ZEJULA 100 mg sert kapsül Kısa Ürün Bilgisi
{ Niraparib }
1. BEŞERİ TIBBİ ÜRÜNÜN ADI
ZEJULA 100 mg sert kapsül
Sitotoksik
2. KALİTATİF VE KANTİTATİF BİLEŞİM
Etkin madde
Her bir sert kapsül 100 mg niraparibe eşdeğer 159,4 mg niraparib tosilat monohidrat içermektedir.
Yardımcı maddeler
Her bir sert kapsül 254,5 mg laktoz monohidrat (sığır sütünden elde edilir) içermektedir.
Her bir sert kapsül kabuğu ayrıca renklendirici ajan tartrazin (E 102) [0,0172 mg] içermektedir.
Yardımcı maddelerin tam listesi için Bölüm 6.1'e bakınız.
3. FARMASÖTİK FORMU
Sert kapsül.
Sert kapsüller yaklaşık 22 mm × 8 mm boyutlarındadır ve siyah mürekkeple "100 mg" baskılı beyaz gövdeye ve beyaz mürekkeple "Niraparib" baskılı mor kapağa sahiptir.
4. KLİNİK ÖZELLİKLER
4.1. Terapötik endikasyonlar
ZEJULA
Birinci basamak over kanseri idame tedavisinde
FIGO evre 3 veya 4, yüksek gradlı seröz epitelyal over, fallop tüpü veya primer peritoneal kanser hastası olan ve birinci basamak platin temelli kemoterapinin tamamlanmasından sonra yanıt veren (tam veya kısmi), ECOG performans durumu 0-1 olan yetişkin hastalarda idame tedavisi için monoterapi olarak endikedir. Tedavi süresi hastalık ilerlemesine veya tolere edilemeyen toksisiteye kadar veya maksimum 3 yıldır.
Rekürren (Tekrarlayan) over kanseri idame tedavisinde
Platinli tedavinin tamamlanmasından en az 6 ay veya sonrasında nüks eden platin duyarlı, yüksek gradlı seröz epitelyal over, fallop tüpü veya primer peritoneal kanser hastası olan ve nüks nedeniyle uygulanan platin temelli kemoterapiye yanıt veren (tam veya kısmi), ECOG performans durumu 0-1 olan yetişkin hastalarda idame tedavisi için monoterapi olarak kullanımda endikedir.
4.2. Pozoloji ve uygulama şekli
ZEJULA tedavisi, kanser tedavi ürünlerinin kullanımında deneyimli bir hekim tarafından başlatılmalı ve bu hekimin gözetiminde yürütülmelidir.
Pozoloji
Birinci basamak over kanseri idame tedavisi
Önerilen ZEJULA başlangıç dozu günde bir kez 200 mg'dır (iki adet 100 mg kapsül). Bununla birlikte 77 kg veya daha fazla kiloda olan ve bazal trombosit sayımı 150.000 mcgL ve daha fazla olan hastalarda, önerilen ZEJULA başlangıç dozu günde bir kez 300 mg'dır (üç adet 100 mg kapsül) (bkz. Bölüm 4.4 ve 4.8).
Rekürren (Tekrarlayan) over kanseri idame tedavisi
Doz, toplam günlük doz olarak 300 mg'a eşdeğer, günde bir kez üç adet 100 mg sert kapsüldür.
Hastalar dozlarını her gün yaklaşık aynı saatte almaya teşvik edilmelidir. Uyku öncesinde kullanım, bulantının yönetilmesinde potansiyel bir yöntem olabilir.
Hastalıkta ilerleme veya toksisite olana kadar tedaviye devam edilmesi önerilmektedir.
Dozun kaçırılması
Hasta bir dozu kaçırırsa, sıradaki dozu düzenli olarak planlanmış saatinde almalıdır.
Advers reaksiyonlar için doz ayarlaması
Advers reaksiyonlar için önerilen doz değişiklikleri Tablo 1, 2 ve 3'te listelenmektedir.
Genel olarak, advers reaksiyonun geçmesi için hastalara zaman vermek amacıyla tedaviye ara verilmesi (ardışık 28 günden uzun olmamak kaydıyla) ve daha sonra tedaviye aynı dozda devam edilmesi önerilmektedir. Advers reaksiyonun tekrar gerçekleşmesi durumunda, tedaviye ara verilmesi ve daha düşük bir dozda devam edilmesi önerilmektedir. 28 günlük aradan sonra da advers reaksiyonların devam etmesi durumunda, ZEJULA'ya devam edilmemesi önerilmektedir. Eğer advers reaksiyonlar, dozlara ara verilmesi ya da dozun azaltılması gibi yöntemlerle yönetilemiyorsa, ZEJULA kullanımının kesilmesi önerilmektedir.
Tablo 1: Advers reaksiyonlar için önerilen doz değişiklikleri
Başlangıç doz düzeyi | 200 mg | 300 mg |
Birinci doz azaltma | 100 mg/gün | 200 mg/gün (iki adet 100 mg kapsül) |
İkinci doz azaltma | İlaç kullanımı kesilmelidir. | 100 mg/gün* (bir adet 100 mg kapsül) |
Dozun 100 mg/gün'ün altına azaltılması gerekiyorsa, ZEJULA kullanımı kesilmelidir.
Tablo 2: Hematolojik olmayan advers reaksiyonlar için doz değişikliği
Profilaksisin uygulanamayacağının düşünüldüğü veya advers reaksiyonun tedaviye karşın devam ettiği, hematolojik olmayan CTCAE* ≥ 3. derece tedaviyle ilişkili advers reaksiyon | İlk kez gerçekleştiğinde: |
İkinci kez gerçekleştiğinde: | |
Hastaya ZEJULA 100 mg/gün verilirken 28 günden uzun süren CTCAE ≥ 3. derece tedaviyle ilişkili advers reaksiyon | Tedavi durdurulmalıdır. |
ZEJULA'ya maksimum 28 gün boyunca olmak kaydıyla veya advers reaksiyon sona erinceye ya da ortadan kalkıncaya kadar ara verilmesi.
Tablo 1'de belirtildiği gibi, ZEJULA'nın daha düşük bir doz düzeyinde devam ettirilmesi.
ZEJULA'ya maksimum 28 gün boyunca olmak kaydıyla veya advers reaksiyon sona erinceye ya da ortadan kalkıncaya kadar ara verilmesi.
Tablo 1'de belirtildiği gibi, ZEJULA'nın daha düşük bir doz düzeyinde devam ettirilmesi veya kullanımının kesilmesi.
4.3. Kontrendikasyonlar
Etkin madde
Emzirme (bkz. Bölüm 4.6).
4.4. Özel kullanım uyarıları ve önlemleri
Hematolojik advers reaksiyonlar
ZEJULA ile tedavi edilen hastalarda hematolojik advers reaksiyonlar (trombositopeni, anemi, nötropeni) bildirilmiştir (bkz. Bölüm 4.8). Vücut ağırlığı veya bazal trombosit sayımı düşük olan hastalarda 3. derece ve üzeri trombositopeni riski daha yüksek olabilir (bkz. Bölüm 4.2).
Tedavi sırasında tüm hematolojik parametrelerin herhangi birindeki klinik açıdan anlamlı değişimleri izlemek için, ilk ay boyunca haftalık tam kan sayımı, ardından izleyen 10 aylık tedavi boyunca aylık izlem ve bu süreden sonra da periyodik izlem önerilmektedir (bkz. Bölüm 4.2).
Hasta, ara verildikten sonraki 28 gün içinde geçmeyen pansitopeni dahil olmak üzere şiddetli persistan hematolojik toksisite geliştirirse, ZEJULA kullanımı durdurulmalıdır.
Trombositopeni riski nedeniyle, antikoagülanlar ve trombosit sayımını düşürdüğü bilinen tıbbi ürünler dikkatli kullanılmalıdır (bkz. Bölüm 4.8).
Miyelodisplastik sendrom/akut miyeloid lösemi
Klinik çalışmalarda ve pazara verilme sonrasında ZEJULA monoterapisi veya kombinasyon tedavisiyle tedavi edilen hastalarda miyelodisplastik sendrom/akut miyeloid lösemi (MDS/AML) olguları, ölümle sonuçlanan vakalar dahil, gözlemlenmiştir (bkz. Bölüm 4.8).
Klinik çalışmalarda hastalarda MDS/AML gelişmeden önceki ZEJULA tedavisi süresi 0,5 ay ila >4,9 yıl aralığındadır. Olgular sekonder, kanser tedavisiyle ilişkili MDS/AML açısından tipiktir. Tüm hastalara platin içeren kemoterapi rejimleri uygulanmış ve birçoğuna aynı zamanda DNA'ya zarar veren başka ajanlar ve radyoterapi uygulanmıştır. Bazı hastalarda
kemik iliği baskılanması öyküsü vardır. NOVA çalışmasında, MDS/AML insidansı gBRCAmut kohortunda (%7,4), gBRCAmut olmayan kohorta (%1,7) göre daha yüksektir.
Şüpheli MDS/AML veya uzun süreli hematolojik toksisiteler için, hasta ileri değerlendirme için bir hematoloğa sevk edilmelidir. MDS/AML tanısı doğrulanırsa, ZEJULA tedavisi durdurulmalı ve hasta uygun şekilde tedavi edilmelidir.
