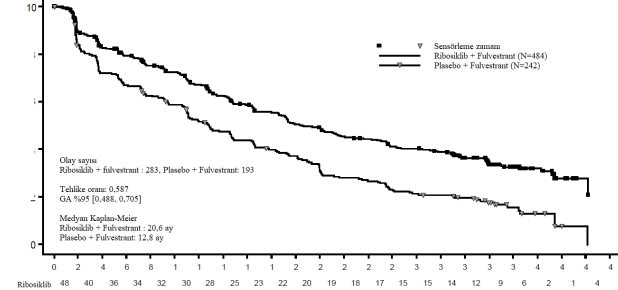
Olays�zl�k oran� (%)
�ekil 7 MONALEESA-3 (F2301)– PFS i�in Ara�t�rmac� de�erlendirmesine dayal� Kaplan-Meier grafi�i (Veri kesme tarihi: 3 Haziran 2019)
Genel yan�t oran� (ORR) ve RECIST v1.1'e dayal� ara�t�rmac� de�erlendirmesine g�re klinik fayda oran� (CBR) i�in etkililik sonu�lar� Tablo 15'te verilmektedir.
Tablo 15 MONALEESA-3 – Ara�t�rmac� de�erlendirmesine dayal� etkililik sonu�lar� (ORR, CBR) (3 Kas�m 2017 veri kesme)
Analiz | Ribosiklib + fulvestrant (%, %95 GA) | Plasebo + fulvestrant (%, %95 GA) |
Tam analiz seti | N=484 | N=242 |
Genel yan�t oran� (ORR) | 32,4 (28,3; 36,6) | 21,5 (16,3;26,7) |
Klinik fayda oran� (CBR) | 70,2 (66,2; 74,3) | 62,8 (56,7;68,9) |
�l��lebilir hastal��� olan hastalar | N=379 | N=181 |
Genel yan�t oran� | 40,9 (35,9; 45,8) | 28,7 (22,1; 35,3) |
Klinik fayda oran� | 69,4 (64,8;74) | 59,7 (52,5; 66,8) |
Ribosiklib art� fulvestrant ile tedavi edilen hastalar�n �nceden tan�mlanm�� alt grup analizine dayal� tehlike oranlar� ya�, �nceki tedavi (erken veya ilerlemi�), �nceki adjuvan/neoadjuvan kemoterapi veya hormonal tedaviler, karaci�er ve/veya akci�er tutulumu ve sadece kemi�i tutan metastatik hastal��� i�eren farkl� alt gruplar genelinde tutarl� fayda g�stermi�tir.
Genel Sa�kal�m Analizi
�kinci genel sa�kal�m analizinde, �al��ma ikincil sonlan�m noktas�n� sa�layarak, genel sa�kal�mda istatistiksel olarak anlaml� bir iyile�me g�stermi�tir.
Genel �al��ma pop�lasyonunda ve alt gruplar analizinde bu nihai genel sa�kal�m analizinden elde edilen bulgular Tablo 16 ve �ekil 8'de sunulmaktad�r.
Tablo 16 MONALEESA-3 (F2301) etkililik bulgular� (OS) (Veri kesme tarihi: 03- Haz-2019)
| Ribosiklib + fulvestrant | Plasebo + fulvestrant |
Genel �al��ma pop�lasyonu | N=484 | N=242 |
Olay say�s� – n [%] | 167 (34,5) | 108 (44,6) |
Medyan OS [ay] (%95 GA) | NE, (NE;NE) | 40 (37;NE) |
HR (%95 GA) | 0,724 (0,568; 0,924) | |
p-de�eri | 0,00455 | |
Birinci basamak alt grubu | n=237 | n=128 |
Olay say�s� – n [%] | 63 (26,6) | 47 (36,7) |
HR (%95 GA) | 0,700 (0,479;1,021) | |
�kinci basamak veya erken relaps alt grubu | n=237 | n=109 |
Olay say�s� – n [%] | 102 (43) | 60 (55) |
HR (%95 GA) | 0,730 (0,530; 1,004) | |
NE = Hesaplanamaz | ||
ve 0,025'lik bir genel anlam d�zeyi i�in Lan-DeMets (O'Brien-Fleming) alfa-harcama fonksiyonu ile tayin edildi�i �zere 0,01129'luk bir e�i�e kar�� k�yaslan�r.
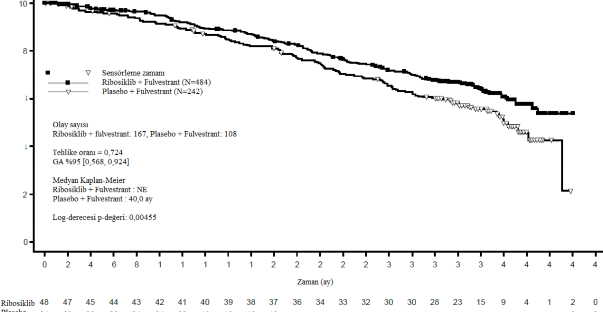
�ekil 8 MONALEESA-3 (F2301) OS Kaplan-Meier grafi�i (tam analiz seti [FAS]) (Veri kesme tarihi: 3 Haziran 2019)
Log-s�ra testi ve Cox modeli akci�er ve/veya karaci�er metastazlar�, ileri hastal�k i�in �nceki kemoterapi ve IRT'ye g�re endokrin kombinasyonu partnerine g�re tabakaland�r�l�r.
Ribosiklib kolundaki hastalarda bir sonraki basamak tedavide progresyona veya �l�me (PFS2) kadar ge�en s�re, genel �al��ma pop�lasyonunda plasebo kolundaki hastalara k�yasla daha uzundur (HR: 0,670 [%95 GA: 0,542, 0,830]). Medyan PFS2, ribosiklib kolu i�in 39,8 ay (%95
GA: 32,5, NE), plasebo kolunda 29,4 ayd�r (%95 GA: 24,1, 33,1).
Ya�l� hastalar
MONALEESA-2 ve MONALEESA-3 �al��malar�nda ribosiklib alan t�m hastalar�n temsili oranlar� ≥65 ya� ve ≥75 ya� aras�ndad�r (bkz. B�l�m 5.1). Bu hastalar ile daha gen� hastalar aras�nda ribosiklibin g�venlili�i ve etkilili�i bak�m�ndan genel farkl�l�klar g�zlenmemi�tir (bkz. B�l�m 4.2).
B�brek yetmezli�i olan hastalar
�� pivot �al��mada (MONALEESA-2, MONALEESA-3 ve MONALEESA-7), normal b�brek fonksiyonu olan 510 (% 53,8) hasta, hafif b�brek yetmezli�i olan 341 (%36) hasta ve orta derecede b�brek yetmezli�i olan 97 (% 10,2) hasta ribosiklib ile tedavi edilmi�tir. �iddetli b�brek yetmezli�i olan hastalar, �al��malara dahil edilmemi�tir. Normal b�brek fonksiyonu olanlara k�yasla, 600 mg ba�lang�� dozunda ribosiklib alan hafif ve orta derecede b�brek yetmezli�i olan hastalarda progresyonsuz sa�kal�m sonu�lar� tutarl�d�r. G�venlilik profili, renal kohortlar aras�nda genel olarak tutarl�d�r (bkz. B�l�m 4.8).
5.2. Farmakokinetik �zellikler
Genel �zelliklerRibosiklibin farmakokineti�i, ilerlemi� meme kanseri olan hastalarda 50 mg ila 1200 mg'l�k g�nl�k oral dozlar�ndan sonra incelenmi�tir. Sa�l�kl� g�n�ll�lere 400 mg ile 600 mg aras�nda tek oral dozlar veya tekrarl� 400 mg dozlar� (8 g�n) verilmi�tir.
Emilim:
Ribosiklibin mutlak biyoyararlan�m� bilinmemektedir.
Ribosiklib oral kullan�m�n ard�ndan C'a ula�ma zaman� (T) 1-4 saat aras�nda olmu�tur. Ribosiklib, test edilen doz aral���nda (50 - 1200 mg) maruziyette (Cve EAA) oransal de�erin biraz �zerinde art��lar sergilemi�tir. Tekrarlanan g�nl�k tek doz uygulamalar� sonras�nda kararl� durumuma genellikle 8 g�n sonra ula��lm��t�r ve ribosiklib, 2,51 geometrik ortalama birikme oran� (aral�k: 0,97-6,4) ile birikme g�stermi�tir.