Hipertansif kriz dahil olmak üzere hipertansiyon
ZEJULA kullanımı sırasında hipertansif kriz dahil olmak üzere hipertansiyon bildirilmiştir (bkz. Bölüm 4.8). ZEJULA tedavisine başlamadan önce, var olan hipertansiyon yeterli düzeyde kontrol altına alınmalıdır. İki ay boyunca en az haftada bir kez, ardından ilk yıl boyunca ayda bir kez, daha sonra ZEJULA tedavisi sırasında periyodik olarak kan basıncı izlemi yapılmalıdır. Uygun hastalarda kan basıncında artış olması durumunda doktorlarına başvurma talimatı verilerek evde kan basıncı izlemi düşünülebilir.
Hipertansiyon gerekirse hem antihipertansif tıbbi ürünlerle hem de ZEJULA dozunun ayarlanmasıyla tıbbi olarak yönetilmelidir (bkz. Bölüm 4.2). Klinik programda, hastalar ZEJULA tedavisi görürken her bir 28 günlük döngünün 1. gününde kan basıncı ölçümü yapılmıştır. Çoğu olguda, hipertansiyon ZEJULA dozunda ayarlama yapılarak veya yapılmaksızın standart antihipertansif tedavi kullanılarak yeterli düzeyde kontrol edilmiştir (bkz. Bölüm 4.2). Hipertansif kriz durumunda veya tıbbi açıdan anlamlı hipertansiyonun antihipertansif tedaviyle yeterli düzeyde kontrol edilemediği durumlarda ZEJULA kullanımı durdurulmalıdır.
Posterior Reversibl Ensefalopati Sendromu (PRES)
ZEJULA alan hastalarda Posterior Reversibl Ensefalopati Sendromu (PRES) raporları bildirilmiştir (bkz. Bölüm 4.8). PRES, seyrek görülen, geri dönüşlü (reversibl) bir nörolojik hastalıktır ve ilişkili hipertansiyon varlığında veya yokluğunda nöbet, baş ağrısı, zihinsel durum değişikliği, görme bozukluğu veya kortikal körlük dahil olmak üzere hızla değişen semptomlarla ortaya çıkabilir. PRES tanısı konması için, tercihen manyetik rezonans görüntülemesiyle (MRI) beyin görüntüleme yapılarak doğrulanmasıgerekmektedir.
PRES olgularında, ZEJULA kullanımının durdurulması ve hipertansiyon dahil olmak üzere spesifik semptomların tedavi edilmesi önerilmektedir. Geçmişte PRES deneyimleyen hastalarda ZEJULA tedavisinin tekrar başlatılmasının güvenliliği bilinmemektedir.
Gebelik/kontrasepsiyon
ZEJULA gebelik döneminde veya çocuk doğurma potansiyeli olan ancak tedavi sırasında ve son ZEJULA dozundan sonra 6 ay boyunca yüksek etkili kontrasepsiyon kullanmak istemeyen kadınlarda kullanılmamalıdır (bkz. Bölüm 4.6). Tedaviden önce çocuk doğurma potansiyeli olan tüm kadınlarda gebelik testi yapılmalıdır.
Karaciğer yetmezliği:
Şiddetli karaciğer yetmezliği olan hastalarda, orta karaciğer yetmezliği olan hastalardan elde edilen verilere göre, niraparib maruziyeti daha yüksek olabilir ve bu hastalar dikkatlice takip edilmelidir (bkz. Bölüm 4.2 ve 5.2).
Yardımcı maddeler
Bu tıbbi ürün laktoz içermektedir. Galaktoz intoleransı, total laktaz eksikliği ya da glukoz- galaktoz malabsorpsiyonu gibi nadir kalıtımsal sorunları olan hastalar bu ilacı kullanmamalıdır.
Bu tıbbi ürün tartrazin (E 102) içermektedir. Alerjik reaksiyonlara sebep olabilir.
4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri
Farmakodinamik etkileşimler
Niraparibin aşılar veya immünosupresan ajanlarla kombinasyonu incelenmemiştir.
Niraparibin sitotoksik tıbbi ürünlerle birlikte kullanımıyla ilişkili veriler sınırlıdır. Bu nedenle niraparib; aşılar, immünosupresan ajanlar veya diğer sitotoksik tıbbi ürünlerle birlikte kullanıldığı zaman dikkatli olunmalıdır.
Farmakokinetik etkileşimler
Diğer tıbbi ürünlerin niraparibin üzerindeki etkileri CYP substratı olarak niraparib (CYP1A2 ve CYP3A4)
Niraparib, in vivo karboksilesterazlar (CE) ve UDP-glukuronosil transferazların (UGT) bir
substratıdır.
Niraparibin oksidatif metabolizması in vivo ortamda minimaldir. CYP enzimlerini inhibisyona uğrattığı (örn. itrakonazol, ritonavir ve klaritromisin) veya indüklediği (örn. rifampin, karbamazepin ve fenitoin) bilinen tıbbi ürünlerle eşzamanlı olarak uygulandığında ZEJULA için doz ayarlaması gerekmemektedir.
Dışa akış taşıyıcılarının substratı olarak niraparib (P-gp, BCRP, BSEP, MRP2 ve MATE1/2)
Niraparib, P-glikoproteini (P-gp) ve Meme Kanseri Direnç Proteininin (Breast Cancer Resistance Protein) (BCRP) bir substratıdır. Ancak, yüksek geçirgenliği ve biyoyararlanımı nedeniyle, bu taşıyıcıları inhibisyona uğratan tıbbi ürünlerle klinik açıdan anlamlı etkileşim riski düşüktür. Bu nedenle, P-gp'yi (örn. amiodaron, verapamil) veya BCRP'yi (örn. osimertinib, velpatasvir ve eltrombopag) inhibisyona uğrattığı bilinen tıbbi ürünlerle eşzamanlı olarak uygulandığında ZEJULA için doz ayarlaması gerekmemektedir.
Niraparib safra tuzu eksport pompası (substrate of bile salt export pump) (BSEP) veya çoklu ilaç direnciyle ilişkili protein 2'nin (MRP2) bir substratı değildir. Majör birincil metabolit olan M1, P-gp, BCRP, BSEP veya MRP2'nin bir substratı değildir. Niraparib çoklu ilaç ve toksin ekstrüzyonu (MATE)-1 veya 2'nin substratı değilken, M1 bunların ikisinin de substratıdır.
Hepatik alım taşıyıcılarının substratı olarak niraparib (OATP1B1, OATP1B3 ve OCT1)
Niraparib veya M1 organik anyon taşıyıcı polipeptit 1B1 (OATP1B1), 1B3 (OATP1B3) veya organik katyon taşıyıcı 1'in (OCT1) substratı değildir. OATP1B1 veya 1B3 (örn. gemfibrozil, ritonavir) veya OCT1 (örn. dolutegravir) alım taşıyıcılarını inhibisyona uğrattığı bilinen tıbbi ürünlerle eşzamanlı olarak uygulandığında ZEJULA için doz ayarlaması gerekmemektedir.
Renal alım taşıyıcılarının substratı olarak niraparib (OAT1, OAT3 ve OCT2)
Niraparib veya M1 organik anyon taşıyıcısı 1 (OAT1), 3 (OAT3) ve organik katyon taşıyıcısı 2'nin (OCT2) substratı değildir. OAT1 (örn. probenesid) veya OAT3 (örn. probenesid, diklofenak) veya OCT2 alım taşıyıcılarını (örn. simetidin, kinidin) inhibisyona uğrattığı bilinen tıbbi ürünlerle eşzamanlı olarak uygulandığında ZEJULA için doz ayarlaması gerekmemektedir.
Niraparibin diğer tıbbi ürünler üzerindeki etkisi
CYP'lerin (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 ve CYP3A4)
inhibisyonu
Niraparib veya M1, etkin maddeyi metabolize eden CYP enzimlerinin (CYP1A1/2, CYP2B6,
CYP2C8, CYP2C9, CYP2C19, CYP2D6 ve CYP3A4/5) herhangi birinin inhibitörü değildir.
CYP3A4'ün karaciğerde inhibisyonu beklenmese de, ilgili niraparib konsantrasyonlarında CYP3A4'ün bağırsak düzeyinde inhibisyon potansiyeli kanıtlanmamıştır. Bu nedenle, niraparibin metabolizmasının CYP3A4'e bağımlı olduğu ve özellikle dar terapötik aralığa sahip etkin maddelerle (örn. siklosporin, takrolimus, alfentanil, ergotamin, pimozid, ketiapin ve halofantrin) kombinasyon halinde kullanıldığı durumlarda dikkat edilmesi önerilmektedir.
UDP-glukuronosil transferazların (UGT) inhibisyonu
Niraparib in vitro 200 µM'a kadar dozlarda UGT izoformlarına karşı (UGT1A1, UGT1A4, UGT1A9 ve UGT2B7) inhibitör etki göstermemiştir. Bu nedenle, UGT'lerin niraparib tarafından klinik açıdan ilgili inhibisyona uğratılma potansiyeli minimaldir.
CYP'lerin indüksiyonu (CYP1A2 ve CYP3A4)
Niraparib veya M1 in vitro CYP3A4 indükleyicisi değildir. İn vitro, niraparib yüksek konsantrasyonlarda CYP1A2'yi zayıf şekilde indüklemektedir ve bu etkinin klinik önemi tamamen dışlanamamaktadır. M1 bir CYP1A2 indükleyicisi değildir. Bu nedenle, niraparibin, metabolizması CYP1A2'ye bağımlı ve özellikle terapötik aralığı dar olan (örn. klozapin, teofilin ve ropinirol) etkin maddelerle kombinasyon halinde dikkatli kullanılması önerilmektedir.