Besin etkisi
A�l�k durumu ile kar��la�t�r�ld���nda, ribosiklib film kapl� tabletin tek bir 600 mg dozunun y�ksek oranda ya� i�eren y�ksek kalorili bir ���n ile birlikte oral yolla uygulanmas�n�n, ribosiklibin emilim h�z� ve �l��s� �zerinde herhangi bir etkisi olmam��t�r.
Da��l�m:
Ribosiklibin insan plazma proteinlerine ba�lanmas� in vitro ortamda yakla��k %70 bulunmu�tur ve konsantrasyondan ba��ms�z olmu�tur (10 - 10000 ng/ml). Ribosiklib k�rm�z� kan h�creleri ile plazma aras�nda e�it da��lm��, in vivo ortamda kan-plazma oran� 1,04 bulunmu�tur. Pop�lasyon farmakokinetik analizine dayan�larak kararl� durumda g�r�n�r da��l�m hacmi (Vss/F) 1090 litredir.
Biyotransformasyon:
In vitro ve in vivo �al��malar ribosiklibin insanda a��rl�kl� olarak CYP3A4 arac�l���yla olmak �zere geni� �l��de hepatik metabolizma ile eliminasyona u�rad���n� g�stermi�tir. [C] ribosiklibin 600 mg'l�k tek dozunun insanlara oral uygulanmas�ndan sonra ribosiklib i�in ana metabolik yolaklar, oksidasyonu (dealkilasyon, C ve/veya N-oksijenasyon, oksidasyon (-2H)) ve bunlar�n kombinasyonlar�n� i�ermi�tir. Ribosiklib faz 1 metabolitlerinin faz II konj�gatlar� N-asetilasyon, s�lfasyon, sistein konj�gasyonu, glikozilasyon ve glukuronidasyonu i�ermi�tir. Ribosiklib, plazmadaki ba�l�ca dola�an ila� kaynakl� entite olmu�tur. Dola��mdaki ba�l�ca metabolitler metabolit M13 (CCI284, N-hidroksilasyon), M4 (LEQ803, N-demetilasyon) ve M1'i (ikincil glukuronid) i�ermi�tir. Ribosiklibin klinik aktivitesi (farmakolojik ve g�venlilik) ba�ta ana ila� kaynakl� olmu�, dola��mdaki metabolitlerin g�z ard� edilebilir katk�s� oldu�u g�r�lm��t�r.
Ribosiklib geni� �l��de metabolize olmu� olup, de�i�memi� ila� fe�es ve idrarda s�ras�yla
%17,3 ve %12,1'e kar��l�k gelmi�tir. Metabolit LEQ803 d��k�da �nemli bir metabolit olmu� ve fe�es ve idrarda s�ras�yla uygulanan dozun yakla��k %13,9 ve %3,74'�n� temsil etmi�tir. Gerek fe�es gerekse idrarda say�s�z ba�ka metabolit de min�r miktarlarda tespit edilmi�tir (uygulanan dozun ≤ %2,78'i).
Eliminasyon:
�lerlemi� kanseri olan hastalarda 600 mg'da kararl� durumda geometrik ortalama plazma efektif yar�lanma �mr� (birikme oran�na dayal�) 32 saat (%63 CV) ve geometrik ortalama g�r�n�r oral klirens (CL/F) 25,5 l/saat (%66 CV) bulunmu�tur. Sa�l�kl� g�n�ll�lerdeki �al��malarda 600 mg
dozunda ribosiklibin geometrik ortalama g�r�n�r plazma terminal yar�lanma �mr� (T) 29,7 ile 54,7 saat, ribosiklibin geometrik ortalama CL/F de�eri ise 39,9 ila 77,5 l/saat aral���nda olmu�tur.
Ribosiklib ve metabolitleri renal yola��n k���k bir katk�s� ile a��rl�kl� olarak fe�es ile elimine olur. 6 sa�l�kl� erkek g�n�ll�de, tek bir oral [C] ribosiklib dozunun uygulanmas�ndan sonra uygulanan toplam radyoaktivitenin %91,7'si 22 g�n i�inde tespit edilmi�tir; fe�es ana at�lma yolu olup (%69,1) dozun %22,6's� idrarda tespit edilmi�tir.
Do�rusall�k/do�rusal olmayan durum:
Ribosiklib, test edilen 50 ila 1200 mg doz aral���nda hem tek hem de tekrarl� dozlardan sonra maruziyette (Cve EAA) oransal de�erin biraz �zerinde art��lar sergilemi�tir. Bu analiz doz gruplar�n�n �o�u i�in k���k �rneklem b�y�kl�kleri ile s�n�rl�d�r ve verilerin �o�u 600 mg doz grubundan gelmektedir.
Hastalardaki karakteristik �zellikler
�zel pop�lasyonlar B�brek bozuklu�u
B�brek fonksiyonunun ribosiklib farmakokineti�i �zerindeki etkisi 400 mg'l�k tek bir ribosiklib dozunda, normal b�brek fonksiyonuna sahip (mutlak GFR [aGFR] ≥90 ml/dakika)14 sa�l�kl� g�n�ll� ile hafif b�brek yetmezli�i (aGFR 60 ila <90 ml/dakika) olan 8, orta derece b�brek yetmezli�i olan (aGFR 30 ile <60 ml/dakika) 6, �iddetli b�brek yetmezli�i olan (aGFR 15 ila
<30 ml/dakika) 7 g�n�ll�n�n ve son d�nem b�brek hastal��� (aGFR <15 ml/dakika) olan (SDBY) 3 hastan�n dahil edildi�i bir b�brek yetmezli�i �al��mas�nda de�erlendirilmi�tir.
Normal b�brek fonksiyonu olan g�n�ll�lerde maruz kalmaya g�re hafif, orta ve �iddetli b�brek yetmezli�i olan hastalarda EAA1,6 kat, 1,9 kat ve 2,7 kat ve C1,8 kat, 1,8 kat ve 2,3 kat artm��t�r. Ribosiklibin etkililik ve g�venlilik �al��malar�, hafif b�brek yetmezli�i olan hastalar�n b�y�k bir k�sm�n� kapsad���ndan (bkz. B�l�m 5.1), b�brek yetmezli�i �al��mas�nda orta veya �iddetli b�brek yetmezli�i olan g�n�ll�lerden elde edilen veriler, normal b�brek fonksiyonu olan ve hafif renal yetmezli�i olan g�n�ll�lere ait birle�tirilmi� verilerle kar��la�t�r�lm��t�r. Normal b�brek fonksiyonu ve hafif b�brek yetmezli�i olan g�n�ll�lere ait birle�tirilmi� verilerle kar��la�t�r�ld���nda, orta ve �iddetli b�brek yetmezli�i olan hastalarda EAAs�ras�yla 1,6 kat ve 2,2 kat ve Cise 1,5 kat ve 1,9 kat artm��t�r. Az say�da denek nedeniyle SDBY'li denekler i�in bir kat fark� hesaplanmam��t�r, ancak sonu�lar �iddetli b�brek yetmezli�i olan deneklere k�yasla ribosiklib maruziyetinde benzer veya biraz daha b�y�k bir art��a i�aret etmektedir.
B�brek fonksiyonunun ribosiklib farmakokineti�i �zerindeki etkisi, hastalara 600 mg ba�lang�� dozunun verildi�i etkinlik ve g�venlik �al��malar�na dahil edilen kanser hastalar�nda da de�erlendirilmi�tir (bkz. B�l�m 5.1). 600 mg ribosiklibin tek doz veya tekrarl� dozlar halinde oral uygulamas�n� takiben kanser hastalar�nda yap�lan �al��malardan elde edilen farmakokinetik verilerin bir alt grup analizinde, hafif (n=57) veya orta (n=14) b�brek yetmezli�i olan hastalarda ribosiklibin EAAve Cde�erleri, normal b�brek fonksiyonu olan hastalardaki (n=86) EAAve Cile kar��la�t�r�labilir olup, hafif veya orta derecede b�brek yetmezli�inin ribosiklib maruziyeti �zerinde klinik olarak anlaml� bir etkisi olmad���n� d���nd�rmektedir.