Dışa akış taşıyıcılarının inhibisyonu (P-gp, BCRP, BSEP, MRP2 ve MATE1/2)
Niraparib, BSEP veya MRP2'nin inhibitörü değildir. İn vitro, niraparib P-gp'yi çok zayıf düzeyde ve BCRP'yi , sırasıyla IC= 161 µM ve 5,8 µM değerleriyle inhibisyona uğratmaktadır. Bu nedenle, bu dışa akış taşıyıcılarının inhibisyona uğramasıyla ilişkili klinik açıdan anlamlı bir etkileşim olasılığı düşük olsa da göz ardı edilemez. Niraparibin BCRP substratlarıyla (irinotekan, rosuvastatin, simvastatin, atorvastatin, metotreksat) kombinasyon halinde kullanıldığı durumlarda dikkat edilmesi önerilmektedir.
Niraparib, MATE1 ve 2'nin inhibitörüdür ve ICdeğerleri sırasıyla 0,18 µM ve ≤0,14 µM'dir. Bu taşıyıcıların substratları olan tıbbi ürünlerle (örn. metformin) birlikte uygulandığında bu ürünlerin plazma konsantrasyonlarında artış olasılığı göz ardı edilemez.
Majör primer metabolit M1, P-gp, BCRP, BSEP, MRP2 veya MATE1/2'nin inhibitörü gibi
görülmemektedir.
Hepatik alım taşıyıcılarının inhibisyonu (OATP1B1, OATP1B3 ve OCT1)
Niraparib veya M1 organik anyon taşıyıcısı polipeptit 1B1 (OATP1B1) veya 1B3'ün (OATP1B3) inhibitörü değildir.
İn vitro, niraparib organik katyon taşıyıcısı 1'i (OCT1) IC= 34,4 µM değeriyle zayıf düzeyde inhibisyona uğratmaktadır. Niraparibin metformin gibi OCT1 yoluyla alım taşınımına maruz kalan etkin maddelerle kombinasyon halinde kullanıldığı durumlarda dikkat edilmesi önerilmektedir.
Renal alım taşıyıcılarının inhibisyonu (OAT1, OAT3 ve OCT2)
Niraparib veya M1, organik anyon taşıyıcısı 1 (OAT1), 3 (OAT3) ve organik katyon taşıyıcısı 2'yi (OCT2) inhibisyona uğratmaz.
Tüm klinik çalışmalar yalnızca yetişkinlerde gerçekleştirilmiştir.
Özel popülasyona ilişkin ek bilgiler
Veri bulunmamaktadır.
Pediyatrik popülasyon
Tüm klinik çalışmalar yalnızca yetişkinlerde gerçekleştirilmiştir.
4.6. Gebelik ve laktasyon
Gebelik kategorisi: X
Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon)
Çocuk doğurma potansiyeli olan kadınlar tedavi sırasında gebe kalmamalı ve tedavi başlangıcında gebe olmamalıdır. Tedaviden önce çocuk doğurma potansiyeli olan tüm kadınlarda gebelik testi yapılmalıdır. Çocuk doğurma potansiyeli olan kadınlar tedavi sırasında ve son ZEJULA dozundan sonra 6 ay boyunca yüksek etkili doğum kontrol yöntemleri kullanmalıdır.
Gebelik dönemi
Niraparibin gebe kadınlarda kullanımıyla ilgili veri bulunmamaktadır veya sınırlı miktardadır. Hayvanlarda üreme veya gelişim toksisitesi çalışması yapılmamıştır. Ancak, etki mekanizmasına göre, niraparib gebe kadınlara uygulandığında embriyo-letal ve teratojenik etkiler dahil olmak üzere embriyonik veya fetal hasara yol açabilir. ZEJULA gebelik sırasında kullanılmamalıdır.
Laktasyon dönemi
Niraparib veya metabolitlerinin insan sütüne geçip geçmediği bilinmemektedir. ZEJULA uygulaması sırasında ve son dozun alınmasından sonraki 1 ay boyunca emzirme kontrendikedir (bkz. Bölüm 4.3).
Üreme yeteneği/Fertilite
Fertiliteyle ilgili klinik veri bulunmamaktadır. Sıçanlarda ve köpeklerde spermatojenezde reversibl bir düşüş gözlemlenmiştir (bkz. Bölüm 5.3).
4.7. Araç ve makine kullanımı üzerindeki etkiler
ZEJULA'nın araç veya makine kullanımı üzerindeki etkisi orta düzeydedir. ZEJULA alan hastalarda asteni, halsizlik, baş dönmesi yaşayabilir veya konsantrasyon güçlüğü görülebilir. Bu semptomları yaşayan hastalar araç veya makine kullanırken dikkat etmelidir.
4.8. İstenmeyen etkiler
Güvenlilik profilinin özeti
Havuzlanmış PRIMA (başlangıç dozu 200 mg veya 300 mg) ve NOVA çalışmalarında ZEJULA monoterapisi alan 851 hastanın ≥% 10'unda görülen tüm derecelerdeki advers reaksiyonlar (ADR) bulantı, anemi, trombositopeni, halsizlik, kabızlık, kusma, baş ağrısı, insomnia, trombosit sayımında düşüş, nötropeni, karın ağrısı, iştah azalması, diyare, dispne, hipertansiyon, asteni, baş dönmesi, nötrofil sayımında düşüş, öksürük, artralji, sırt ağrısı, beyaz kan hücresi sayımında düşüş ve sıcak basmasıdır.
En yaygın ciddi advers reaksiyonlar % 1'den fazla (tedavide ortaya çıkan sıklıklar) trombositopeni ve anemidir.
Advers reaksiyonların tablolanmış listesi
ZEJULA monoterapisi alan hastalarda klinik araştırmalara ve pazarlama sonrası gözleme dayalı olarak aşağıdaki advers reaksiyonlar görülmüştür (bkz. Tablo 4). Advers reaksiyonların görülme sıklıkları, hasta maruziyetinin bilindiği PRIMA ve NOVA klinik çalışmalarından (300 mg/günlük sabit başlanıç dozu) elde edilen havuzlanmış advers olay verilerine dayanmaktadır ve şu şekilde tanımlanmıştır: Çok yaygın (≥1/10); yaygın (≥1/100 ila <1/10); yaygın olmayan (≥1/1.000 ila <1/100); seyrek (≥1/10.000 ila <1/1.000); çok seyrek (<1/10.000); bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). Her bir sıklık gruplamasında, istenmeyen etkiler, azalan ciddiyet sırasına göre bildirilmektedir.
Tablo 4: Advers reaksiyonların tablolanmış listesi
Sistem Organ Sınıfı | Tüm CTCAE* derecelerinin sıklığı | 3. veya 4. CTCAE* derecelerinin sıklığı |
Enfeksiyonlar ve enfestasyonlar | Çok yaygın İdrar yolu enfeksiyonu Yaygın Bronşit, konjonktivit | Yaygın olmayan İdrar yolu enfeksiyonu, bronşit |
(Kist ve polipler de dahil olmak üzere) iyi huylu, kötü huylu ve tanımlanmamış neoplazmalar | Yaygın Miyelodisplastik sendrom/ akut miyeloid lösemi** | Yaygın Miyelodisplastik sendrom/ akut miyeloid lösemi** |
Kan ve lenf sistemi hastalıkları | Çok yaygın Trombositopeni, anemi, nötropeni, lökopeni Yaygın olmayan Pansitopeni, febril nötropeni | Çok yaygın Trombositopeni, anemi, nötropeni Yaygın Lökopeni Yaygın olmayan Pansitopeni, febril nötropeni |
Bağışıklık sistemi hastalıkları | Yaygın Aşırı duyarlılık | Yaygın olmayan Aşırı duyarlılık |
Metabolizma ve beslenme hastalıkları | Çok yaygın İştah azalması Yaygın Hipokalemi | Yaygın Hipokalemi Yaygın olmayan İştah azalması |
Psikiyatrik hastalıklar | Çok yaygın Insomnia Yaygın Anksiyete, depresyon, bilişsel bozukluk Yaygın olmayan Konfüzyon | Yaygın olmayan Insomnia, anksiyete, depresyon, konfüzyon |
Sistem Organ Sınıfı | Tüm CTCAE* derecelerinin sıklığı | 3. veya 4. CTCAE* derecelerinin sıklığı |
Sinir sistemi hastalıkları | Çok yaygın Baş ağrısı, baş dönmesi Yaygın Tat alma duygusunda bozukluk (disgüzi) Seyrek Posterior Reversibl Ensefalopati Sendromu (PRES)** | Yaygın olmayan Baş ağrısı |
Kardiyak hastalıklar | Çok yaygın Palpitasyon Yaygın Taşikardi |
|
Vasküler hastalıklar | Çok yaygın Hipertansiyon Seyrek Hipertansif kriz | Yaygın Hipertansiyon |
Solunum, göğüs bozuklukları ve mediastinal hastalıklar | Çok yaygın Dispne, öksürük, nazofarenjit Yaygın Epistaksis Yaygın olmayan Pnömonit | Yaygın olmayan Dispne, epistaksis, pnömonit |
Gastrointestinal hastalıklar | Çok yaygın Bulantı, kabızlık, kusma, karın ağrısı, diyare, dispepsi Yaygın Ağız kuruluğu, abdominal distensiyon, mukozal enflamasyon (mukoza iltihabı), stomatit | Yaygın Bulantı, kusma, karın ağrısı Yaygın olmayan Diyare, kabızlık, mukozal enflamasyon (mukoza iltihabı), stomatit, ağız kuruluğu |
Deri ve deri altı doku hastalıkları | Yaygın Fotoduyarlılık, döküntü | Yaygın olmayan Fotoduyarlılık, döküntü |
Kas-iskelet bozuklukları, bağ doku ve kemik hastalıkları | Çok yaygın Sırt ağrısı, artralji Yaygın Miyalji | Yaygın olmayan Sırt ağrısı, artralji, miyalji |
Genel bozukluklar ve uygulama bölgesine ilişkin hastalıkları | Çok yaygın Halsizlik, asteni Yaygın Periferik ödem | Yaygın Halsizlik, asteni |
Sistem Organ Sınıfı | Tüm CTCAE* derecelerinin sıklığı | 3. veya 4. CTCAE* derecelerinin sıklığı |
Araştırmalar | Yaygın Gamma-glutamil transferaz artışı, AST artışı, kan kreatinin artışı, ALT artışı, kan alkalin fosfataz artışı, vücut ağırlığı azalması | Yaygın Gamma-glutamil transferaz artışı, ALT artışı Yaygın olmayan AST artışı, kan alkalin fosfataz artışı |
* CTCAE = Advers Olaylar İçin Ortak Terminoloji Kriterleri versiyon 4.02.