Karaci�er bozuklu�u
Karaci�er bozuklu�u olan kansersiz g�n�ll�lerde y�r�t�len bir farmakokinetik �al��maya dayan�larak, hafif karaci�er bozuklu�unun ribosiklib maruziyetine herhangi bir etkisi olmam��t�r (bkz. b�l�m 4.2). Ribosiklib i�in ortalama maruziyet, orta �iddetli (geometrik ortalama oran [GMR]: Ci�in 1,44; EAAi�in 1,28) ve �iddetli (GMR: Ci�in 1,32; EAAi�in 1,29) karaci�er bozuklu�u olan hastalarda iki kattan az art�� g�stermi�tir (bkz. B�l�m 4.2).
Normal karaci�er fonksiyonuna sahip 160 meme kanserli hasta ve hafif karaci�er bozuklu�una sahip 47 hastay� i�eren bir pop�lasyon farmakokineti�i analizine dayan�larak, hafif karaci�er bozuklu�unun ribosiklib maruziyetine herhangi bir etkisi olmayarak spesifik karaci�er bozuklu�u �al��mas�n�n bulgular�n� desteklemi�tir. Ribosiklib orta �iddette veya �iddetli hepatik bozuklu�u olan meme kanseri hastalar�nda ara�t�r�lmam��t�r.
Ya�, a��rl�k, cinsiyet ve �rk�n etkisi
Pop�lasyon farmakokinetik analizi ya�, beden a��rl��� veya cinsiyetin ribosiklibin sistemik maruziyetinde doz ayarlamas� gerektirebilecek bir etkisinin olmad���n� g�stermi�tir. Farmakokinetikte �rka ba�l� de�i�ikliklerin verileri sonuca varabilmek i�in �ok s�n�rl�d�r.
In vitro etkile�imler
Ribosiklibin sitokrom P450 enzimleri �zerindeki etkisi
�n vitro, ribosiklib klinik a��dan ilgili konsantrasyonlarda CYP1A2, CYP2E1 ve CYP3A4/5'in geri d�n���ml� inhibit�r� ve CYP3A4/5'in zamana ba��ml� inhibit�r�d�r. �n vitro de�erlendirmeler ribosiklibin klinik a��dan ilgili konsantrasyonlarda CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19 ve CYP2D6'n�n aktivitelerini inhibe etme potansiyeline sahip olmad���n� g�stermi�tir. Ribosiklib CYP1A2, CYP2C9 ve CYP2D6'n�n zamana ba��ml� inhibisyonu i�in potansiyele sahip de�ildir.
�n vitro veriler ribosiklibin UGT enzimlerini veya CYP enzimleri CYP2C9, CYP2C19 ve CYP3A4'� PXR ile ind�kleme potansiyeline sahip olmad���n� g�stermektedir. Bu nedenle, VALAMOR'un bu enzimlerin substratlar�n� etkilemesi olas� de�ildir. �n vitro veriler CAR arac�l���yla CYP2B6'y� ind�kleme potansiyelini d��lamak i�in yeterli de�ildir.
Transporterlerin ribosiklib �zerindeki etkisi
Ribosiklib, in vitroda P-gp i�in bir substratt�r; fakat k�tle denge verilerine dayal� olarak, P-gp veya BCRP inhibisyonunun terap�tik dozlarda ribosiklib maruziyetini etkileme olas�l��� yoktur. Ribosiklib in vitroda, karaci�er al�m transporterleri OATP1B1, OATP1B3 veya OCT-1'nin bir substrat� de�ildir.
Ribosiklibin transporterler �zerindeki etkisi:
�n vitro de�erlendirmeler, ribosiklibin, ila� transporterleri P-gp, BCRP, OATP1B1/1B3, OCT1, OCT2, MATE1 ve BSEP'nin aktivitelerini inhibe etme potansiyeline sahip oldu�una i�aret etmi�tir. Ribosiklib in vitroda klinik a��dan �nemli konsantrasyonlarda OAT1, OAT3 veya MRP2'yi inhibe etmemi�tir.
5.3. Klinik �ncesi g�venlilik verileri
G�venlilik farmakolojisi
K�peklerdeki in vivo kardiyak g�venlilik �al��malar�nda, �nerilen 600 mg dozundan sonra hastalarda elde edilmesi beklenecek maruziyette doz ve konsantrasyon ile ili�kili QTc aral���
uzamas� g�sterilmi�tir. Artm�� maruziyetlerde (beklenen klinik C'�n yakla��k 5 kat�) premat�r ventrik�ler kontraksiyon (PVC'ler) olaylar�n� tetikleme potansiyeli de mevcuttur.
Tekrarl� doz toksisitesi
S��anlarda 27 haftaya ve k�peklerde 39 haftaya varan tekrarl� doz toksisitesi �al��malar� (3 haftal�k tedavi/1 haftal�k tedavisiz tedavi plan�), ribosiklib toksisitesinin ana hedef organ� olarak hepatobiliyer sistemi g�stermi�tir (proliferatif de�i�iklikler, kolestaz, kum benzeri safra ta�� ve koyu safra). Tekrarl� doz �al��malar�nda ribosiklibin farmakolojik etkisi ile ili�kili hedef organlar kemik ili�ini (hiposell�lerite), lenfoid sistem (lenfoid deplesyonu), intestinal mukoza (atrofi), deri (atrofi), kemik (azalm�� kemik formasyonu), b�brek (t�b�ler epitel h�crelerde e� zamanl� dejenerasyon ve rejenerasyon) ve testisi (atrofi) i�ermi�tir. Testiste g�r�len geri d�n��l�l�k e�ilimi g�stermi� atrofik de�i�ikliklerin yan� s�ra di�er t�m de�i�iklikler 4 haftal�k tedavisiz periyottan sonra tamamen geri d�n��l� olmu�tur. Bu etkiler, testik�ler germ h�creler �zerinde, seminifer�z t�b�llerde atrofi ile sonu�lanan do�rudan anti-proliferatif etki ile ba�lant�l� olabilir. Toksisite �al��malar�nda hayvanlarda ribosiklibe maruziyet genellikle 600 mg/g�n (EAA baz�nda) �oklu dozlar� alan hastalarda g�zlenenden k���k veya e�it olmu�tur.
�reme toksisitesi/Fertilite
Ribosiklib, s��anda veya tav�anda maternal toksisite g�stermeyen dozlarda fetotoksisite ve teratojenisite g�stermi�tir. Prenatal maruziyetten sonra EAA baz�nda en y�ksek �nerilen doz olan 600 mg/g�n dozunda, s�ras�yla insandaki maruziyetten daha d���k d�zeylerde veya insandaki maruziyetin 1,5 kat�nda ribosiklib ile s��anlarda daha y�ksek implantasyon sonras� kay�p ve azalm�� fetal a��rl�k insidanslar� g�zlenmi�tir ve ribosiklib tav�anlarda teratojeniktir.
S��anlarda, ge�ici olarak de�erlendirilmi� ve/veya d���k fetal a��rl�kla ili�kilendirilmi� iskelet de�i�ikliklerinin e�lik etti�i azalm�� fetal a��rl�k tespit edilmi�tir. Tav�anlarda, embriyofetal geli�im �zerine, fetal anomaliler (malformasyonlar ve d��, i� organ ve iskelet varyantlar�) ve fetal b�y�me (daha d���k fetal a��rl�klar) insidans�nda art�� ile kan�tlanm�� olan advers etkiler olmu�tur. Bu bulgular azalm��/k���k akci�er loblar�n� ve aort ark�nda ek damar� ve diyafram f�t���n�, aksesuar lobun olmamas�n� veya (k�smen) kayna�m�� akci�er loblar�n� ve azalm��/k���k aksesuar akci�er lobunu (30 ve 60 mg/kg), ekstra/ rudimenter on���nc� kaburgay� ve �ekilsiz hiyoid kemi�ini ve polekste azalm�� parmak kemi�i say�s�n� i�ermi�tir. Herhangi bir embriyofetal mortalite g�stergesi s�z konusu olmam��t�r.