** Niraparib klinik çalışma verilerine dayanmaktadır. Pivot ENGOT-OV16 monoterapi çalışmasıyla sınırlı değildir.
†Aşırı duyarlılık, ilaca aşırı duyarlılık, anafilaktoid reaksiyon, ilaç erüpsiyonu, anjiyoödem ve
ürtiker dahildir.
††Hafıza bozukluğu, konsantrasyon bozukluğu içerir.
Bazal ağırlık veya trombosit sayımına göre 200 mg ZEJULA başlangıç dozu uygulanan hasta grubunda görülen advers reaksiyonlar, sabit başlangıç dozu 300 mg uygulanan grupla karşılaştırıldığında benzer veya daha düşük sıklıkta görülmüştür (Tablo 4).
Trombositopeni, anemi ve nötropeni sıklığıyla ilgili özgün bilgiler için aşağıdaki bölüme bakınız.
Seçili advers reaksiyonların tanımı
Klinik tanılar ve/veya laboratuvar bulguları dahil olmak üzere hematolojik advers reaksiyonlar (trombositopeni, anemi, nötropeni) genellikle niraparib tedavisinin erken dönemlerinde gerçekleşmiş ve zaman içinde görülme oranları azalmıştır.
NOVA ve PRIMA çalışmalarında, ZEJULA tedavisine uygun olan hastaların bazal hematolojik parametreleri şöyledir: Tedaviden önce mutlak nötrofil sayısı (MNS) ≥ 1.500 hücre/µL; trombosit ≥ 100,000 hücre/µL ve hemoglobin ≥ 9 g/dL (NOVA) veya ≥ 10 g/dL (PRIMA). Klinik programda, hematolojik advers reaksiyonlar laboratuvar izlem ve doz değişiklikleriyle yönetilmiştir (bkz. Bölüm 4.2).
PRIMA'da, bazal ağırlık veya trombosit sayımına göre ZEJULA başlangıç dozu uygulanan hastalarda, ≥3. derece trombositopeni, anemi ve nötropeni, 300 mg sabit başlangıç dozu verilen grupla karşılaştırıldığında sırasıyla % 48'den % 21'e, % 36'dan % 23'e, % 24'ten % 15'e düşmüştür. Hastaların sırasıyla % 3, % 3 ve % 2'sinde trombositopeni, anemi ve nötropeni nedeniyle tedavi bırakılmıştır.
Trombositopeni
PRIMA'da, ZEJULA ile tedavi edilen hastaların % 39'unda, plaseboyla tedavi edilen hastaların
% 0,4'ünde 3./4. derece trombositopeni görülmüştür ve ilk dozdan ilk başlangıca kadar geçen medyan süre 22 gün (aralık: 15 ila 335 gün), medyan süre 6 gündür (aralık: 1 ila 374 gün). Niraparib alan hastaların % 4'ünde trombositopeni nedeniyle tedavi bırakılmıştır.
NOVA'da, ZEJULA alan hastaların yaklaşık % 60'ında herhangi bir derecede trombositopeni ve hastaların % 34'ünde 3./4. derece trombositopeni görülmüştür. Bazal trombosit sayımı 180
× 10/L'den düşük olan hastalarda, herhangi bir derecede ve 3./4. derece trombositopeni hastaların sırasıyla % 76'sı ve % 45'inde gerçekleşmiştir. Derecesinden bağımsız olarak trombositopeni ve 3./4. derece trombositopeninin başlangıcına kadar geçen medyan süre sırasıyla 22 ve 23 gündür. 4. döngüden itibarren tedavinin ilk 2 ayında yapılan yoğun doz
değişikliklerinden sonra yeni trombositopeni görülme oranı % 1,2'dir. Herhangi bir derecedeki trombositopeni olaylarının medyan süresi 23 gündür ve 3./4. derece trombositopeni olaylarının medyan süresi 10 gündür. ZEJULA ile tedavi edilen hastalarda hemoraji riski yükselebilir. Klinik programda, trombositopeni laboratuvar izlemi, doz modifikasyonu ve uygun olan yerlerde trombosit transfüzyonuyla yönetilmiştir (bkz. Bölüm 4.2). Hastaların yaklaşık % 3'ü trombositopeni olayları (trombositopeni ve trombosit sayımında düşüş) nedeniyle tedaviyi bırakmıştır.
NOVA çalışmasında, 367 hastanın 48'inde (% 13) eşzamanlı trombositopeniyle birlikte kanama görülmüştür; pansitopeni ciddi advers olayıyla eşzamanlı olarak gözlenlenen bir 3. derece peteşi ve hematom olayı dışında, trombositopeniyle eşzamanlı tüm kanama olaylarının şiddeti
1. veya 2. derecedir. Trombositopeni bazal trombosit sayımı 180 × 10/L'den düşük olan hastalarda daha sık gerçekleşmiştir. Bazal trombosit sayımı düşük olan (<180 × 10/L) ve ZEJULA alan hastaların yaklaşık % 76'sında herhangi bir dereceden trombositopeni ve hastaların % 45'inde 3./4. derece trombositopeni görülmüştür. Niraparib alan hastaların <% 1'inde pansitopeni gözlenmiştir.
Anemi
PRIMA'da, ZEJULA ile tedavi edilen hastaların % 31'inde, plaseboyla tedavi edilen hastaların
% 2'sinde 3./4. derece anemi görülmüştür ve ilk dozdan ilk başlangıca kadar geçen medyan süre 80 gün (aralık: 15 ila 533 gün), medyan süre 7 gündür (aralık: 1 ila 119 gün). Niraparib alan hastaların % 2'sinde anemi nedeniyle tedavi bırakılmıştır.
NOVA'da, hastaların yaklaşık % 50'sinde herhangi bir derecede anemi, % 25'inde 3./4. derece anemi görülmüştür. Herhangi bir derecede anemi başlangıcına kadar geçen medyan süre 42 gündür ve 3./4. derece olaylar için 85 gündür. Herhangi bir derecede aneminin medyan süresi 63 gün, 3./4. derece olaylarda 8 gündür. ZEJULA tedavisi sırasında herhangi bir derecede anemi devam edebilir. Klinik programda, anemi laboratuvar izlemi, doz değişikliği (bkz. Bölüm 4.2) ve uygun olan yerlerde alyuvar transfüzyonuyla yönetilmiştir. Hastaların % 1'inde anemi nedeniyle tedavi bırakılmıştır.
Nötropeni
PRIMA'da, ZEJULA ile tedavi edilen hastaların % 21'inde, plaseboyla tedavi edilen hastaların
% 1'inde 3./4. derece nötropeni görülmüştür ve ilk dozdan ilk başlangıca kadar geçen medyan süre 29 gün (aralık: 15 ila 421 gün), medyan süre 8 gündür (aralık: 1 ila 42 gün). Niraparib alan hastaların % 2'sinde nötropeni nedeniyle tedavi bırakılmıştır.
NOVA'da, hastaların yaklaşık %30'unda herhangi bir derecede nötropeni, % 20'sinde 3./4. derece nötropeni görülmüştür. Herhangi bir derecede anemi başlangıcına kadar geçen medyan süre 27 gündür ve 3./4. derece olaylar için 29 gündür. Herhangi bir derecede aneminin medyan süresi 26 gün, 3./4. derece olaylarda 13 gündür. Ayrıca, niraparible tedavi edilen hastaların yaklaşık % 6'sına nötropeni için eşzamanlı tedavi olarak Granülosit-Koloni Uyarıcı Faktör (G- CSF) uygulanmıştır. Hastaların % 2'sinde nötropeni nedeniyle tedavi bırakılmıştır.
Miyelodisplastik sendrom/Akut miyeloid lösemi
Klinik çalışmalarda, ZEJULA ile tedavi edilen hastaların %1'inde MDS/AML meydana gelmiş ve vakaların %41'inde ölümle sonuçlanmıştır. İnsidansın, daha önce 2 veya daha fazla platin kemoterapi almış ve 75 aylık sağkalım takibinin ardından gBRCAmut ile tekrarlayan yumurtalık kanseri olan hastalarda daha yüksek olduğu görülmüştür. Daha önce platin ajanlarla kemoterapi almış tüm hastaların MDS/AML gelişimine katkıda bulunan potansiyel faktörleri vardır. Birçoğu ayrıca diğer DNA'ya zarar veren ajanlar ve radyoterapi almıştır. Raporların
çoğu gBRCAmut taşıyıcılarına aittir. Hastalardan bazılarının önceden kanser veya kemik iliği baskılanması öyküsü olduğu bilinmektedir.
PRIMA çalışmasında, MDS/AML insidansı ZEJULA alan hastalarda % 0,8 ve plasebo alan
hastalarda % 0,4 olmuştur.
Daha önce en az iki basamak platin kemoterapi almış nükseden yumurtalık kanseri hastalarında yapılan NOVA çalışmasında, 75 aylık takipte genel MDS/AML insidansı ZEJULA alan hastalarda % 3,8 ve plasebo alan hastalarda % 1,7 olmuştur. gBRCAmut ve gBRCAmut olmayan kohortlarda, MDS/AML insidansı sırasıyla ZEJULA alan hastalarda % 7,4 ve % 1,7 ve plasebo alan hastalarda %3,1 ve %0,9 olmuştur.
Hipertansiyon
PRIMA'da, ZEJULA ile tedavi edilen hastaların % 6'sında, plaseboyla tedavi edilen hastaların
% 1'inde 3./4. derece hipertansiyon görülmüştür ve ilk dozdan ilk başlangıca kadar geçen medyan süre 50 gün (aralık: 1 ila 589 gün), medyan süre 12 gündür (aralık: 1 ila 61 gün). Niraparib alan hastaların % 0'ında hipertansiyon nedeniyle tedavi bırakılmıştır.
NOVA'da, ZEJULA ile tedavi edilen hastaların yaklaşık % 19,3'ünde herhangi bir derecede hipertansiyon görülmüştür. Hastaların % 8,2'sinde 3./4. derece hipertansiyon görülmüştür. Hipertansiyon anti-hipertansif tıbbi ürünlerle kolayca yönetilmiştir. Hastaların % 1'inden azında hipertansiyon nedeniyle tedavi bırakılmıştır.
Pediyatrik popülasyon:
Pediyatrik hastalarda hiçbir çalışma yapılmamıştır.
Şüpheli advers reaksiyonların raporlanması
Ruhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar / risk dengesinin sürekli olarak izlenmesine olanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu Türkiye Farmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e- posta:tufam@titck.gov.tr; tel: 0 800 314 00 08; faks: 0 312 218 35 99).
4.9. Doz aşımı ve tedavisi
ZEJULA doz aşımı durumu için spesifik bir tedavi yoktur ve doz aşımı semptomları belirlenmemiştir. Doz aşımı durumunda, hekimler genel destekleyici önlemleri izlemeli ve semptomatik tedavi uygulamalıdır.
5. FARMAKOLOJİK ÖZELLİKLER
5.1. Farmakodinamik özellikler
Farmakoterapötik grup: Diğer antineoplastik ajanlar, Poli (ADP-riboz) polimeraz (PARP) inhibitörleri
ATC kodu: L01XK02
Etki mekanizması ve farmakodinamik etkiler
Niraparib, DNA onarımında rol oynayan poli(ADP-riboz) polimeraz (PARP) enzimleri PARP- 1 ve PARP-2'nin bir inhibitörüdür. İn vitro çalışmalarda, niraparible indüklenen sitotoksisiteye PARP enzimatik aktivitesinin inhibisyonu ve DNA hasarı, apoptoz ve hücre ölümü ile sonuçlanan PARP-DNA komplekslerinin oluşumunda artışın dahil olabileceği gösterilmiştir. Meme kanseri (BRCA) 1 ve 2 tümör baskılayıcı genlerinde eksiklik olan veya olmayan tümör
hücre hatlarında artan niraparib kaynaklı sitotoksisite gözlenmiştir. Farelerde büyütülen ortotopik yüksek dereceli seröz over kanseri hasta kaynaklı ksenograft tümörlerinde (PDX) niraparibin; BRCA 1 ve 2 mutantlarında, homolog rekombinasyon (HR) eksikliği olan BRCA vahşi tipte ve tespit edilebilir HR eksikliği olmayan BRCA vahşi tip tümörlerde tümör büyümesini azalttığı görülmüştür.
Klinik etkililik ve güvenlilik
Birinci basamak over kanseri idame tedavisi
PRIMA, Faz 3 çift kör, plasebo kontrollü bir çalışmadır ve birinci basamak platin temelli kemoterapiye tam veya kısmi yanıt veren hastalar (n=733) 2:1 oranında ZEJULA veya eşleştirilmiş plaseboya randomize edilmiştir. PRIMA, 475 hastada (317'si niraparib koluna, 158'i plasebo koluna randomize edilmiştir) devam eden 28 günlük döngülerde günde tek doz 300 mg başlangıç dozuyla başlatılmıştır. PRIMA'nın başlangıç dozu Protokolün 2. Değişikliği ile değiştirilmiştir. Bu noktadan sonra, bazal vücut ağırlığı ≥77 kg ve bazal trombosit sayımı ≥ 150.000/µL olan hastalara günlük ZEJULA 300 mg (3×100 mg kapsül) (n=34) veya plasebo (3 kapsül) (n=21) tedavisi uygulanırken, bazal vücut ağırlığı <77 kg ve bazal trombosit sayımı < 150.000/µL olan hastalara günlük ZEJULA 200 mg (2×100 mg kapsül) (n=122) veya plasebo (2 kapsül) (n=61) tedavisi uygulanmıştır.
Hastalar birinci basamak platin temelli kemoterapi yanı sıra ameliyat oldu ise, ameliyatın tamamlanmasından sonra randomize edilmiştir. Gönüllüler kemoterapinin son döngüsünün ilk gününden sonraki 12 hafta içinde randomize edilmiştir. Gönüllülere ≥6 ve ≤9 platin temelli tedavi döngüsü uygulanmıştır. Tümör hacminin azaltıldığı ara ameliyatlardan sonra, gönüllülere ≥2 postoperatif platin temelli tedavi döngüsü uygulanmıştır. Kemoterapiyle birlikte bevasizumab verilen ama idame tedavisi olarak bevasizumab alamayan hastalar çalışma dışında tutulmamıştır. Hastalar ZEJULA dahil olmak üzere geçmiş PARP inhibitörü (PARPi) tedavisi görmemiş olmalıdır. Neoadjuvan kemoterapi ve sonrasında tümör hacminin azaltıldığı ara ameliyat geçiren hastalarda görünür rezidüel hastalık olabilir veya rezidüel hastalık olmayabilir. Hastalığı 3. evrede olan ve tümör hacminin azaltıldığı primer ameliyattan sonra tam sitoredüksiyon uygulanan (yani görünür rezidüel hastalığı olmayan) hastalar çalışmanın dışında bırakılmıştır. Randomizasyon, birinci basamak platin rejimi sırasında verdiği en iyi yanıt (tam yanıt ya da kısmi yanıt), neoadjuvan kemoterapi (NAKT) (Evet ya da Hayır) ve homolog rekombinasyon eksikliği (HR eksikliği) durumuna [pozitif (HR eksikliği) ya da negatif (HR eksikliği yok) veya belirlenememiş] göre tabakalaştırılmıştır. HR eksikliği testi, ilk tanı tarihinde alınan tümör dokusunda yapılan HR eksikliği testi kullanılarak yapılmıştır. CA-125 düzeyleri hastanın birinci basamak tedavisi sırasında normal aralıkta (veya >% 90 CA- 125 düşüşü) olmalı ve en az 7 gün boyunca stabil devam etmelidir.
Hastalar 1. döngü/1. günde (C1/D1), devamlı 28 günlük döngülerde günde tek doz uygulanan ZEJULA 200 veya 300 mg veya karşılık gelen plasebo ile tedaviyle başlamıştır. Her bir döngüde klinik ziyaretleri gerçekleştirilmiştir (4 hafta ±3 gün).
Birincil sonlanım noktası, RECIST'e (versiyon 1.1) kör bağımsız merkezi inceleme (BICR) ile belirlenen progresyonsuz (ilerlemesiz) sağkalımdır (PFS). Genel sağkalım (OS) anahtar ikincil hedeftir. PFS testi, ilki HR eksikliği olan popülasyonda ve sonra genel popülasyonda olmak üzere hiyerarşik olarak yapılmıştır: Medyan yaş 62'dir ve ZEJULA'ya randomize edilen hastalarda 32 ila 85 yaş aralığında, plaseboyla randomize edilen hastalarda 33 ila 88 yaş aralığındadır. Tüm hastaların % 89'u beyazdır. ZEJULA ile randomize edilen hastaların % 69'u, plaseboyla tedavi edilen hastaların % 71'inin çalışmanın başlangıcındaki ECOG skoru 0'dır. Genel popülasyonda, hastaların % 65'inde 3. evre hastalık, % 35'inde 4. evre hastalık vardır. Genel popülasyonda, çoğu hastanın (≥% 80) primer tümör bölgesi overdir; çoğu hastada (>% 90) seröz histolojiye sahip tümör vardır. Hastaların % 67'si neoadjuvan kemoterapi NAKT
almıştır. Hastaların % 69'u birinci basamak platin temelli kemoterapiye tam yanıt vermiştir. Niraparib verilen toplam 6 hasta over kanseri için önceden bevasizumab tedavisi görmüştür.