Di�i s��anlardaki bir fertilite �al��mas�nda ribosiklib, 300 mg/kg/g�ne kadarki dozlarda (olas�l�kla, EAA baz�nda en y�ksek �nerilen doz olan 600 mg/g�n dozunda hastalardaki klinik maruziyetten daha d���k veya e�it bir maruziyette) �reme fonksiyonunu, fertiliteyi ya da erken d�nem embriyonik geli�imi etkilememi�tir.
Ribosiklib erkek fertilitesi �al��malar�nda de�erlendirilmemi�tir. Bununla birlikte, s��an ve k�pek toksisite �al��malar�nda, EAA baz�nda en y�ksek �nerilen doz olan 600 mg/g�n dozundaki maruziyetin alt�nda veya bu maruziyete e�it d�zeylerde, testislerde atrofik de�i�iklikler bildirilmi�tir.
Bu etkiler, testik�ler germ h�creler �zerinde, seminifer�z t�b�llerde atrofi ile sonu�lanan do�rudan anti-proliferatif etki ile ba�lant�l� olabilir.
Ribosiklib ve metabolitleri s��an s�t�ne kolayl�kla ge�mi�tir. Ribosiklibe maruziyet s�tte plazmad�kinden daha y�ksektir.
Genotoksisite
Bakteriyel in vitro sistemlerde ve memeli in vivo ve in vitro sistemlerde metabolik aktivasyon i�eren veya i�ermeyen genotoksisite �al��malar�, ribosiklibin genotoksik potansiyeline ili�kin herhangi bir kan�t ortaya koymam��t�r.
Karsinojenez
Ribosiklib, s��anlarda yap�lan 2 y�ll�k bir �al��mada karsinojenite a��s�ndan de�erlendirilmi�tir.
Ribosiklibin 2 y�l boyunca oral yoldan ≥300 mg/kg/g�n dozlarda verilmesi, di�i s��anlar�n uterusunda/serviksinde endometriyal epitelyal t�m�rler ve gland�ler ve skuam�z hiperplazi insidans�nda art��a ve 50 mg/kg/g�n dozunda verilmesi ise erkek s��anlar�n tiroid bezinde folik�ler t�m�rlerde insidansta art��a neden olmu�tur. Neoplastik de�i�ikliklerin g�r�ld��� di�i ve erkek s��anlarda kararl� durumda (EAA) ortalama maruziyet, hastalarda �nerilen 600 mg/g�n dozunda elde edilenin s�ras�yla 1,2 ve 1,4 kat�d�r. Neoplastik de�i�ikliklerin g�r�ld��� di�i ve erkek s��anlarda kararl� durumda ortalama maruziyet (EAA), hastalarda 400 mg/g�n dozunda elde edilenin s�ras�yla 2,2 ve 2,5 kat� olmu�tur.
Ek neoplastik olmayan proliferatif de�i�iklikler, artan karaci�er de�i�tirilmi� odaklar� (bazofilik ve berrak h�creli) ve erkek s��anlarda testik�ler interstisyel (Leydig) h�cre hiperplazisini, s�ras�yla ≥5 mg/kg/g�n ve 50 mg/kg/g�n dozlar�nda i�ermi�tir.
Rahim/serviks ve testik�ler interstisyel (Leydig) h�creler �zerindeki etkiler, hipofiz bezinde laktotrofik h�cre fonksiyonunun CDK4 inhibisyonuna ba�l� olarak hipotalamus-hipofiz- gonadal ekseni de�i�tiren uzun s�reli hipoprolaktinemi ile ili�kili olabilir.
Erkeklerde tiroid bulgular� i�in potansiyel mekanizmalar, karaci�erde kemirgenlere �zg� bir mikrozomal enzim ind�ksiyonunu ve/veya persistan hedefli (on-target) bir hipoprolaktinemiye ikincil olarak hipotalamus-hipofiz-testis-tiroid ekseninin disreg�lasyonunu i�erir.
Bu mekanizma ile insanlarda �strojen/progesteron oran�ndaki herhangi bir potansiyel art��, ribosiklibin insanlarda �strojen seviyesini azaltan tedavilerle kombinasyon halinde kullan�m endikasyonu oldu�u gibi, �strojen sentezi �zerindeki e� zamanl� anti-�strojen tedavisinin bir inhibit�r etkisi ile telafi edilecektir.