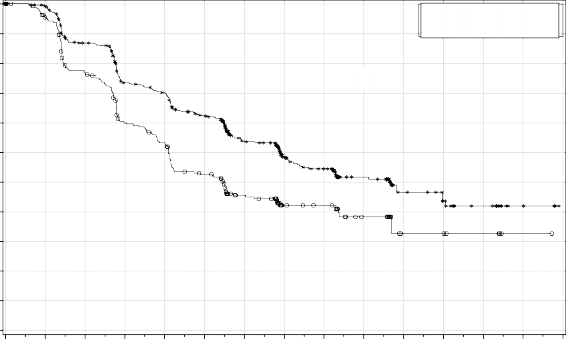
PRIMA çalışmasında, HR (Homolog Rekombinasyonu) eksikliği (HR eksikliği) olan ve genel popülasyonda plaseboyla karşılaştırıldığında ZEJULA'ya randomize edilen hastalarda PFS'de istatistiksel açıdan anlamlı iyileşme görülmüştür (Tablo 5, Şekil 1 ve 2).
İkincil etkililik sonlanım noktaları arasında, ilk izleyen tedaviden sonra PFS (PFS2) ve OS bulunmaktadır (Tablo 5).
Tablo 5: Etkililik sonuçları - PRIMA (BICR belirlemesine göre)
| HR eksikliği olan popülasyon | Genel popülasyon | ||
ZEJULA (N=247) | plasebo (N=126) | ZEJULA (N=487) | plasebo (N=246) | |
Medyan PFS (% 95 GA) | 21,9 (19,3; NE) | 10,4 (8,1; 12,1) | 13,8 (11,5; 14,9) | 8,2 (7,3; 8,5) |
Tehlike oranı (% 95 GA) | 0,43 (0,31; 0,59) |
| 0,62 (0,5; 0,76) |
|
p değeri | <0,0001 |
| <0,0001 |
|
| ||||
PFS2 Tehlike oranı (% 95 GA) | 0,84 (0,485; 1,453) |
| 0,81 (0,577; 1,139) |
|
| ||||
OS* Tehlike oranı (% 95 GA) | 0,61 (0,265; 1,388) |
| 0,7 (0,44; 1,11) |
|
PFS = progresyonsuz sağkalım; GA = güven aralığı; NE = değerlendirilemez; OS = genel
sağkalım; PFS2 = sonraki ilk tedaviden sonra progresyonsuz sağkalım.
* Primer PFS analizi sırasında, genel popülasyonda ZEJULA alan hastaların %84'ünde, plasebo alan hastaların % 77'sinde randomizasyondan iki yıl sonra tahmini sağkalım.
PFS2 ve OS verileri henüz olgunlaşmamıştır.
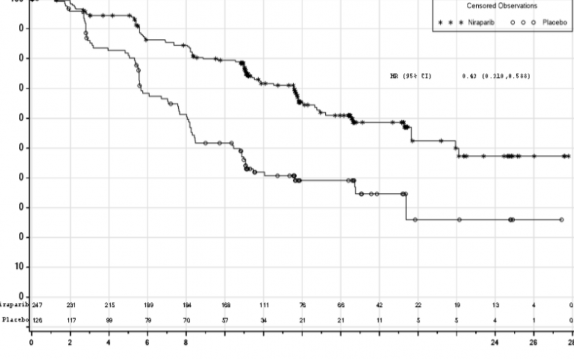
Şekil 1: HR eksikliği olan tümörlere sahip hastalarda progresyonsuz sağkalım - PRIMA (ITT popülasyonu, N=373)
Şekil 2: Genel popülasyonda progresyonsuz sağkalım – PRIMA (ITT popülasyonu, N=733)
![]()
![]()
Alt grup analizleri
Homolog Rekombinasyon (HR) eksikliği olan popülasyonda, BRCA (+) over kanserli hastalardan oluşan alt grupta 0,4 tehlike oranı (% 95 GA 0,27; 0,62) gözlenmiştir (N=223). HR eksikliği BRCA (-) hastalardan oluşan alt grupta (N=150), 0,5 tehlike oranı (%95 GA 0,31; 0,83) gözlenmiştir. HR eksikliği olmayan popülasyonda (N=249); 0,68 tehlike oranı (% 95 GA 0,49; 0,94) gözlenmiştir.
Bazal vücut ağırlığı veya trombosit sayımına göre 200 veya 300 mg ZEJULA uygulanan hastaların keşifsel alt grup analizlerinde, benzer etkililik gözlenmiştir (araştırmacının değerlendirdiği PFS) ve tehlike oranı HR eksikliği olan popülasyonda 0,54 (% 95 GA 0,33; 0,91) ve genel popülasyonda 0,68'dir (% 95 GA 0,49; 0,94). HR eksikliği olmayan alt grupta, 200 mg dozunun tedavi etkisinin 300 mg dozuyla karşılaştırıldığında daha düşük olduğu görülmüştür.
Platine duyarlı rekürren (tekrarlayan) over kanseri idame tedavisi
İdame tedavisi olarak niraparib güvenliliği ve etkililiği, nükseden ağırlıklı olarak yüksek derecede seröz epitelyal over, fallop tüpü veya birincil periton kanseri hastası olan ve altı aydan uzun süre sondan önceki platin temelli tedavilerine kadar tam yanıt (CR) veya kısmi yanıt (PR) tanımlarına göre platin duyarlı hastalarda gerçekleştirilen Faz 3 randomize, çift kör, plasebo kontrollü uluslararası bir çalışmada (NOVA) incelenmiştir. Hastaların niraparib tedavisine uygun olması için son platin temelli kemoterapinin tamamlanmasından sonra yanıt vermesi (CR veya PR) gerekmektedir. Son platin tedavisinden sonra CA 125 seviyeleri normal (veya CA 125'te başlangıca göre > %90 azalma) olmalı ve en az 7 gün boyunca stabil kalmalıdır. Hastalar daha önce ZEJULA dahil olmak üzere hiçbir PARPi tedavisi almamış olmalıdır. Uygun hastalar germline BRCA (gBRCA) mutasyon testinin sonucuna göre iki kohorttan birine atanmıştır. Her kohort içinde, hastalar 2:1 oranında niraparib ve plaseboya randomize edilmiştir. Hastalar randomizasyondan önce gBRCA analizi için alınan kan numunelerine göre gBRCA mutasyonlu kohortuna atanmıştır. Tümör BRCA (tBRCA) mutasyonu ve HR eksikliği testleri, ilk tanı konduğu tarihte veya tekrar gerçekleşme tarihinde alınan tümör dokusu üzerinde yapılan HR eksikliği testi kullanılarak gerçekleştirilmiştir.
Tüm kohortlardaki randomizasyon, çalışmaya katılmadan önceki platin tedavisinden sonra ilerlemeye kadar geçen zamana (6 ila <12 ay ve ≥12 ay); sondan önceki veya son platin rejimiyle birlikte bevasizumab kullanılmadığına ve en yakın geçmişteki platin rejimi sırasındaki en iyi yanıta (tam yanıt ve kısmi yanıt) göre tabakalaştırılmıştır.
Döngü 1/1.gündeki (C1/D1) hastalar , 28 günlük sürekli döngüler halinde günde tek doz uygulanan niraparib 300 mg veya eşleştirilmiş plasebo ile tedaviye başlamıştır. Her döngüde klinik ziyaretleri yapılmıştır (4 hafta ± 3 gün).
NOVA çalışmasında, 1. döngüde hastaların % 48'inin dozuna ara verilmiştir. Hastaların yaklaşık % 47'si 2. döngüde daha düşük bir dozla tekrar başlamıştır.
NOVA çalışmasında niraparible tedavi edilen hastalarda en yaygın olarak kullanılan doz 200 mg'dır.
Progresyonsuz sağkalım (PFS), RECIST 1'e (Solid Tümörlerde Yanıt Değerlendirme, versiyon 1.1) veya klinik belirtiler ve semptomlara ve CA-125 artışına göre belirlenmiştir. PFS, (kemoterapi rejiminin tamamlanmasından 8 haftaya kadar sonraki) randomizasyon tarihinden hastalık ilerlemesi veya ölüme kadar ölçülmüştür.
PFS için primer etkililik analizi, körleştirilmiş merkezi bağımsız değerlendirmeyle belirlenmiştir ve gBRCA mutasyonlu kohort ve gBRCA mutasyonsuz kohort için ayrıca ileriye dönük şekilde tanımlanmış ve değerlendirilmiştir. Genel sağkalım (OS) analizleri, ikincil sonuç ölçütleridir.
İkincil etkililik sonlanım noktaları arasında, kemoterapisiz ara (CFI), izleyen ilk tedaviye kadar geçen süre (TFST), izleyen ilk tedaviden sonra geçen süre (PFS2)ve OS'bulunmaktadır.
gBRCA mutasyonlu (n=203) ve gBRCA mutasyonsuz kohortlarda (n=350) niraparib ve plasebo kolları arasında demografik, bazal hastalık karakteristikleri ve geçmiş tedavi öyküsü genellikle dengelidir. Medyan yaşlar tüm tedaviler ve kohortlarda 57 ila 63 aralığındadır. Tüm kohortlarda çoğu hastadaki (>% 80) birincil tümör bölgesi overlerdir; çoğu hastada (>% 84) seröz histolojiye sahip tümörler vardır. Her iki kohorttaki her iki tedavi kolunda bulunan hastaların yüksek bir oranı daha önce 3 veya daha fazla kemoterapi basamağı almıştır; bunlara gBRCA mutasyonlu ve gBRCA mutasyonsuz kohortlardaki niraparib hastalarının sırasıyla %49'u ve
%34'ü dahildir. Çoğu hasta 18 ila 64 yaşındadır (% 78), beyazdır (% 86) ve ECOG performans skoru 0'dır (% 68).
gBRCA mutasyonlu kohortta, tedavi döngüsünün medyan sayısı niraparib kolunda plasebo koluna göre daha yüksektir (sırasıyla 14 ve 7 döngü). Niraparib grubunda plasebo grubuyla karşılaştırıldığında daha fazla sayıda hasta (sırasıyla % 54,4 ve % 16,9) 12 aydan uzun süre tedaviye devam etmiştir.
Genel gBRCA mutasyonsuz kohortta, tedavi döngüsünün medyan sayısı niraparib kolunda plasebo koluna göre daha yüksektir (sırasıyla 8 ve 5 döngü). Niraparib grubunda plasebo grubuyla karşılaştırıldığında daha fazla sayıda hasta (sırasıyla % 34,2 ve % 21,1) 12 aydan uzun süre tedaviye devam etmiştir.
Çalışma gBRCA mutasyonlu kohortta ve gBRCA mutasyonsuz kohortta plaseboyla karşılaştırıldığında niraparib idame tedavisi için PFS'de istatistiksel açıdan anlamlı iyileşme olan esas amacına ulaşmıştır. Tablo 6 ve Şekil 3'te, primer etkililik popülasyonlarında PFS birincil sonlanım noktası için sonuçlar görülmektedir (gBRCA mutasyonlu kohort ve genel gBRCA mutasyonsuz kohort).
Tablo 6: NOVA çalışmasındaki birincil nesnel sonuçların özeti
| gBRCA mutasyonlu kohort | gBRCA mutasyonsuz kohort | ||
| Niraparib (N=138) | plasebo (N=65) | niraparib (N=234) | plasebo (N=116) |
PFS medyan (% 95 GA*) | 21 (12,9; NR) | 5,5 (3,8; 7,2) | 9,3 (7,2; 11,2) | 3,9 (3,7; 5,5) |
P değeri | <0,0001 | <0,0001 | ||
Tehlike oranı (TO) (Niraparib:plasebo) (% 95 GA*) | 0,27 (0,173; 0,41) | 0,45 (0,338; 0,607) | ||
PFS = progresyonsuz sağkalım;GA = güven aralığı; NE = değerlendirilemez
Estimated Survival Function
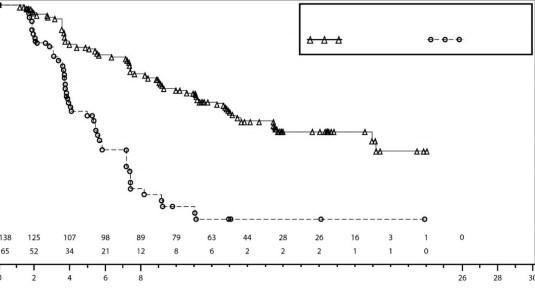
Time since Randomization (Months)
![]()
Şekil 3: IRC değerlendirmesine göre gBRCA mutasyonlu kohortta progresyonsuz sağkalım için Kaplan-Meier grafiği – NOVA (ITT popülasyonu, n=203)
Şekil 4: IRC değerlendirmesine göre gBRCA mutasyonsuz kohortta progresyonsuz sağkalım için Kaplan-Meier grafiği – NOVA (ITT popülasyonu, n=350)

NOVA'da ikincil etkililik sonlanım noktaları
Son analizde, gBRCAmut kohortunda medyan PFS2, niraparib ile tedavi edilen hastalarda 29,9 ay iken, plasebo alan hastalarda 22,7 aydır (HR = 0,70; % 95 GA: 0,50, 0,97). gBRCAmut olmayan kohorttaki medyan PFS2, niraparib ile tedavi edilen hastalarda 19,5 ay iken, plasebo alan hastalarda 16,1 aydır (HR = 0,80; %95 GA: 0,63, 1,02).
Genel sağkalımın son analizinde, gBRCAmut kohortunda (n = 203) medyan OS, niraparib ile tedavi edilen hastalarda 40,9 ay, plasebo alan hastalarda ise 38,1 ay (HR = 0,85; %95 GA: 0,61, 1,20) olduğu görülmüştür. gBRCAmut kohortu için kohort olgunluğu %76'dır. gBRCAmut olmayan kohortta (n = 350) medyan OS, niraparib ile tedavi edilen hastalarda 31.0 ay, plasebo alan hastalarda ise 34.8 ay (HR = 1.06; %95 GA: 0.81, 1.37) olduğu görülmüştür. gBRCAmut olmayan kohort için kohort olgunluğu %79'dur.
Doğrulanmış anket yöntemleriyle (FOSI ve EQ-5D) elde edilen hasta beyanlı sonuç (PRO) verileri, niraparible tedavi edilen hastaların yaşam kalitesiyle ilişkilendirilen ölçümlerde plasebodan farklı olmadığını göstermiştir.
Pediyatrik popülasyon
Avrupa İlaç Kurumu (EMA), ZEJULA ile over karsinomda (rabdomiyosarkoma ve germ hücreli tümörler hariç) pediyatrik popülasyon tüm alt gruplarında çalışma sunma yükümlülüğü konusunda muafiyet getirmiştir.
5.2. Farmakokinetik özellikler
Genel özelliklerEmilim:
Açlık koşullarında tek doz 300 mg niraparib uygulandıktan sonra, niraparib plazmada 30 dakika içinde ölçülebilir düzeydedir ve niraparib için ortalama pik plazma konsantrasyonuna (C) yaklaşık 3 saatte ulaşılmıştır [804 ng/mL (% VK: % 50,2]. Günde bir kez 30 mg ila 400 mg aralığında çoklu oral niraparib dozlarından sonra, niraparib birikimi yaklaşık 2 ila 3 kattır.
Niraparib dozu 30 mg'dan 400 mg'a arttırıldığında niraparibe sistemik maruziyetler (Cve EAA) dozla orantılı şekilde artmıştır. Niraparibin mutlak biyoyararlanımı yaklaşık % 73'tür ve ilk geçiş etkisinin minimal olduğuna işaret etmektedir. Niraparibin popülasyon farmakokinetik analizinde, biyoyararlanım açısından bireyler arası değişkenliğin değişim katsayısı (VK) % 31'dir.
300 mg niraparib uygulamasından sonra, eşzamanlı yüksek yağlı öğün tüketilmesi niraparibin farmakokinetiğini anlamlı düzeyde etkilememiştir.
Tablet ve kapsül formülasyonlarının biyoeşdeğer olduğu gösterilmiştir. Açlık koşulları altında kitle tümörü olan 108 hastada bir 300 mg tablet veya üç 100 mg niraparib kapsülünün uygulanmasını takiben, tablet için Cmaks, EAAve EAAiçin kapsüllere kıyasla geometrik ortalama oranlarının %90 güven aralıkları biyoeşdeğerlik limitlerinde olmuştur (0,80 ve 1,25).
Dağılım:
Niraparib insan plazmasında proteinlere orta düzeyde (% 83), esas olarak serum albüminle
bağlanmaktadır. Niraparibin popülasyon farmakokinetik analizinde, kanser hastalarında (VK
% 116) görülen dağılım hacmi (V/F) 1,331 L'dir (70 kg hasta ağırlığına göre) ve niraparibin
dokulara yaygın olarak dağıldığına işaret etmektedir.
Biyotransformasyon:
Niraparib ağırlıklı olarak karboksilesterazlar (CE) tarafından metabolize edilir ve majör inaktif metabolit olan M1 oluşur. Bir kütle denkliği çalışmasında, M1 ve M10 (sonradan oluşturulan M1 glukuronidleri) dolaşımdaki majör metabolitlerdir.
Eliminasyon:
Tek oral 300 mg niraparib dozundan sonra, niraparibin ortalama terminal yarı ömrü (t) 48 ila 51 saattir (yaklaşık 2 gün). Popülasyon farmakokinetik analizinde, niraparibin görünür toplam klirensi (CL/F) kanser hastalarında 16,5 L/h'dir (VK % 23,4).
Niraparib esas olarak hepatobiliyer ve renal yollardan elimine edilmektedir. Tek 300 mg [C]- niraparib dozu uygulandıktan sonra, dozun ortalama % 86,2'si (aralık % 71 ila % 91) 21 günlük bir sürede idrar ve dışkıda geri kazanılmıştır. İdrardaki radyoaktivite geri kazanımı dozun % 47,5'i (aralık % 33,4 ila % 60,2) ve dışkıda % 38,8'idir (aralık: % 28,3 ila % 47). 6 günlük bir sürede toplanan havuzlanmış numunelerde, dozun % 40'ı idrarda ağırlıklı şekilde metabolit olarak, dozun % 31,6'sı dışkıda esas olarak değişmemiş niraparib olarak geri kazanılmıştır.