Prolaktin sentezi ve rol� a��s�ndan kemirgenler ve insanlar aras�ndaki �nemli farkl�l�klar g�z �n�ne al�nd���nda, bu etki bi�iminin insanlarda sonu�lar� olmas� beklenmemektedir.
6. FARMAS�T�K �ZELL�KLER
6.1. Yard�mc� maddelerin listesi
Tablet bile�enleri:
Mikrokristalin sel�loz Krospovidon (Tip A)
D���k s�bstit�e hidroksipropilsel�loz Magnezyum stearat
Kolloidal silikon dioksit
Film kaplama bile�enleri:
Siyah demir oksit (E172) K�rm�z� demir oksit (E172) Soya lesitin
Polivinil alkol (k�smen hidrolize)
Talk
Titanyum dioksit (E171) Ksantan sak�z�
6.2. Ge�imsizlikler
Ge�erli de�ildir.
6.3. Raf �mr�
36 ay
6.4. Saklamaya y�nelik �zel tedbirler
30C'nin alt�nda oda s�cakl���nda saklay�n�z.
6.5. Ambalaj�n niteli�i ve i�eri�i
21 film kapl� tablet i�eren PCTFE/PVC blisterler
Ambalaj b�y�kl���: 63 film kapl� tablet
6.6. Be�eri t�bbi �r�nden arta kalan maddelerin imhas� ve di�er �zel �nlemler
Kullan�lmam�� olan �r�nler ya da at�k materyaller “T�bbi At�klar�n Kontrol� Y�netmeli�i” ve “Ambalaj At�klar�n�n Kontrol� Y�netmeli�i”ne uygun olarak imha edilmelidir.
 Deri Kanseri
Deri kanseri �ok rastlanan bir hastal�kt�r. �� ana t�r� bulunur ;genelde kemirici �lser olarak bilinen bazal h�creli karsinom, yass� h�creli karsinom ve k�t� huylu t�m�r.
Deri Kanseri
Deri kanseri �ok rastlanan bir hastal�kt�r. �� ana t�r� bulunur ;genelde kemirici �lser olarak bilinen bazal h�creli karsinom, yass� h�creli karsinom ve k�t� huylu t�m�r. |
 G�da Alerjisi
Her y�l milyonlarca insan yiyeceklere alerji g�steriyor.
G�da Alerjisi
Her y�l milyonlarca insan yiyeceklere alerji g�steriyor. |
 |
HIV ve Aids HIV, Human Immunodeficiency Virus’d�r (�nsanlarda Ba����kl�k Sistemini Bozan Vir�sd�r). Bu vir�s AIDS hastal���na sebep olur. |
 |
En Yayg�n Alerji T�rleri Ba����kl�k sistemi, polen, ar� zehiri veya evcil hayvan gibi yabanc� bir maddeye veya �o�u insanda reaksiyona neden olmayan bir yiyece�e tepki g�sterdi�inde alerjiler meydana gelir. |
 |
Artrit Artrit, olduk�a yayg�n bir hastal�kt�r ancak iyi anla��lamam��t�r. Asl�nda �artrit� tek bir hastal���n ad� de�ildir; eklem a�r�s� veya eklem hastal�klar�n� adland�rman�n gayri resmi yoludur. |
�LA� GENEL B�LG�LER�
Farmanova Sa�l�k Hizmetleri Ltd. �ti
| Sat�� Fiyat� | 19712.74 TL [ 10 Jul 2023 ] |
| �nceki Sat�� Fiyat� | 19354.33 TL [ 3 Jul 2023 ] |
| Original / Jenerik | Original �la� |
| Re�ete Durumu | Normal Re�eteli bir ila�d�r. |
| Barkodu | 8699074090721 |
| Etkin Madde | Ribosiklib |
| ATC Kodu | L01EF02 |
| Birim Miktar | 200 |
| Birim Cinsi | MG |
| Ambalaj Miktar� | 63 |
| Antineoplastik ve �mm�nomod�lat�r Ajanlar |
| �thal ( ref. �lke : Fransa ) ve Be�eri bir ila�d�r. |