Hastalardaki karakteristik özellikler Böbrek yetmezliği
Popülasyon farmakokinetik analizinde, hafif (kreatinin klirensi 60-90 mL/dk.) ve orta dereceli (30-60 ml/dk.) böbrek yetmezliği olan hastalarda, böbrek işlevi normal (hafif yetmezlikte % 7- 17 daha yüksek maruziyet ve orta dereceli yetmezlikte % 17-38 daha yüksek maruziyet) olan bireylerle karşılaştırıldığında niraparib klirensi hafifçe düşmüştür. Maruziyet farkının doz ayarlaması gerektirdiği düşünülmemektedir. Klinik çalışmalarda önceden şiddetli böbrek yetmezliği veya son dönem böbrek hastalığı olan ve hemodiyaliz gören hiçbir hasta belirlenmemiştir (bkz. Bölüm 4.2).
Karaciğer yetmezliği
Hastalarda yapılan klinik çalışmalarda elde edilen verilerin popülasyon farmakokinetik analizinde, önceden var olan hafif dereceli karaciğer yetmezliği (n=155) niraparibin klirensini etkilememiştir. Karaciğer yetmezliğinin derecesini sınıflandırmak için NCI-ODWG kriterlerini kullanan, kanser hastalarına yönelik bir klinik çalışmada, tek seferde 300 mg'lık dozun uygulanmasını takiben, orta dereceli karaciğer yetmezliği olan hastalarda (n=8) niraparib EAA, normal karaciğer fonksiyonuna sahip hastaların (n=9) 1,56 (%90 GA: 1,06 ila 2,30) katı olmuştur. Orta derecede karaciğer yetmezliği olan hastalarda niraparib doz ayarlaması önerilmektedir (bkz. Bölüm 4.2). Orta derecede karaciğer yetmezliğinin niraparib Cveya niraparib protein bağlanması üzerinde bir etkisi olmamıştır. Niraparibin farmakokinetiği şiddetli karaciğer yetmezliği olan hastalarda değerlendirilmemiştir (bkz. Bölüm 4.2 ve 4.4).
Kilo, yaş ve ırk
Popülasyon farmakokinetik analizinde kilo artışının niraparibin dağılım hacmini arttırdığı görülmüştür. Ağırlığın niraparib klirensi veya genel maruziyet üzerinde hiçbir etkisi görülmemiştir. Farmakokinetik açıdan vücut ağırlığına göre doz ayarlaması gerekmemektedir.
Popülasyon farmakokinetik analizinde yaş artışının niraparib klirensini azalttığı görülmüştür. 91 yaşındaki bir hastadaki ortalama maruziyetin, 30 yaşındaki bir hastadan % 23 daha yüksek olacağı öngörülmüştür. Yaşın etkisinin doz ayarlaması gerektirdiği düşünülmemektedir.
Irkın niraparibin farmakokinetiği üzerindeki etkisiyle ilgili bir sonuca varmak için, ırklarda yeterli veri bulunmamaktadır.
Pediyatrik popülasyon
Niraparibin pediyatrik hastalardaki farmakokinetiğini incelemek için henüz çalışma yapılmamıştır.
5.3. Klinik öncesi güvenlilik verileri
Güvenlilik farmakolojisi
İn vitro ortamda, niraparib insan maruziyet düzeylerinin altındaki konsantrasyonlarda dopamin taşıyıcısı DAT'ı inhibisyona uğratmıştır. Farelerde, tek doz niraparib, korteksteki hücre içi dopamin ve metabolit düzeylerini arttırmıştır. Farelerde yapılan iki tek doz çalışmasından birinde lokomotor aktivitede düşüş görülmüştür. Bu bulguların klinik önemi bilinmemektedir. Beklenen terapötik maruziyet düzeylerine benzer veya bunların altında olması beklenen tahmini MSS Merkezi Sinir Sistemi (MSS) maruziyeti düzeylerinde sıçanlar ve köpeklerde yapılan yinelenen dozlu toksisite çalışmalarında davranışsal ve/veya nörolojik parametreler üzerinde hiçbir etki gözlemlenmemiştir.
Yinelenen doz toksisitesi
Klinik olarak görülen maruziyet düzeylerinde sıçanlar ve köpeklerde spermatojenezde düşüş gözlenmiştir ve dozun kesilmesini izleyen 4 hafta büyük oranda geri dönüşlüdür.
Genotoksisite
Niraparib bakteriyel ters mutasyon tayininde (Ames) mutajenik değildir ama in vitro memeli kromozom aberasyon analizinde ve in vivo sıçan kemik iliği mikronükleus tayininde klastojeniktir. Bu klastojenite, niraparibin esas farmakolojisinden kaynaklanan genomik instabiliteyle tutarlıdır ve insanlarda genotoksisite potansiyeli olduğuna işaret etmektedir.
Üreme toksikolojisi
Niraparible üreme ve gelişim toksisitesi çalışması yapılmamıştır. Karsinojenisite
Niraparible karsinojenisite çalışması yapılmamıştır.
6. FARMASÖTİK ÖZELLİKLER
6.1. Yardımcı maddelerin listesi
Kapsül içeriği:
Magnezyum stearat
Laktoz monohidrat (sığır sütünden elde edilir)
Baskılı sert jelatin kapsül kabuğu (jelatin sığırdan elde edilmektedir) Kapsül kabuğu ve baskı mürekkebi:
Titanyum dioksit (E 171)
Jelatin
SİYAH SW-9049 mürekkebi Şellak
Susuz alkol İzopropil alkol Butil alkol
Propilen glikol (E 1520) Arıtılmış su
Güçlü amonyak çözeltisi Potasyum hidroksit (E 525) Siyah demir oksit (E 172) FD&C mavi#1
FD&C kırmızı #3
Tartrazine (E 102)
BEYAZ SB-0007P mürekkebi Sodyum hidroksit (E 524) Povidon (E 1201)
6.2. Geçimsizlikler
Geçerli değildir.
6.3. Raf ömrü
36 ay.
6.4. Saklamaya yönelik özel tedbirler
25°C'nin altındaki oda sıcaklığında saklayınız.
6.5. Ambalajın niteliği ve içeriği
84 × 1, 56 × 1 ve 28 × 1 sert kapsül içeren kutularda Aclar/PVC/alüminyum folyoda delikli
birim doz blisterler.
Tüm ambalaj boyutları pazara sunulmamış olabilir.
6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler
Kullanılmamış olan ürünler ya da atık materyaller “Tıbbi Atıkların Kontrolü Yönetmeliği†ve Ambalaj Atıklarının Kontrolü Yönetmeliğiâ€ne uygun olarak imha edilmelidir.
 Omurilik zedelenmeleri
Omurilik zedelenmesini takip eden birkaç gün içinde, hiçkimse hasarin ne kadar olacagini tahmin edemez. Buradaki sorun, omuriligin herhangi bir zedelenmesinden hemen sonra, bir omurilik sokunun olusmasidir.
Omurilik zedelenmeleri
Omurilik zedelenmesini takip eden birkaç gün içinde, hiçkimse hasarin ne kadar olacagini tahmin edemez. Buradaki sorun, omuriligin herhangi bir zedelenmesinden hemen sonra, bir omurilik sokunun olusmasidir. |
 Tiroid Kanseri
En sık görülen tiroid kanseri türü olan papiller tiroid kanseri, tüm tiroid kanserlerinin yaklaşık %70'ini oluşturur.
Tiroid Kanseri
En sık görülen tiroid kanseri türü olan papiller tiroid kanseri, tüm tiroid kanserlerinin yaklaşık %70'ini oluşturur. |
 |
HIV ve Aids HIV, Human Immunodeficiency Virus’dür (İnsanlarda Bağışıklık Sistemini Bozan Virüsdür). Bu virüs AIDS hastalığına sebep olur. |
 |
Dış Gebelik Dış gebelik, her 100 gebelikten birini etkileyen, sık görülen ve ölüme sebep olabilecek bir durumdur. Bu, döllenen yumurta, rahimin dışına yerleşirse, oluşan bir durumdur. Gebelik ilerledikçe, ağrıya ve kanamalara sebep olur. |
 |
Sırt Ağrısı Sırt ağrısı birden bire ortaya çıkıp şiddetli (akut) olabilir veya zamanla gelişip daha uzun süreli sorunlara (kronik) neden olabilir. |
İLAÇ GENEL BİLGİLERİ
Glaxo Smith Kline İlaçları San.Ve Tic.A.Ş
| Satış Fiyatı | 100457.98 TL [ 25 Apr 2025 ] |
| Önceki Satış Fiyatı | 100457.98 TL [ 18 Apr 2025 ] |
| Original / Jenerik | Original İlaç |
| Reçete Durumu | Normal Reçeteli bir ilaçdır. |
| Barkodu | 8699522154210 |
| Etkin Madde | Niraparib |
| ATC Kodu | L01XK02 |
| Birim Miktar | 100 |
| Birim Cinsi | MG |
| Ambalaj Miktarı | 56 |
| Antineoplastik ve İmmünomodülatör Ajanlar > Diğer Kanser İlaçları |
| İthal ( ref. ülke : Yunanistan ) ve Beşeri bir ilaçdır. |